


Abstract
A new family of peptide receptors, the incretin receptor family, overexpressed on many neuroendocrine tumors (NETs) is of great importance because it may enable the in vivo peptide-based receptor targeting of a category of NETs that does not express the somatostatin receptor. Impressive in vivo diagnostic data were published for glucagonlike peptide 1 receptor–targeting radiopeptides. Recently, promising in vitro data have appeared for the second member of the incretin family, the glucose-dependent insulinotropic polypeptide (GIP) receptor. This prompted us to develop and evaluate a new class of radioligands with the potential to be used for the in vivo targeting of GIP receptor–positive tumors. Methods: GIP(1–42) was modified C-terminally, and the truncated peptides [Lys30(aminohexanoic acid [Ahx]-DOTA)]GIP(1–30)NH2 (EG1), [Lys16(Ahx-DOTA)]GIP(1–30)NH2 (EG2), and [Nle14, Lys30(Ahx-DOTA)]GIP(1–30)NH2 (EG4) were conjugated with Ahx-DOTA via the Lys16 and Lys30 side chains. Their inhibitory concentration of 50% (IC50) was determined using [125I-Tyr10]GIP(1–30) as radioligand and GIP(1–30) as control peptide. The DOTA conjugates were labeled with 111In and 68Ga. In vitro evaluation included saturation and internalization studies using the pancreatic endocrine cell line INR1G9 transfected with the human GIP receptor (INR1G9-hGIPr). The in vivo evaluation consisted of biodistribution and PET imaging studies on nude mice bearing INR1G9-hGIPr tumors. Results: Binding studies (IC50 and saturation studies) showed high affinity toward GIP receptor for the GIP conjugates. Specific in vitro internalization was found, and almost the entire cell-associated activity was internalized (>90% of the cell-bound activity), supporting the agonist potency of the 111In-vectors. 111In-EG4 and 68Ga-EG4 were shown to specifically target INR1G9-hGIPr xenografts, with tumor uptake of 10.4% ± 2.2% and 17.0% ± 4.4% injected activity/g, 1 h after injection, respectively. Kidneys showed the highest uptake, which could be reduced by approximately 40%–50% with a modified-fluid-gelatin plasma substitute or an inhibitor of the serine protease dipeptidyl peptidase 4. The PET images clearly visualized the tumor. Conclusion: The evaluation of EG4 as a proof-of-principle radioligand indicated the feasibility of imaging GIP receptor–positive tumors. These results prompt us to continue the development of this family of radioligands for imaging of a broad spectrum of NETs.
- glucose-dependent insulinotropic polypeptide receptor (GIPr)
- neuroendocrine tumors (NETs)
- 111In
- 68Ga
- imaging
The regulatory peptides and their receptors have gained an important role in the targeted imaging and radionuclide therapy of cancer over the last few decades. Overexpression of the receptors on the surface of cancer cells, in combination with their considerably low expression in surrounding healthy tissues, is the key to successful application of these methods in the field of nuclear medicine (1,2).
The somatostatin receptor was the first to be defined for in vivo targeting. The overexpression of somatostatin receptor on neuroendocrine tumors (NETs) has allowed tumor localization, staging, therapy follow-up, and targeted radionuclide therapy (3). Radiolabeled somatostatin analogs can be used to localize NETs with high specificity and sensitivity, translating into successful targeted radionuclide therapy or to PET imaging (4–7). Although the overexpression of somatostatin has proven to be an important tool for the targeting of NETs, there are limitations since not all NETs express somatostatin (8). Therefore, the identification of other families of receptors that would enable the in vivo targeting of those tumors is of high importance (9). A new and promising family of G-protein–coupled receptors is the incretin receptor family (8,10,11). The imaging of tumors overexpressing the glucagonlike peptide 1 (GLP-1) receptor was proven to be successful in insulinoma preclinically (12–17) and clinically (18,19). Recently, it was found that the second member of the incretin receptor family, the glucose-dependent insulinotropic polypeptide (GIP) receptor, is overexpressed in specific NETs. Most somatostatin-negative NETs (∼90%) and GLP-1 receptor–negative malignant insulinomas are GIP receptor–positive (20).
GIP is a 42-amino-acid polypeptide synthesized by K cells of the duodenum and small intestine. Like its sister incretin hormone GLP-1, GIP is secreted by the intestine, causing a glucose-dependent release of insulin from the β cells and inhibition of glucagon from the α cells (21,22). GIP is rapidly inactivated to GIP(3–42) by the widespread serine protease dipeptidyl peptidase (DPP) 4, with a plasma half-life of approximately 5–7 min (23,24). The intact N-terminal and parts of the first 30 amino acids are necessary for biologic activity and binding to the GIP receptor–binding pocket with high affinity (25–27). GIP receptor belongs to the subfamily B1 of the G protein–coupled receptor superfamily, specifically the glucagon receptor family, and is expressed in low density throughout the human body (22,25). In pathologic conditions, GIP receptor is overexpressed in functional (such as insulinomas and gastrinomas) and nonfunctional pancreatic, ileal, and bronchial NETs. Malignant insulinomas are an interesting case of cancer since they all seem to preserve GIP receptor expression while seldom expressing somatostatin and GLP-1 receptors. The high expression of GIP receptor in these cases of NETs, in contrast to the low physiologic expression as proven by autoradiographic studies and messenger RNA analysis, may lead to high tumor-to-background ratios and fulfill the prerequisite for successful in vivo peptide receptor targeting (20).
