


Abstract
Tumor-specific targeting ligands were recently exploited to deliver both imaging and therapeutic agents selectively to cancer tissues in vivo. Because the cholecystokinin 2 receptor (CCK2R) is overexpressed in various human cancers (e.g., lung, medullary thyroid, pancreatic, colon, and gastrointestinal stromal tumors) but displays limited expression in normal tissues, natural ligands of CCK2R were recently explored for use in the imaging of CCK2R-expressing cancers. Unfortunately, the results from these studies revealed not only that the peptidic CCK2R ligands were unstable in vivo but also that the ligands that mediated good uptake by tumor tissues also promoted a high level of retention of the radioimaging agent in the kidneys, probably because of capture of the conjugates by peptide-scavenging receptors. In an effort to reduce the normal organ retention of CCK2R-targeted drugs, we synthesized a nonpeptidic ligand of CCK2R and examined its specificity for CCK2R both in vitro and in vivo. Methods: Nonpeptidic agonists and antagonists of CCK2R described in the literature were evaluated for their affinities and specificities for CCK2R. Z-360, a benzodiazepine-derived CCK2R antagonist with subnanomolar affinity, was selected for complexation to 99mTc via multiple spacers. After synthesis and purification, 4 complexes with different physicochemical properties were evaluated for binding to CCK2R-transfected HEK 293 cells. The best conjugate, termed CRL-3–99mTc, was injected into mice bearing CCK2R tumor xenografts and examined by γ scintigraphy and SPECT/CT. The uptake of the conjugate in various organs was also quantified by tissue resection and γ counting. Results: CRL-3–99mTc was shown to bind with low nanomolar affinity to CCK2R in vitro and was localized to tumor tissues in athymic nu/nu mice implanted with CCK2R-expressing tumors. At 4 h after injection, tumor uptake was measured at 12.0 ± 2.0 percentage injected dose per gram of tissue. Conclusion: Because the uptake of CRL-3–99mTc by nonmalignant tissues was negligible and retention in the kidneys was only transient, we suggest that CRL-3–99mTc may be a useful radioimaging agent for the detection, sizing, and monitoring of CCK2R-expressing tumors.
- tumor-targeted therapy
- radioimaging of tumors
- cholecystokinin 2/gastrin receptor
- cancer imaging
- nonpeptide
- antagonist
The overexpression of specific receptors on pathologic cell surfaces has been exploited for the functional imaging of disease activity in both humans (1–4) and animal models of human disease (5–7). One such receptor, the cholecystokinin (CCK) 2 receptor (gastrin receptor; CCK2R), is a G-protein–coupled receptor that is normally expressed in the central nervous system and the cells of the gastric mucosa (8). In the central nervous system, CCK regulates appetite, pain, anxiety, and wakefulness (9,10). In the gastrointestinal tract, the closely related gastrin peptides modulate the growth and differentiation of the gastric mucosa and the secretion of acid, bile, and digestive enzymes (11).
CCK2R is also overexpressed in several cancers, in which it has been reported to mediate the stimulation of cancer cell growth and migration (12,13), metastasis (14), and survival (15). Cancers shown to upregulate CCK2R include malignancies of the thyroid (16); lung (17); pancreas, ovary, and brain (18); stomach and gastrointestinal stroma (19); and colon (20). Recently, a novel splice variant of CCK2R that is tumor-specific and often coexpressed with the normal CCK2R splice form in malignant cells (20,21) was also identified. This splice variant, designated CCK2i4svR, results from the retention of intron 4, which yields a modified receptor containing a 64-amino-acid insertion in the third cytoplasmic domain. Importantly, this modified CCK2R is constitutively active (22), leading to increased basal cell proliferation and enhanced tumorigenicity, even in the absence of a ligand (23).
The limited expression of CCK2R in normal tissues, coupled with the overexpression of CCK2R and CCK2i4svR in various cancers, has rendered the receptor an attractive candidate for targeted imaging and therapy of malignant disease. Indeed, radioactive conjugates of CCK and gastrin have already been explored for use in the imaging of CCK2R-overexpressing tumors in mice (24–26). Unfortunately, the results from these studies revealed inadequacies that suggested the need for improved CCK2R targeting ligands. First, gastrin peptides that exhibit high levels of tumor uptake are also compromised by high levels of retention in the kidneys, whereas CCK peptides that display low levels of kidney uptake also show little retention in tumors (27–29). As a consequence, the development of CCK2R-targeted peptides for radiotherapeutic applications has been hampered by concerns that the targeted radiation may simultaneously cause damage to the kidneys. Second, the fact that gastrin and CCK, like most other peptide hormones, are rapidly degraded by peptidases in the serum and on cell surfaces (30) has raised concern that attached cytotoxic cargos may be released during circulation, resulting in their nonspecific distribution into CCK2R-negative tissues. Third, CCK- and gastrin-related peptides are prone to oxidation at a methionine residue, leading to the loss of receptor binding affinity and their consequent dissemination into receptor-negative tissues (30). Finally, CCK- and gastrin-related peptides can stimulate the growth, proliferation, and survival of cancer cells, rendering the tumor-targeted imaging agent an unwanted promoter of tumor growth.
To avoid the unwanted consequences associated with the use of a peptidic agonist for tumor imaging, we designed a nonpeptidic CCK2R targeting ligand and used it to deliver a 99mTc-based radioimaging agent to CCK2R-expressing cancers in tumor-bearing mice. We report here that this CCK2R targeting ligand promoted the tumor-specific accumulation of an attached radioimaging agent with a low level of accumulation in normal tissues, suggesting a possible use for the targeted imaging agent in the detection, localization, and staging of CCK2R-expressing tumors.
MATERIALS AND METHODS
Chemicals
All amino acids and resins were purchased from Chem Impex Intl. Sodium pertechnetate (99mTc) was obtained from Cardinal Health. HC Matrigel was purchased from BD Biosciences. Stannous chloride, sodium glucoheptonate, diisopropylethylamine (DIPEA), piperidine, dimethylformamide (DMF), isopropyl alcohol, and all other reagents were purchased from Sigma-Aldrich.
