


Abstract
Recent studies have highlighted the high sensitivity of PET imaging with 68Ga-labeled octreotide derivatives for the detection and staging of neuroendocrine tumors. A somatostatin receptor ligand that is easily radiolabeled with 18F-fluoride could improve the availability of PET imaging of neuroendocrine tumors. We report an alkyltrifluoroborate–octreotate conjugate that is radiolabeled in a 1-step 18F exchange reaction in high yield and with high specific activity. Methods: We conjugated a new alkyltrifluoroborate to octreotate to obtain AMBF3-TATE, which was stored in 50-nmol aliquots for radiolabeling. 18F labeling was performed by 18F-19F isotope exchange with 18F-fluoride, and the tracer was purified by C18 cartridge separation. The radiochemical yield was 20%–25%. PET imaging and biodistribution were performed on mice bearing AR42J tumor xenografts. Results: AMBF3-TATE bound the somatostatin receptor subtype 2 with high affinity (inhibition constant, 0.13 ± 0.03 nM). Starting with 29.6–37 GBq (0.8–1 Ci) of 18F-fluoride, more than 7.4 GBq (>200 mCi) of 18F-AMBF3-TATE were obtained in 25 min (n = 5) with greater than 99% radiochemical purity at high specific activity (>111 GBq [3 Ci]/μmol). 18F-AMBF3-TATE was stable in plasma. PET imaging and biodistribution showed rapid renal excretion with low liver activity. High tumor uptake (10.11% ± 1.67% injected dose/g, n = 5) was detected at 60 min after injection. Bone uptake was negligible. Tumor-to-liver, tumor-to-blood, tumor-to-muscle, and tumor-to-bone ratios (at 60 min) were 26.2 ± 0.8, 25.1 ± 1.0, 89.0 ± 3.1, and 21.3 ± 3.6, respectively. Conclusion: 18F-AMBF3-TATE was radiolabeled in high yield and at high specific activity, did not require high-performance liquid chromatography purification, exhibited unexpectedly high binding affinity to somatostatin receptor subtype 2, and showed excellent pharmacokinetic properties in vivo, with high tumor uptake and high contrast ratios.
The somatostatin receptor subtype 2 (sstr2) is overexpressed in many neuroendocrine tumors. Hence, over the past 30 y there has been considerable interest in developing high-affinity somatostatin-derived ligands that bind sstr2, notably for radionuclide therapy (1). To diagnose and monitor patients with sstr2-positive tumors, radiotracers based on the somatostatin family of peptides (2–5), notably octreotate (TATE) and octreotide, have been labeled with various radioisotopes for noninvasive imaging. 111In-diethylenetriaminepentaacetic acid-pentetreotide (Octreoscan; Mallinckrodt) is the current clinical standard for imaging neuroendocrine tumors (6–8). 99mTc derivatives such as 99mTc-depreotide (9) and 99mTc-hydrazinonicotinyl-Tyr3-octreotide have also been used (10) but are not commercialized in North America.
For PET imaging, 68Ga, 64Cu, and 18F along with various radioprosthetics have been conjugated to various octreotide derivatives (11–16). Of these, certain 68Ga ligands such as 68Ga-DOTATOC, 68Ga-DOTATATE, and 68Ga-DOTANOC have shown promise for neuroendocrine tumor imaging (17–19) and are used in clinical trials as well as under the local practice of pharmacy, particularly in Europe. Nevertheless, 68Ga-PET imaging is not widely available because of the limited daily availability of 68Ga (∼50 mCi) and the lack of Food and Drug Administration–approved 68Ge/68Ga generators (20).
18F-fluoride presents several attractive properties for imaging (21,22) and is produced on a daily basis in large quantities in hundreds of cyclotrons in hospitals and radiopharmacies worldwide. Yet the challenges of labeling peptides with 18F-fluoride are significant: the low chemical reactivity of 18F-fluoride in water (23) and short half-life (109.8 min) challenge 18F labeling of peptides that are generally soluble only in water or aqueous cosolvents. Hence, fluoride must be dried and reacted in dry solvents at high temperature to radiolabel a radioprosthetic that is then conjugated to the peptide in at least 1 additional step. Although such multistep 18F-labeling reactions are commonplace (24), the relatively short half-life of 18F-fluoride often impedes the clinical application of multistep reactions, particularly in terms of ensuring specific activity greater than 37 GBq/μmol (>1 Ci/μmol) (25). Given these challenges, an sstr2 ligand that is easily labeled with 18F-fluoride in high yield and at high specific activity would facilitate sstr2 imaging by PET. Toward these ends, new 18F-octreotate derivatives, such as 18F-SiFA and Al-18F-NOTA, have been labeled in 1 step and imaged with relative success (26–28).