Cell Culture and Animal Husbandry
Two HEK 293 cell lines, one transfected with CCK2R (HEK 293 CCK2R cells) and the other expressing CCK2i4svR (HEK 293 CCK2i4svR cells), were kind gifts from Dr. Mark Hellmich (University of Texas Medical Branch). Cells were cultured in Dulbecco modified Eagle medium (GIBCO) supplemented with 10% fetal bovine serum, G418 disulfate (Sigma-Aldrich; 400 μg/mL), and 1% penicillin–streptomycin at 37°C in a humidified 95% air–5% CO2 atmosphere. Athymic female nu/nu mice were purchased from Harlan Laboratories, housed in a sterile environment on a standard 12-h light–dark cycle, and maintained on normal rodent chow. All animal procedures were approved by the Purdue Animal Care and Use Committee in accordance with National Institutes of Health guidelines.
Synthesis of Radioimaging Agents
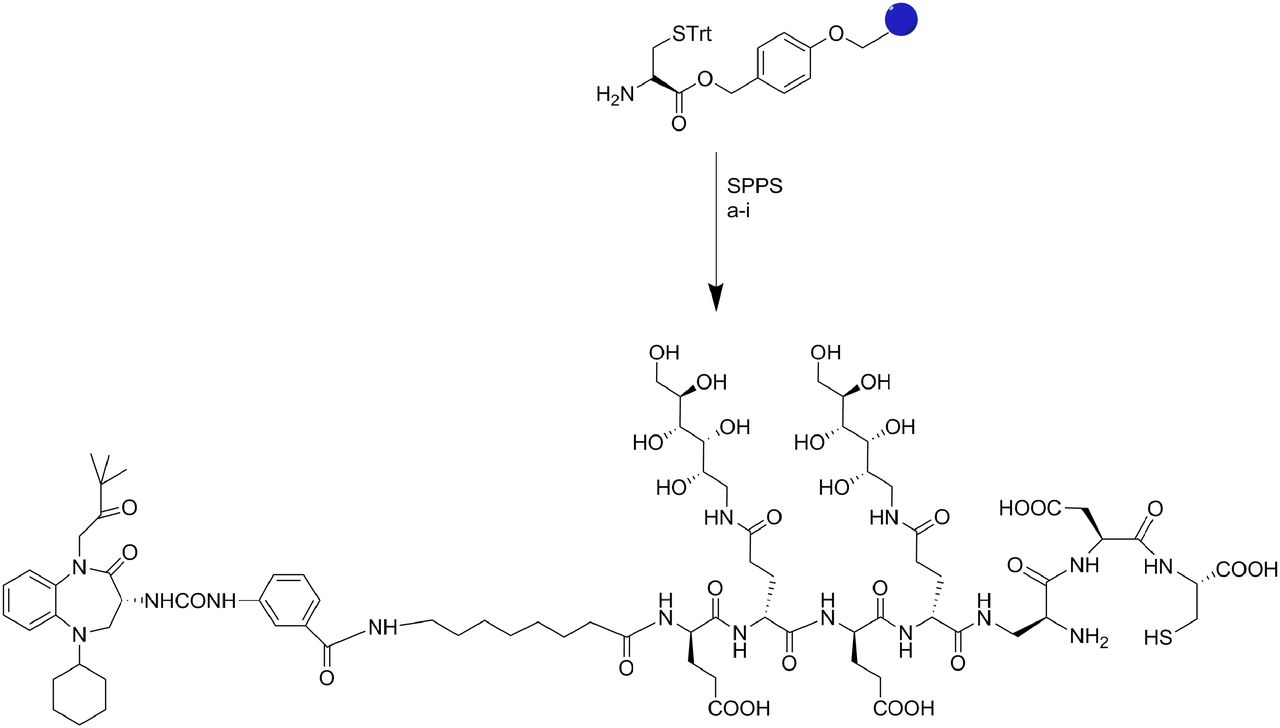
Z-360, a CCK2R antagonist, was synthesized in accordance with previously reported methods (31,32) and linked via the desired spacer to a 99mTc chelating agent after solid-phase synthesis of the chelate–spacer conjugate. For this purpose, the chelator, comprising the peptide sequence β-l-diaminopropionic acid, l-aspartic acid (l-Asp), and l-cysteine (l-Cys), was first prepared by solid-phase methodology (33); the desired spacer was then added (Fig. 1).

Solid-phase synthesis of CRL-3. All reactions were conducted under N2 or Ar at room temperature. Reagents and conditions in reactions a–i were as follows: (a) Fmoc-Asp(OtBu)-OH, HATU, DIPEA, 4 h; (b) (i) 20% piperidine–DMF, 10 min; (ii) Fmoc-diaminopropionic (DAP) acid, HATU, DIPEA, 4 h; (c) (i) 20% piperidine–DMF, 10 min; (ii) 3,4,5,6-di-isopropylidene-1-amino-deoxy(Fmoc-Glu-OH)-d-glucitol, HATU, DIPEA, 4 h; (d) (i) 20% piperidine–DMF, 10 min; (ii) Fmoc-Glu(OtBu)-OH, HATU, DIPEA, 4 h; (e) (i) 20% piperidine–DMF, 10 min; (ii) 3,4,5,6-di-isopropylidene-1-amino-deoxy(Fmoc-Glu-OH)-d-glucitol, HATU, DIPEA, 4 h; (f) (i) 20% piperidine–DMF, 10 min; (ii) Fmoc-Glu(OtBu)-OH, HATU, DIPEA, 4 h; (g) (i) 20% piperidine–DMF, 10 min; (ii) Fmoc-8-amino-octanoic acid, HATU, DIPEA, 4 h; (h) (i) 20% piperidine–DMF, 10 min; (ii) Z-360, HATU, DIPEA, overnight; (i) cleavage in trifluoroacetic acid–H2O–TIPS–EDT (92.5:2.5:2.5:2.5), 30 min. EDT = ethanedithiol; SPPS = solid-phase peptide synthesis; STrt = trityl protected cysteine; TIPS = triisopropylsilane.
Next, the Z-360 ligand was linked to the terminal amine, and the final conjugate was cleaved from the resin and purified as described later. In brief, acid-sensitive Wang resin (Sigma-Aldrich) loaded with 0.106 mmol of H-carbonyl-trityl-l-cysteine [H-l-Cys(Trt)-OH] was reacted with Fmoc-Asp(OtBu)-OH (0.265 mmol), 2-(1H-7-azabenzotriazol-1-yl)–1,1,3,3-tetramethyl uranium hexafluorophosphate (HATU; 0.265 mmol), and diisopropylethylamine (1.06 mmol); this step was followed by the addition of β-l-diaminopropionic acid (0.265 mmol), HATU (0.265 mmol), and diisopropylethylamine (1.06 mmol) to yield the 99mTc chelating moiety. The chelator was then conjugated to Z-360 via various spacers that were selected for their abilities both to render the final conjugate water-soluble and to reduce nonspecific binding to receptor-negative cells. The monomeric components of these spacers were derived from protected amino acids and a peptidosaccharide construct described by others (34). All conjugation reactions were performed under an argon atmosphere. Fmoc-protecting groups were removed after each coupling step under standard conditions (20% piperidine in DMF). Release of the partially deprotected conjugate from the polymeric support was finally accomplished by treatment with a solution of 92.5% trifluoroacetic acid, 2.5% 1,2-ethanedithiol, 2.5% triisopropylsilane, and 2.5% deionized water. This reaction also resulted in the simultaneous removal of all t-butyl–, t-butoxycarbonyl–, and trityl–protecting groups.
The crude product was purified by preparative reverse-phase high-performance liquid chromatography with a gradient mobile phase of 20 mM ammonium acetate buffer and acetonitrile and a solvent gradient of 5% acetonitrile to 80% acetonitrile over 30 min (xTerra C18; Waters; 10 μm; 19 × 250 mm). Elution of the conjugate was monitored at a wavelength of 280 nm, and the identities of the eluted compounds were analyzed by liquid chromatography–mass spectrometry and matrix-assisted laser desorption ionization. Formulation and radiolabeling of the conjugates with 99mTc were performed in accordance with previously described methods (33).
Binding Affinity and Specificity of CCK2R Ligand–99mTc-Labeled Conjugates
HEK 293 CCK2R and HEK 293 CCK2i4svR cells were seeded onto 24-well plates and grown to 80%–90% confluence for 48–72 h. Spent medium in each well was replaced with 0.5 mL of fresh medium containing 0.5% bovine serum albumin, and increasing concentrations of the radioconjugate were added. After incubation for 1 h at 37°C, the cells were rinsed with incubation solution (2 × 1.0 mL) and resuspended in 0.5 mL of 0.25N NaOH before radioactivity was counted with a γ counter. The dissociation constant (Kd) was calculated by plotting cell-bound radioactivity as a function of the concentration of the radiotracer in the medium, with the assumption of a noncooperative single-site-binding equilibrium.
Analysis of Tumor-Bearing Mice
Six-week-old female nu/nu mice were inoculated subcutaneously in their shoulders with either HEK 293 CCK2R or HEK 293 CCK2i4svR cells (in 50% HC Matrigel–Dulbecco modified Eagle medium; 5.0 × 106 cells per mouse) with a 25-gauge needle. The growth of the tumors was measured in 2 perpendicular directions every 2 d with a caliper, and the volumes of the tumors were calculated with the formula 0.5 × L × W2, where L is the measurement of the longest axis and W is the measurement of the axis perpendicular to L, in millimeters. Radiotracer biodistribution studies were performed 12–18 d after tumor cell implantation, when the tumors had reached volumes of approximately 400–500 mm3. The mice were randomly assigned to different treatment groups and injected intravenously with the desired 99mTc-labeled conjugate (10 nmol; 5.55 MBq [150 μCi] in 100 μL of phosphate-buffered saline). At various time points (0.5, 2, 4, 8, and 24 h after injection), the animals were sacrificed by CO2 asphyxiation, and imaging was performed with a KODAK Image Station. The parameters used for radioimaging were as follows: acquisition time, 2 min; f-stop, 4; focal plane, 7; field of view, 200; and binning, 4. For white-light imaging, the parameters were as follows: acquisition time, 0.05 s; f-stop, 11; focal plane, 7; field of view, 200; and no binning. SPECT/CT imaging was performed with micro-SPECT II/CT (MILabs).
Biodistribution of Radioimaging Agents
After radioimaging, each animal was dissected, and selected organs and tissues were collected into preweighed tubes and subjected to γ counting. Counts per minute were decay corrected and converted to percentage injected dose per gram of wet tissue.
RESULTS
Because of the limited expression of CCK2R in normal tissues and the overexpression of CCK2R in various tumors, we elected to develop a CCK2R-targeted 99mTc–chelate complex that would not be encumbered by the specificity and stability problems associated with previous CCK2R-targeted peptidic radioimaging agents. After an extensive literature search, we found a small molecule antagonist, Z-360 (35,36), that binds CCK2R with subnanomolar affinity and high selectivity relative to the cholecystokinin 1 receptor (CCK1R) (Kd, 0.47 nmol/L; selectivity relative to CCK1R, 672). Preferential binding to CCK2R was important because CCK1R is expressed at high levels in several normal tissues (37). Moreover, 99mTc was chosen as the radiolabel because of its widespread availability, low cost, and short half-life (6.02 h).