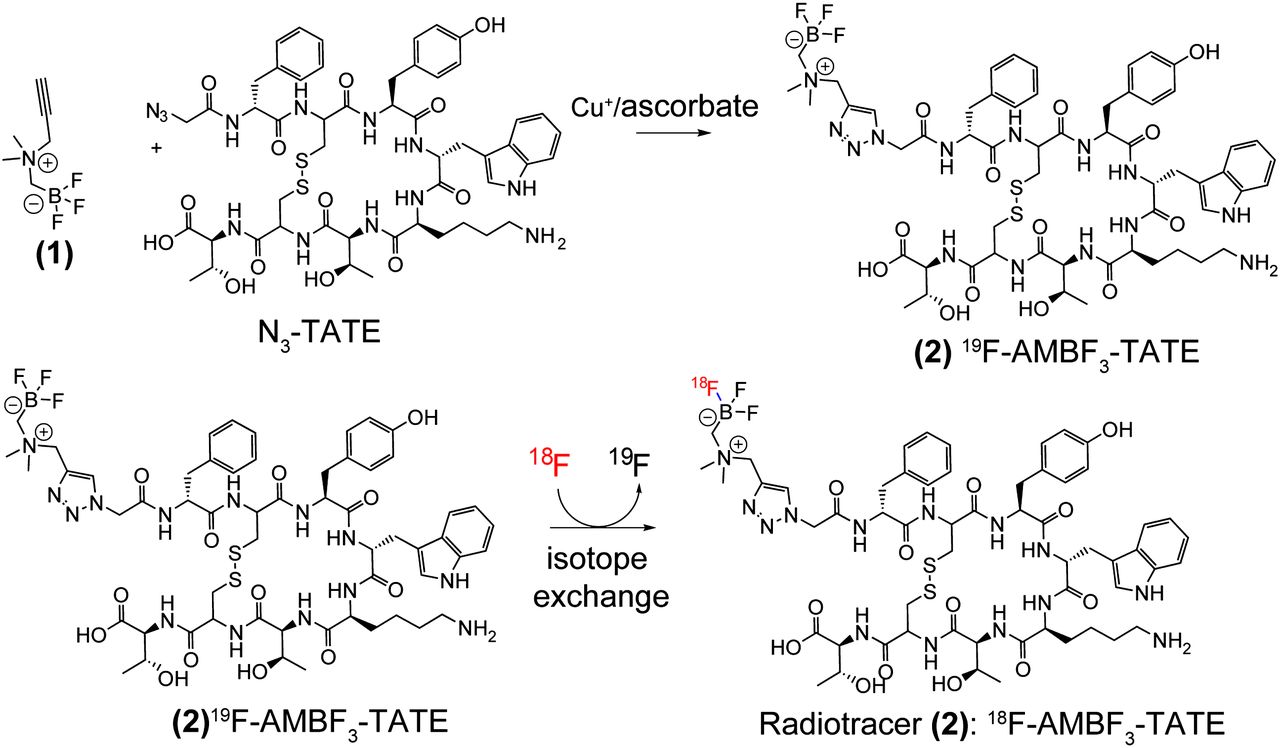
Similarly, aryltrifluoroborate prosthetics, when conjugated to various peptides, allow 1-step aqueous radiofluorination in high yield and very high specific activity (29–31). Recently, we identified a new ammoniomethyl-BF3 (AMBF3) that undergoes facile 18F-19F isotope exchange. To test the efficacy of boron-based 18F labeling, we conjugated AMBF3 to octreotate by means of a simple chemical synthesis to obtain AMBF3-TATE, which as a precursor is 18F-labeled by isotope exchange (Compound 2, Fig. 1). We report the radiosynthesis and preclinical evaluation of this new derivative.
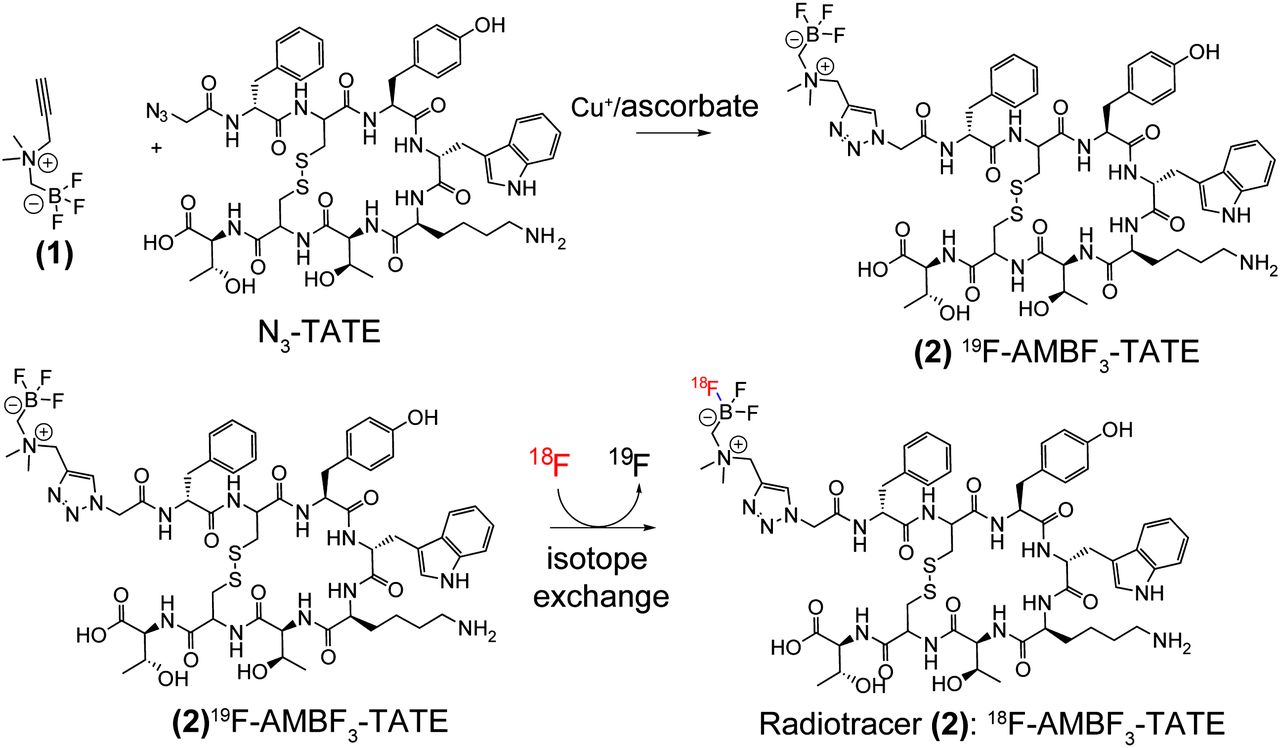
N3-TATE is condensed with N-propargyl-N,N-dimethyl-ammoniomethyltrifluoroborate (1) to provide precursor AMBF3-TATE (2). Precursor 2 is labeled by isotope exchange to provide isotopolog 18F-2 at high specific activity for tracer studies.
MATERIALS AND METHODS
Reagents and solvents were purchased from Advanced Chemtech, Sigma-Aldrich, Combi-Blocks, or Novabiochem. The AR42J cell line was purchased from ATCC. 18F-fluoride Trap & Release Columns were purchased from ORTG Inc., and C18 Sep-Pak cartridges (1 cm3, 50 mg) were obtained from Waters. An Endeavor 90 peptide synthesizer (Aapptec) was applied to synthesize the peptide. Electron-spray ionization low-resolution mass spectroscopy was performed on a Waters ZQ with a single quadrupole detector, attached to a Waters 2695 high-performance liquid chromatography (HPLC) column. All nuclear MR spectra were recorded at room temperature on a Bruker Avance 300 MHz spectrometer.
The following HPLC methods were used for purification and quality control. Method A: Agilent Eclipse XDB-C18 5-μm 9.2 × 250 mm semipreparative column; solvent A, 0.1% trifluoroacetic acid (TFA) water; solvent B, MeCN; 0–15 min, 20%–40% B; 15–20 min, 40%–20% B; flow rate, 4.5 mL/min; column temperature, 19°C–21°C. Method B: Agilent Eclipse XDB-C18 5-μm 9.2 × 250 mm semipreparative column; solvent A, 0.1% TFA water; solvent B, MeCN; 0–2 min, 5%–20% B; 2–5 min, 20%–30% B; 5–20 min, 30%–50%; 20–22 min, 50%–5% B; flow rate, 3 mL/min; column temperature, 19°C–21°C. Method C: Phenomenex Jupiter 10-μm C18 300-Å 4.6 × 250 mm analytic column; solvent A, 0.1% TFA water; solvent B, MeCN; 0–2 min, 5%–5% B; 2–7 min, 5%–20% B; 7–15 min, 20%–100%; 15–20 min, 100%–5% B; flow rate, 2 mL/min; column temperature, 19°C–21°C.