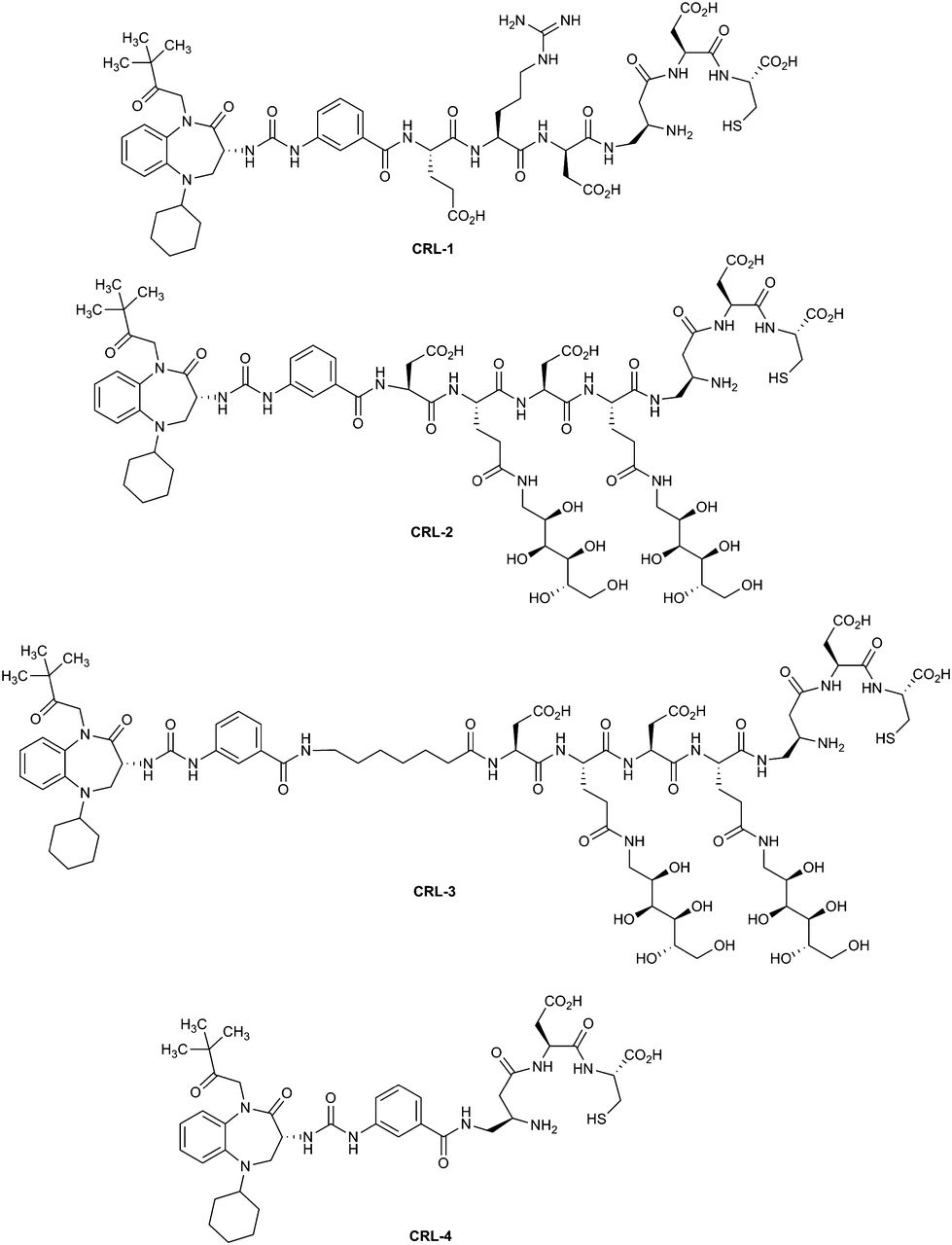
For construction of the optimal CCK2R-targeted radioimaging agent, Z-360 was attached to a previously described 99mTc chelating agent (33) via various spacers designed to both improve water solubility and prevent chelate complex interference with ligand binding. The final conjugates selected for comparison are shown in Figure 2. CCK receptor ligand 1 (CRL-1) contains a simple tripeptide spacer. Because scavenger receptors in the kidneys and liver can bind diverse peptidic conjugates (38,39), the peptide spacer in CRL-1 was replaced with a less readily scavenged peptidosaccharide spacer in CCK receptor ligand 2 (CRL-2) (39). Next, to ensure sufficient separation between the CCK receptor ligand and its tethered radiochelate, CCK receptor ligand 3 (CRL-3) was designed with the same spacer as CRL-2, but an octanoyl moiety was inserted before the peptidosaccharide spacer. Finally, to assist with evaluation of the impact of spacer length, CCK receptor ligand 4 (CRL-4) was prepared with no spacer.

Structures of CCK receptor ligand–targeted radioimaging conjugates.
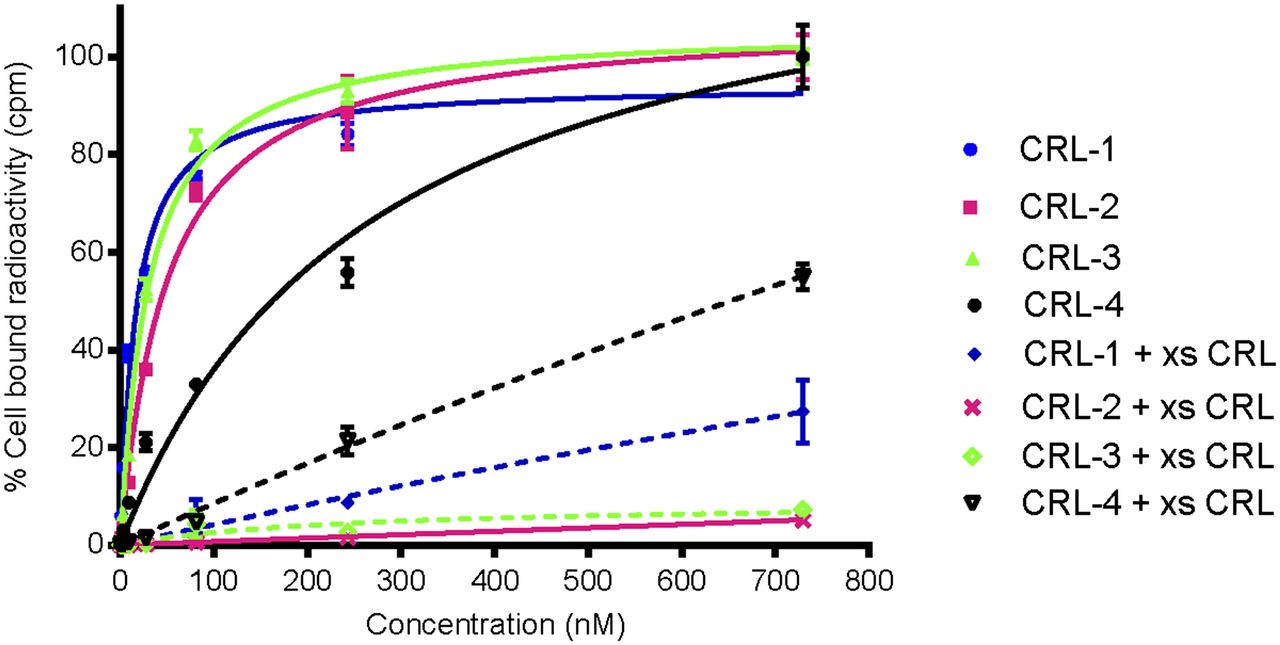
Because the affinity of a ligand for its receptor can often be compromised by attachment to its cargo, the binding affinity of each of the conjugates for both CCK2R and CCK2i4svR was examined in vitro. For this purpose, HEK 293 CCK2R and HEK 293 CCK2i4svR cells were incubated with increasing concentrations of each radioactive conjugate, both in the presence and in the absence of a 100-fold excess of unlabeled CCK receptor ligand. Next, after washing was done to remove unbound conjugate, cell-associated radioactivity was determined by γ counting. As shown in Figure 3 and Table 1, CRL-1, CRL-2, and CRL-3 all showed high affinities for both cell lines, with binding constants in the nanomolar range. In contrast, CRL-4, which contained no spacer, displayed low affinities for both cell lines; this result confirmed the impact of spacer length on ligand affinity.

In vitro binding isotherms of CRL-1, CRL-2, and CRL-3 for HEK 293 CCK2R cells in absence (solid lines) and presence (broken lines) of 100-fold molar excess of unlabeled CCK receptor ligand (xs CRL). Error bars indicate SDs (n = 3).
Binding Affinities of CCK Receptor Ligand–Targeted Radioimaging Agents in HEK 293 Cells
To determine the fraction of conjugate uptake that was receptor-mediated, we performed parallel experiments in the presence of a 100-fold molar excess of unlabeled CCK receptor ligand to competitively block receptor binding. As shown in Figure 3, CRL-2 and CRL-3 showed quantitative competition on the addition of excess ligand, whereas CRL-4 and CRL-1 showed approximately 50% and approximately 20% residual nonspecific binding, respectively. We suspect that the strong hydrophobicity of Z-360 favored membrane association for all conjugates and that only the enhanced steric bulk and hydrophilicity of the peptidosaccharide spacers were sufficient to prevent nonspecific membrane association. The slightly higher affinity of CRL-3 than of CRL-2 may have been due to the increased separation of the binding ligand from the bulky peptidosaccharide spacer; however, additional subsite interactions may also have contributed to this difference in affinity. On the basis of these data, CRL-3 was selected for further testing in mice bearing both CCK2R and CCK2i4svR tumor xenografts.
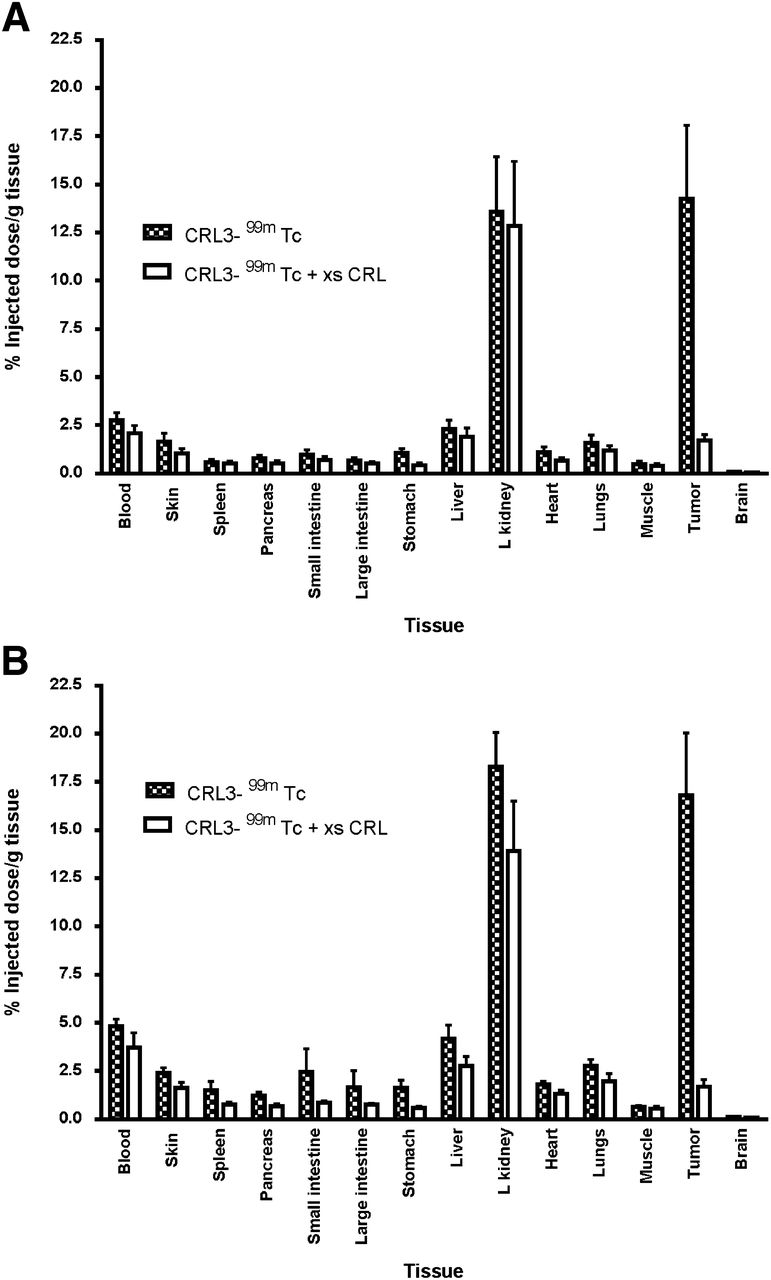
The in vivo targeting and specificity of CRL-3–99mTc were evaluated by injecting CRL-3–99mTc intravenously into athymic nu/nu mice bearing HEK 293 CCK2R and HEK 293 CCK2i4svR tumor xenografts. As shown in Figure 4, CRL-3–99mTc was observed to accumulate in HEK 293 CCK2R and HEK 293 CCK2i4svR tumors, with little or no accumulation in other tissues except the kidneys (14.25 percentage injected dose per gram of tissue 2 h after injection). Preinjection of the mice with a 100-fold excess of unlabeled CCK receptor ligand was found to block tumor uptake, indicating that the accumulation of the radiotracer in the malignant mass was receptor-mediated (Fig. 5). In contrast, the uptake of CRL-3–99mTc in the kidneys was not affected by excess CRL-3 (Fig. 5), suggesting that the presence of CRL-3–99mTc in the kidneys did not depend on CCK2R expression.