To synthesize the precursor for labeling, a suitable TATE was first synthesized as previously described (32) and converted to an azide derivative. The resin (Fmoc-Thr(tBu)-Wang) and growing chain were treated with 20% piperidine (1 × 5 min and 1 × 10 min) in N,N-dimethylformamide to remove the Nα-Fmoc protecting group. The amino acids (3 equivalents [eq.] per coupling) Fmoc-Cys(Acm)-OH, Fmoc-Thr(tBu)-OH, Fmoc-Lys(Boc)-OH, Fmoc-d-Trp(Boc)-OH, Fmoc-Tyr(tBu)-OH, and Fmoc-d-Phe-OH were subsequently coupled in nuclear matrix protein with the standard in situ activating reagent O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate (HBTU) (3 eq.) in the presence of diisopropylethylamine (6 eq.). Thallium(III) trifluoroacetate (2 eq.) in N,N-dimethylformamide deprotected the cysteines and concomitantly induced disulfide formation. Bromoacetic acid (40 eq.) was preactivated with diisopropylcarbodiimide (20 eq.) in dichloromethane for 10 min, filtered, and coupled to the N terminus. NaN3 (27.5 eq.) in dimethyl sulfoxide was added. The peptide was deprotected and simultaneously cleaved from the resin in 90:2.5:2.5:5 TFA:H2O:triisopropylsilane:phenol for 4 h at room temperature. TATE-N3 was purified by HPLC with a semipreparative column using method A to afford pure TATE-N3 in quantities of about 10 mg. The calculated mass was 1,131.2, and the measured mass by electrospray ionization was 1,131.4. The purity of the peptide was greater than 99%. N-propargyl-N,N-dimethyl-ammoniomethyl-boronylpinacolate (alkynyl-AMB(pin)) was first synthesized by condensation of iodomethyl-boronylpinacolate and propargylamine as previously described (33). Briefly, a dry round-bottomed flask was charged with N,N-dimethylpropargylamine (98 μL, 1.0 mmol) and 2 mL of anhydrous diethyl ether under argon, to which iodomethyl-boronylpinacolate (165 μL, 0.9 mmol) was added dropwise at room temperature. On stirring, the solution became cloudy and the desired product was collected as a white precipitate that was filtered and washed with cold Et2O and then dried under high vacuum to give a fluffy white powder in 95% yield. 1H nuclear MR (300 MHz [Bruker], CD3CN): δ 4.40 (d, 2H), 3.31 (s, 2H), 3.22 (s, 6H), 3.21 (t, 1H), 1.27 (s, 12H); electrospray ionization: calculated, 224.1; found, 224.1. N-propargyl-N,N-dimethylammonio-methylboronylpinacolate (5.0 mg, 22.3 μmol) was converted to the trifluoroborate (alkynyl-AMBF3) through the addition of KHF2 (3 M, 30 μL in water), HCl (4 M, 30 μL in water), deionized water (20 μL), and N,N-dimethylformamide (60 μL), 45°C, 2 h, and then quenched by NH4OH (concentration, 10 μL). The crude reaction was directly used for click conjugation to TATE azide without further purification: a mixture of TATE-azide (4.0 mg, 3.4 μmol), CuSO4 (1.0 M, 5.0 μL), sodium ascorbate (1.0 M, 12.5 μL), and 5% NH4OH (1:1 MeCN:H2O, 50 μL) was added, and the mixture was heated to 45°C for 2 h. Purification was performed with method B to isolate 2.3 mg of AMBF3-TATE. Purity was confirmed with liquid chromatography–mass spectrometry (calculated, 1,296.5; obtained, 1,296.4). The purified 19F-AMBF3-TATE was diluted in ethanol and portioned into aliquots of approximately 60 μg (50 nmol) for radiolabeling in kitlike fashion.
After successful synthesis, the activity of 19F-AMBF3-TATE was examined in vitro. Membranes from Chinese hamster ovary K1 cells transfected with sstr1, sstr2, sstr3, sstr5, and [125I]-Tyr-somatostatin-14 were obtained from PerkinElmer. A standard filtration binding assay was performed in 96-well plates (MultiScreen; Millipore) to determine the binding affinities (inhibition constant, or Ki) of AMBF3-TATE against different receptor subtypes. Briefly, membranes (0.25 μL/well) were incubated with the 125I-labeled standard at a concentration of 0.05 nM for sstr2 or 0.2 nM for other subtypes. Increasing concentrations of AMBF3-TATE were added to the wells in buffer (25 mM N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid, pH 7.4; 10 mM MgCl2; 1 mM CaCl2; and 0.5% bovine serum albumin). After incubation (37°C, 1 h), the wells were aspirated and washed 8 times with 50 mM ice-cold buffer (Tris-HCl, pH 7.4) over grade GF/B filters. The filters were removed and counted by a γ counter (Cobra II; Packard). A typical competition curve is shown in Figure 2 (n = 3). Data were fitted to a 1-site competition model (GraphPad Prism 6.1 software) to calculate Ki.

Representative example of competitive binding assay for 19F-AMBF3-TATE. y-axis shows counts bound. Assay was run with triplicate data points. CPM = counts per minute.
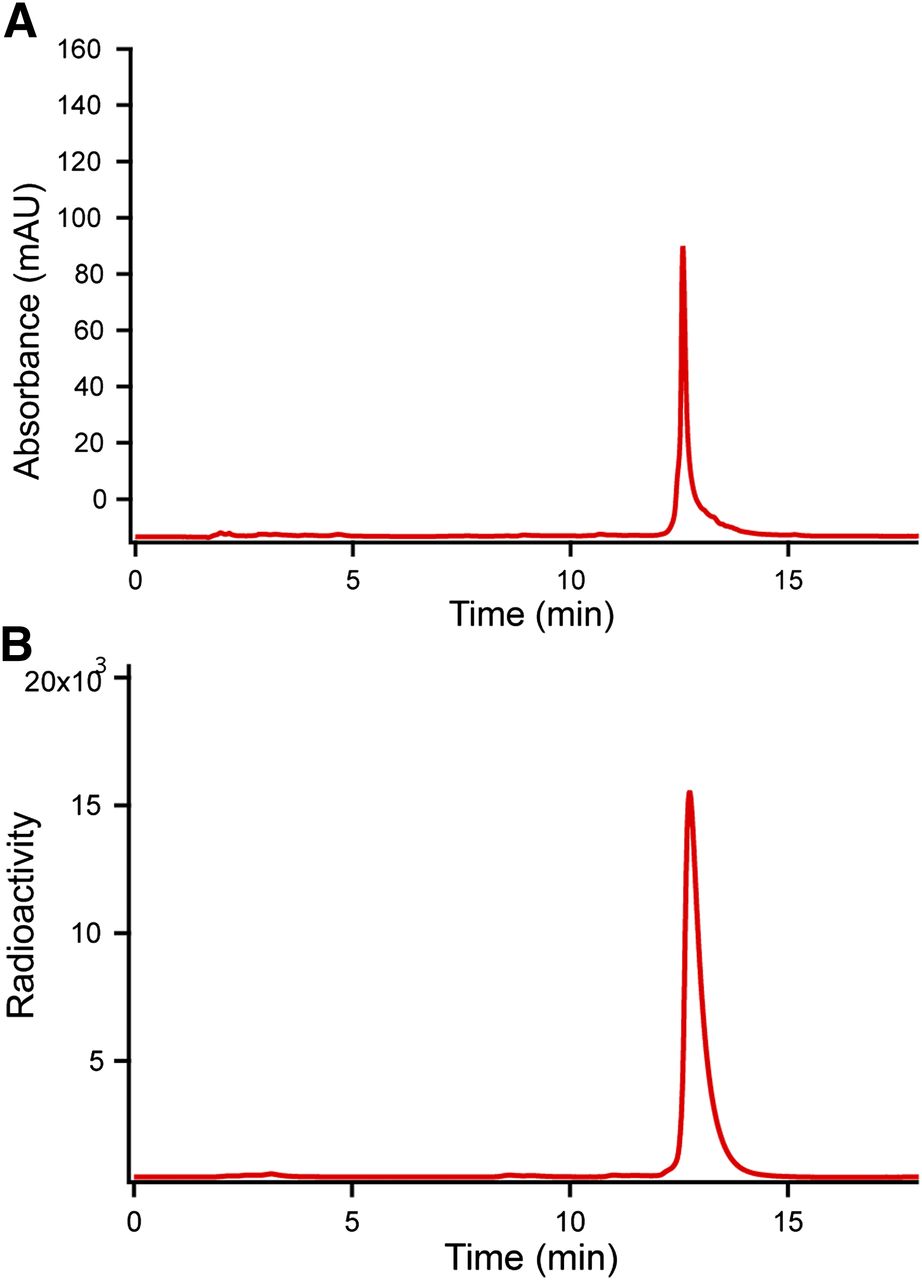
For 18F labeling, AMBF3-TATE (50 nmol) was resuspended in aqueous pyridazine-HCl buffer (∼50 μL, pH 2) in a vial (polypropylene Falcon Tube; Corning) just before labeling. No-carrier-added 18F-fluoride, 29.6–37 GBq (800–1,000 mCi), was obtained by bombardment of H218O with 18-MeV protons, followed by trapping on an anion exchange resin (9 mg, quaternary ammonium, chloride form, prewashed with deionized water). The 18F-fluoride was eluted with 70–100 μL of isotonic saline into the reaction vial containing AMBF3-TATE. The vial was placed in a heating block set at 80°C for 20 min, whereupon the reaction was quenched by the injection of 2 mL of 5% NH4OH in water. The reaction mixture was loaded onto a C18 light cartridge that was preconditioned by wetting with MeCN and washing with distilled water. Impurities (e.g., 18F-fluoride, pyridazine) were removed by flushing with 2 mL of saline. Radiochemically pure 18F-AMBF3-TATE was released into a glass vial with 0.5 mL of 1:1 ethanol:saline to provide 7.4 GBq (200 mCi) of tracer. This solution was formulated in isotonic saline (5 mL). A small sample was removed for quality control analysis by HPLC with mass detection at 277 nm (Fig. 3). Radiochemically pure 18F-AMBF3-TATE, formulated in saline, was assayed for plasma stability. For this assay, 20 μL of 18F-AMBF3-TATE were added to mouse plasma (500 μL) and incubated at 37°C for 0, 60, and 120 min. After incubation at each time point, the reaction was quenched by adding 1 mL of MeCN to precipitate insoluble proteins from the solution. The quenched reactions were centrifuged to remove insoluble material. The supernatant was aspirated, filtered, and analyzed by HPLC using method C.