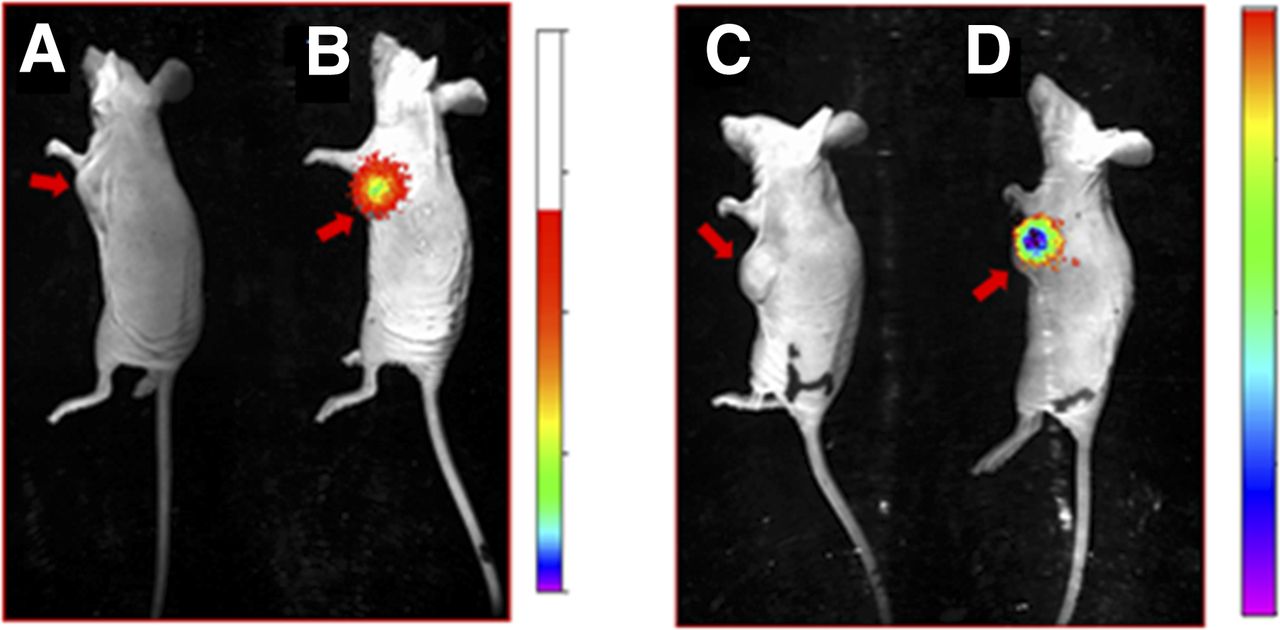
Evaluation of specificity of CRL-3–99mTc for CCK2R-expressing tumors in mice. Overlay of whole-body radioimages (rainbow colors) on white-light photographs of nu/nu mice bearing HEK 293 CCK2R (A and B) and HEK 293 CCK2i4svR (C and D) tumor xenografts in presence (A and C) and absence (B and D) of 100-fold molar excess of CRL-3. At 2 h after tail vein administration of 5.55 MBq of CRL-3–99mTc, mice were euthanized, and kidneys were shielded with lead plate to allow easier visualization of radioconjugate in other tissues. Red arrows indicate locations of tumors.
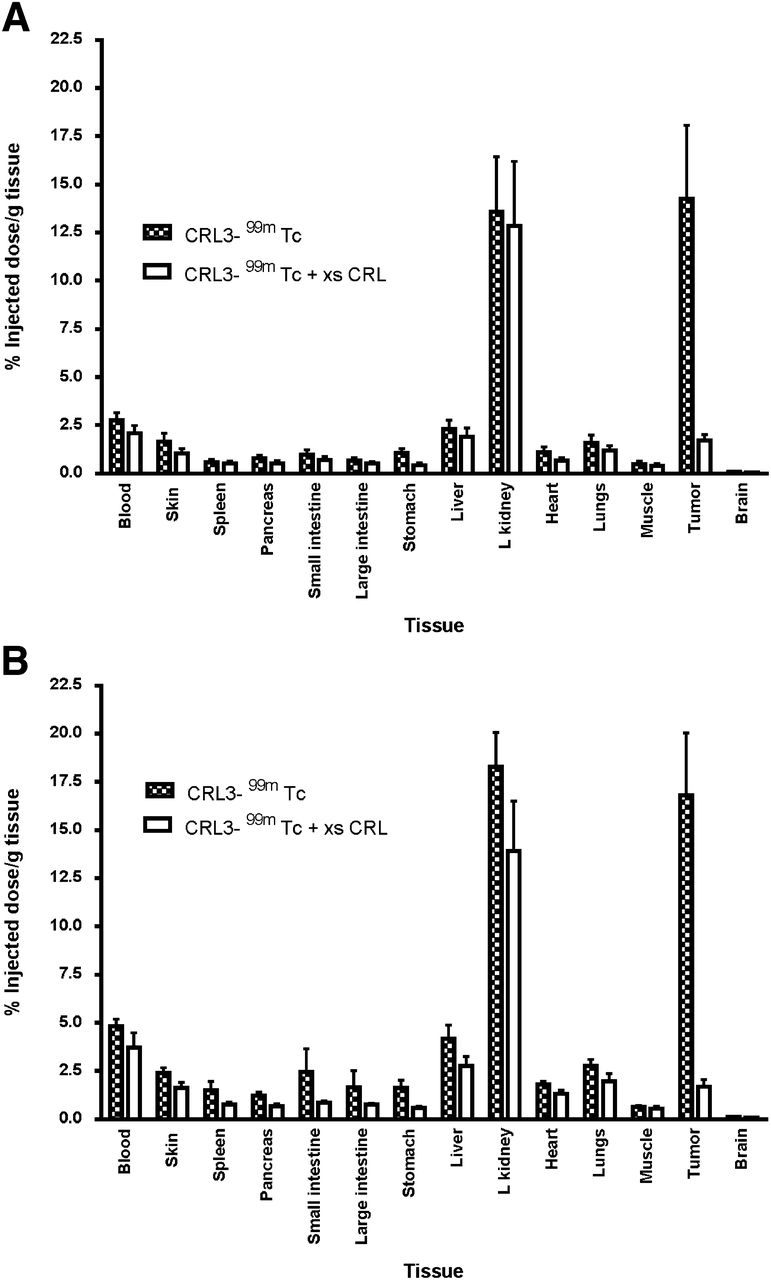
Tissue distribution of CRL-3–99mTc in nu/nu mice bearing HEK 293 CCK2R (A) and HEK 293 CCK2i4svR (B) tumor xenografts 2 h after injection of 5.55 MBq of CRL-3–99mTc. Error bars represent SDs (n = 5 mice per group). L kidney = left kidney; xs CRL = excess of unlabeled CCK receptor ligand.