HPLC traces of Sep-Pak–purified 18F-AMBF3-TATE. (A) Ultraviolet trace measured at 277 nm. (B) Radioactivity trace. AU = arbitrary units.
After labeling, imaging was undertaken. All animal studies were performed in accordance with the Canadian Council on Animal Care guidelines and were approved by the animal care committee of the University of British Columbia. Rat pancreatic adenocarcinoma cells (107 AR42J cells) were freshly expanded in a mixture of phosphate-buffered saline and Matrigel (Corning) and inoculated subcutaneously in female immunocompromised mice (NOD SCID [nonobese diabetic severe combined immunodeficient] IL2r-γ-null, bred in house). The tumors were grown for 2 wk until they reached 5–7 mm in diameter. While under 2% isoflurane anesthesia, the mice were injected via the tail vein with 0.37–0.74 MBq (10–20 μCi, 4–8 pmol) of 18F-AMBF3-TATE (n = 5). To demonstrate the specificity of in vivo uptake in receptor-positive tissues, 100 μg of 69/71Ga-DOTATATE were preinjected 15 min before 18F-AMBF3-TATE injection as a blocking control cohort (n = 4). Sixty minutes after injection, the mice were anesthetized with isoflurane and euthanized by carbon dioxide. The organs were harvested, rinsed with saline, blotted dry, and collected in previously weighed tubes. The tubes containing the organs were counted in a Cobra-II γ counter. The tissue weight and associated count per minute were used to calculate the percentage injected dose per gram of tissue (%ID/g). Images were acquired using a multimodality PET/CT system (Inveon; Siemens). Approximately 3.7 MBq (100 μCi, ∼40 pmol) of radiotracer were injected in the caudal lateral tail vein of tumor-bearing mice. Sixty minutes after radiotracer injection, the animals were anesthetized with isoflurane inhalation and a baseline low-dose CT scan was obtained for localization and attenuation correction, followed by a static PET scan acquired for 10 min. The mice were kept warm by a heated pad on the scanner bed during acquisition. 69/71Ga-DOTATATE (100 μg per mouse) was preinjected as a blocking agent. The images were reconstructed by an iterative reconstruction algorithm (3-dimensional ordered-subsets expectation maximization/maximum a posteriori) using the Inveon Acquisition Workplace Software (Siemens), applying normalization, dead time, random, and attenuation corrections. Uptake into tumor and tissues of interest was determined by regions of interest, and %ID/g was calculated (assuming a tissue density of 1.0 g/cm3). The mean %ID/g was calculated from a region of interest that matched the tumor contours on CT. The peak %ID/g was calculated from the hottest 2 × 2 voxel cluster within the tumor. In 1 animal, a dynamic scan was acquired in list mode for 60 min under continuous isoflurane inhalation, starting concurrently with radiotracer injection. Imaging data from this mouse were not combined with the results of static imaging and were used to obtain the tissue time–activity curves reported in Figure 4.

Time–activity curves indicating blood, liver, and kidney clearance and peak tumor uptake (from hottest voxel cluster in tumor from a single mouse).
RESULTS
In Vitro Affinity
The Ki of AMBF3-TATE using human sstr2 receptors expressed on Chinese hamster ovary membranes was 0.13 ± 0.03 nM. Using identical assay conditions and the same lot of membranes, the Ki for gallium-DOTATATE was 0.7 ± 0.2 nM. A representative competitive binding assay curve is shown in Figure 2. No significant displacement of binding to sstr1 was observed. The inhibition constants for sstr3 and sstr5 were 28.4 ± 8.6 nM and 11.6 ± 2.8 nM, respectively.
Radiosynthesis
Starting with 29.6–37 GBq of no-carrier-added 18F-fluoride (800–1,000 mCi), approximately 7.4 GBq of 18F-AMBF3-TATE were obtained within 25 min (24% ± 4%, n = 5) and reinjected into HPLC for quality analysis (Fig. 3), which showed a single peak in both radioactive and ultraviolet modes. Because approximately 7.4 GBq (∼200 mCi) of 2 were obtained starting with 50 nmol of precursor, the specific activity was 148 GBq/μmol (3 Ci/μmol), with a radiochemical yield of 20%–25% (not corrected for decay). To validate this calculation, a standard curve showed that the specific activity was more than 111 GBq/μmol (>3 Ci/μmol). 18F-AMBF3-TATE was incubated in mouse plasma for 120 min with no detectable decomposition (Fig. 5).

Plasma stability assay of 18F-AMBF3-TATE; radiotraces are shown for 0, 60, and 120 min.
PET/CT Imaging
Uptake in the AR42J tumors was intense and clearly specific as evidenced by the lack of uptake in the tumors of mice receiving unlabeled competitor (Fig. 6). The average of the tumor uptake based on the whole tumor region of interest was 10.2 ± 2.1 %ID/g. The average of the peak tumor uptake based on the hottest voxel cluster was 23.6 ± 3.0 %ID/g. In contrast, the average uptake in the liver, blood pool, and muscle was 0.83 ± 0.16, 0.47 ± 0.12, and 0.09 ± 0.03 %ID/g, respectively. Excretion was predominantly renal, with significant clearance to the bladder and low kidney retention. Some hepatobiliary tract excretion was notably rapid, leading to high tumor-to-liver ratios. Bone uptake was undetectable, and there was low background activity in blood and muscle, resulting in high-contrast images.

18F-AMBF3-TATE PET images of AR42J tumor–bearing mice at 60 min after injection: unblocked (A and C) and blocked (B and D). Upper panels are maximum-intensity-projection images; bottom panels are corresponding fused coronal images. Color bars are calibrated in %ID/g with no background subtracted. Tracer specifically accumulated into tumor (t), whereas remainder rapidly cleared via kidneys (k) to bladder (b). Some gut (g) and gallbladder (gb) accumulation occurred because of rapid hepatobiliary excretion.
Pharmacokinetics (Time–Activity Curve Analysis)
Time–activity curves of uptake in tumor and other tissues from a tumor-bearing mouse are presented in Figure 4. Time-dependent tumor uptake increased to a peak voxel cluster value of approximately 40 %ID/g in a mouse with a fairly large tumor. Uptake in nontarget tissues rapidly declined after reaching the peak value at a time point soon after intravenous administration.
Biodistribution Studies
The ex vivo biodistribution data of 18F-AMBF3-TATE at 1 h (Table 1) corroborate the scanning data. The relative uptake values in various tissues are shown in Figure 7. Uptake in AR42J xenograft tumors in the unblocked mice was 10.11 ± 1.67 %ID/g. As expected, excess competitor caused a substantial reduction in tumor uptake: 0.32 ± 0.21 %ID/g. Hence, blocking efficiency was 97%. Uptake values in blood and muscle were low: 0.40 ± 0.31 %ID/g and 0.11 ± 0.03 %ID/g, respectively, which gave high tumor-to-blood and tumor-to-muscle ratios of 25.1 ± 1.0 and 89.0 ± 3.1, respectively. Bone uptake was negligible (0.46 ± 0.17 %ID/g), indicating no in vivo defluorination.
Biodistribution of 18F-AMBF3-TATE (%ID/g)

Selected organs from biodistribution of 18F-AMBF3-TATE at 60 min after injection in AR42J-bearing mice, showing high receptor-mediated uptake in tumors compared with normal tissues.
DISCUSSION
Isotope exchange on the organotrifluoroborate prosthetic greatly simplified labeling on several accounts. First, only submilligram quantities of precursor were needed for labeling. Second, no time-consuming azeotropic fluoride drying was required, because no-carrier-added 18F-fluoride was eluted in isotonic saline and used directly for an aqueous labeling reaction. Third, labeling was rapid (∼20 min) and provided for high specific activity. Fourth, because the precursor is chemically identical to the product, time-consuming HPLC purification was obviated in favor of a simple C18 Sep-Pak elution to remove free fluoride. Besides the methodologic simplicity, the yields provide multiple human doses in a single run. In light of improvements in cyclotron output to provide multiple-curie levels of 18F-fluoride (34), this methodology is readily applicable to existing production facilities. Moreover, the simplicity of the process should be easily amenable to automation and microfluidic flow technologies.