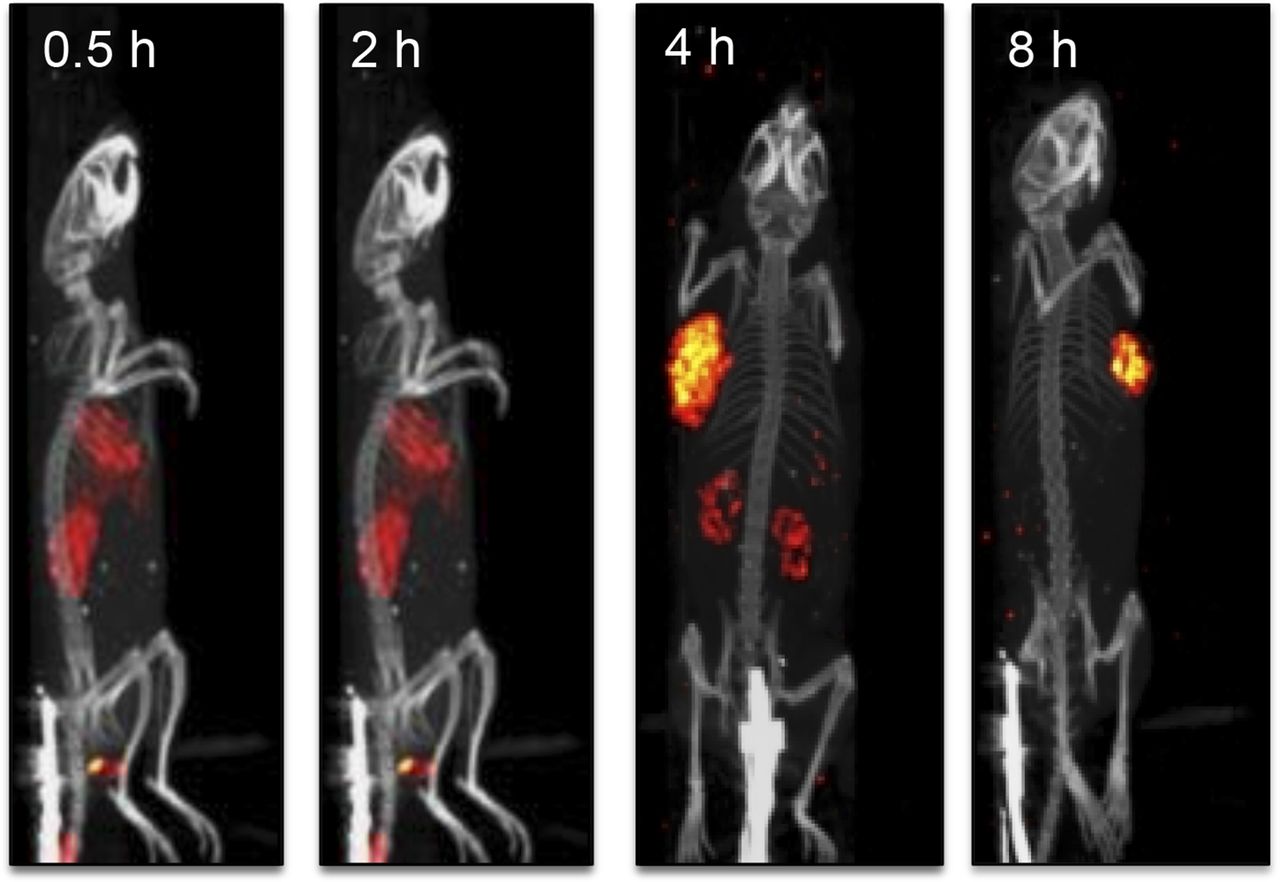
Because small molecules are often excreted via the liver or kidneys (40), we examined whether the uptake of CRL-3–99mTc in the liver and kidneys may have been transient. As shown in the SPECT/CT images in Figure 6, the CRL-3–99mTc content in the liver and kidneys decreased over time, whereas uptake in the tumor mass was relatively stable.