Whereas good radiosynthetic attributes are a prerequisite for use, the real value of a given tracer ultimately lies in the in vivo image data and corroborating biodistribution data. There is an extensive body of literature, which cannot be fully reviewed here, detailing the labeling and imaging of various TATE analogs by SPECT or PET. A brief comparison of the representative radiolabeled TATE derivatives is featured in Table 2. Comparison of receptor binding affinities is difficult because many authors report an inhibitory concentration of 50%, which is dependent on assay conditions. Among the published TATE-based radiotracers, gallium-DOTATATE has the highest affinity for sstr2 reported to date. Under identical conditions, 18F-AMBF3-TATE showed higher affinity than gallium-DOTATATE. This finding was both unanticipated and significant. The sensitivity of somatostatin analogs, either agonists or antagonists, to substitutions at the N terminus and to the radiometal is well documented (35,36). Here, the octreotate was modified with the AMBF3 prosthetic via copper-catalyzed click conjugation; although we expected a decrease in the binding affinity to the sstr2, we instead observed a more than 5-fold higher binding affinity than for 68Ga-DOTATATE as assayed under identical conditions. The inhibition constants to sstr3 and sstr5 also appeared to be lower than the values published for Ga-DOTATATE, suggesting that other zwitterionic moieties at the N terminus may improve affinity.
Comparison of Some Octreotide and Octreotate Derivatives
On the basis of PET/CT imaging, 18F-AMBF3-TATE exhibited low background activity in nontarget tissues and high receptor-mediated uptake in a preclinical murine model of sstr2-positive cancer. Corroborating the in vivo imaging data, ex vivo biodistribution verified the high tumor uptake values. Although liver uptake of radiometallated octreotides typically is low, this is not always the case for 18F-labeled octreotides. Liver uptake, and in particular nonspecific uptake, is often observed and may preclude clinical detection of liver metastasis. Interestingly, the liver uptake of 18F-AMBF3-TATE was low, resulting in a higher tumor-to-liver ratio (26.21 ± 0.79 1 h after injection) than has been reported for other 18F-labeled TATE analogs (0.25–5.0 2 h after injection) (16,26).
A plasma stability assay (37°C) showed negligible decomposition of 18F-AMBF3-TATE after 120 min. Consistent with this finding, minimal bone uptake was observed in both PET/CT and biodistribution, resulting in a high tumor-to-bone ratio of up to 21.3 ± 3.6. This low, nonspecific bone uptake is particularly encouraging for the detection of bone metastasis. Moreover, this result highlights the general stability of such alkyltrifluoroborate radioprosthetics, an observation that should augur well for the development of other peptide tracers based on the same zwitterionic ammoniomethyl-BF3.
CONCLUSION
We report a high-affinity octreotate–organotrifluoroborate conjugate that was radiolabeled with 18F in high yield and high specific activity via a facile isotope exchange reaction using minute quantities of precursor peptide, without HPLC purification. This methodology provides for rapid, multidose tracer production in a single run that should be amenable to automation. In addition to radiosynthetic ease, the biologic evaluation of 18F-AMBF3-TATE indicated that this tracer provides good stability, optimal pharmacokinetics, excellent binding affinity, and high tumor–to–nontarget-tissue ratios for in vivo imaging.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This work was supported by the Canadian Cancer Society Research Institute (grant 20071) and the Canadian Breast Cancer Foundation and support from Genome B.C. A provisional patent application was filed for some of the material presented in this article. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Navjit Hundal, Zhengxing Zhang, Wade English, Julius Klug, and Milan Vuckovic for technical assistance.
Footnotes
↵* Contributed equally to this work.
Published online Jun. 26, 2014.
- © 2014 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication January 22, 2014.
- Accepted for publication May 12, 2014.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- One-Step 18F-Labeling and Preclinical Evaluation of Prostate-Specific Membrane Antigen Trifluoroborate Probes for Cancer Imaging
- Recent Advances in 18F Radiochemistry: A Focus on B-18F, Si-18F, Al-18F, and C-18F Radiofluorination via Spirocyclic Iodonium Ylides
- Trimeric Radiofluorinated Sulfonamide Derivatives to Achieve In Vivo Selectivity for Carbonic Anhydrase IX-Targeted PET Imaging
- In Vivo Evaluation of 18F-SiFAlin-Modified TATE: A Potential Challenge for 68Ga-DOTATATE, the Clinical Gold Standard for Somatostatin Receptor Imaging with PET