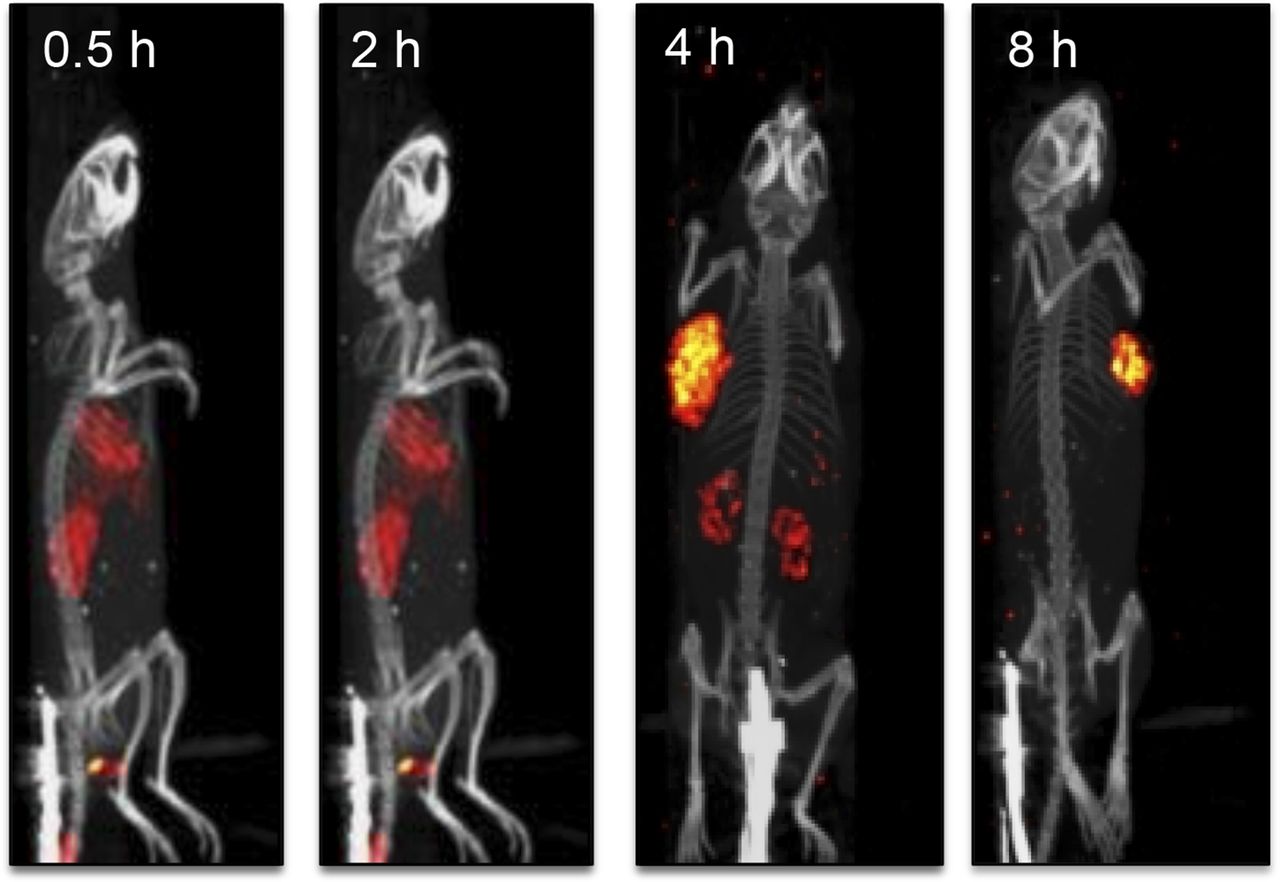
Examination of rate of clearance of CRL-3–99mTc from tumor-bearing mice with SPECT/CT. Mice implanted with HEK 293 CCK2R tumor xenografts were injected in tail vein with 5.55 MBq of CRL-3–99mTc and imaged 0.5, 2, 4, and 8 h after injection.
By 24 h after tail vein injection (Table 2), the tumor-to-normal tissue ratios of CRL-3–99mTc in the muscle, heart, skin, blood, liver, and spleen were 90, 83, 30, 61, 4, and 14, respectively. These data suggest that CCK2R-expressing tissues retained accumulated CRL-3–99mTc in substantially greater quantities and, therefore, that any radiation damage to normal tissues should be minimal.
Tissue Distribution of CRL-3–99mTc After Subcutaneous Injection of Mice with HEK 293 Cells Expressing CCK2R
DISCUSSION
The overexpression of CCK2R in many malignant cells and the limited expression of CCK2R in normal tissues have suggested that CCK2R-targeted radioemitters may be attractive candidates for the radioimaging of many human cancers. However, the very high uptake of natural ligands of CCK2R by the kidneys (in some cases, ∼60% of the injected dose) (29) has strongly discouraged further investigation of peptides for this application. The objective of this study therefore was to explore the development of nonpeptidic ligands for the imaging of CCK2R-expressing tumors. We showed here that a small organic antagonist of CCK2R could be exploited to yield high-resolution images of CCK2R-expressing tumors, especially when time was allowed for the clearance of CCK receptor ligand conjugates from the liver and kidneys. In addition to the avoidance of hepatic and renal retention with nonpeptidic CCK2R ligands, these ligands may enable improved serum stability (35).
To evaluate the effect of spacer chemistry on CCK2R targeting, we synthesized and evaluated 4 different conjugates with distinct spacers. Although each conjugate recognized both CCK2R and CCK2i4svR, CRL-3 displayed the highest affinity for both forms of the receptor, whereas CRL-4 showed the lowest affinity for the 2 types of receptor. Moreover, CRL-4 and, to a lesser extent, CRL-1 were both compromised by a high level of nonspecific binding, suggesting the need for a hydrophilic spacer to offset the hydrophobicity of the core ligand, Z-360. As shown in Figure 3, the binding of both peptidosaccharide-containing spacers could be nearly quantitatively blocked on the addition of excess ligand, suggesting almost exclusive receptor-mediated binding by those conjugates. CRL-3–99mTc also showed favorable pharmacokinetics in vivo, with rapid blood and kidney clearance and sustained tumor retention.
If CCK receptor ligand–targeted radiotherapeutic and chemotherapeutic agents display similar biodistribution profiles, then the possible use of CCK receptor ligands for targeting therapeutic payloads should not be ignored. The observed tissue distribution of CRL-3–99mTc implied that toxicity may be localized to the tumor, with negligible damage to normal tissues. Moreover, because kidney and liver accumulation was only transient, the selection of a radiotherapeutic agent with a longer half-life may allow clearance of the radioactivity from the kidneys before significant decay of the radiotherapeutic conjugate in the tumor has occurred. In both radiotherapeutic and chemotherapeutic applications, prior imaging of patients with CRL-3–99mTc may also prove useful for preselecting patients whose tumors express sufficient CCK2R to benefit from CCK receptor ligand–targeted therapy.
CONCLUSION
In addition to the targeting of radioimaging and chemotherapeutic agents, CCK receptor ligands may also be exploited for the delivery of other imaging modalities (e.g., PET, CT, and optical and MR imaging contrast agents) selectively to CCK2R-expressing tumors. Furthermore, as shown for other tumor-targeted ligands (4,7), CCK receptor ligands may also be developed for the delivery of fluorescent probes for use in fluorescence-guided resection of malignant masses during surgery (32). Fluorescent CCK receptor ligands may similarly be adapted for use in the detection and capture of circulating tumor cells in the blood of cancer patients. In brief, if the tumor selectivity of CRL-3–99mTc is maintained with other conjugates of CCK receptor ligands, then various future clinical applications can be envisioned.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. No potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Dr. Venkatesh Chelvam for help with the KODAK imaging system, Aaron Taylor at the Purdue University Bioimaging Center for assistance with SPECT/CT, and Dr. Mini Thomas for helpful discussions.
Footnotes
Published online Dec. 11, 2014.
- © 2015 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication June 30, 2014.
- Accepted for publication November 7, 2014.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.