In this study, we developed and evaluated a new class of GIP-based radioligands with the potential to image a broad spectrum of NETs. Three GIP-based radioligands, [Lys30(aminohexanoic acid [Ahx]-DOTA)]GIP(1–30)NH2 (EG1), [Lys16(Ahx-DOTA)]GIP(1–30)NH2 (EG2), and [Nle14, Lys30(Ahx-DOTA)]GIP(1–30)NH2 (EG4), were developed and labeled with 68Ga and 111In. In vitro evaluation included determination of inhibitory concentration of 50% (IC50), dissociation constant (Kd), and maximum number of binding sites (Bmax), along with internalization studies using the pancreatic endocrine cell line INR1G9 transfected with the human GIP receptor (INR1G9-hGIPr). With the most promising candidate, EG4, we extended the in vivo evaluation to biodistribution and PET studies in INR1G9-hGIPr xenografts.
MATERIALS AND METHODS
The supplier information for all reagents and details of instruments used are provided in the supplemental data (supplemental materials are available at http://jnm.snmjournals.org).
Radiolabeling with 111In and 68Ga
The radioligands were prepared as described in detail in the supplemental data. Briefly, 111In-conjugates were prepared by dissolving 20 μg (5 nmol) of peptide in 250 μL of ammonium acetate buffer (0.5 mol/L, pH 5.4), followed by incubation with 111InCl3 (35–40 MBq) for 30 min at 95°C. After the labeling with 111In, one equivalent of natInCl3 × 2H2O was added and the final solution was incubated again at 95°C for 30 min to obtain structurally characterized homogeneous ligands, which were used for the saturation binding studies. 68Ga-conjugates were prepared within 10 min at 95°C starting with 40 μg of the conjugates, followed by purification using the Modular-Lab PharmTracer module (Eckert and Ziegler).
Cell Line
Hamster glucagon-secreting pancreatic cells (INR1G9) kindly given by Prof. Jacques Philippe, University of Geneva, Switzerland, were transfected with pcDNA3 vector containing human cDNA encoding short variant of the human GIP receptor using lipofectamine. Selected clones were screened by fluorescence-activated call sorter analysis of binding of fluorescent tagged GIP(1–30) (Alexa Fluor 647 dye; Life Technologies), as previously described (25). The INR1G9-hGIPr cells were cultured at 37°C and 5% CO2 in Dulbecco modified Eagle medium containing fetal bovine serum (10%), penicillin (100 U/mL), streptomycin (100 μg/mL), and the antibiotic G418 (500 μg/mL) for the selection of the stably transfected cells.
Determination of Binding Affinity
The binding affinity profiles (IC50 values) of EG1, EG2, EG4, and GIP(1–30) were determined by in vitro GIP receptor autoradiography on cryostat sections of human insulinoma tissue as described previously (20). The radioligand used was [125I-Tyr10]GIP(1–30) (74,000 GBq [2,000 Ci]/mmol) (Anava) (20,25).
Saturation Binding and Internalization Studies
INR1G9-hGIPr cells were seeded at a density of 0.8–1 million cells per well in 6-well plates and incubated with increasing concentrations of either 111/natIn-EG2 or 111/natIn-EG4 (1–100 nM) for saturation binding studies. For internalization studies, approximately 2.5 pmol of the respective radiopeptide were added to the cell medium followed by incubation for 0.5, 1, 2, 4, and 6 h at 37°C, 5% CO2. Both procedures are described in detail in the supplemental data.
Animal Model
Female athymic nude mice (age, 4–6 wk; weight, 17–20 g) were purchased from Janvier. For implantation, the cells were harvested by trypsinization and 5 × 106 cells in 100 μL of phosphate-buffered saline were inoculated subcutaneously into the right shoulder of the mice. After an average of 2–3 wk, tumors reached 80–100 mg and the animals were used for biodistribution and PET studies. All animal experiments were approved by local authorities and were in compliance with institutional guidelines.
Biodistribution and Renal Uptake Reduction Studies
Ten picomoles/0.03–0.04 MBq of 111In-EG4 and 68Ga-EG4 in 100 μL of NaCl 0.9% were injected into the tail vein of INR1G9-hGIPr xenografts. Animals were sacrificed by isoflurane anesthesia at 1, 4, and 24 h after injection in the case of 111In-EG4 and at 1 and 2 h after injection in the case of 68Ga-EG4.
Three groups of mice (n = 4/group) were intravenously administered 100 μL of a modified-fluid-gelatin plasma substitute (Gelofusine; B. Braun) (40 mg/mL), were orally administered 150 μL (10 μmol/kg) of the DPP-4 inhibitor vildagliptin (Galvus [Novartis], 50 mg) dissolved in water, or were given the combination of the two. Gelofusine was administered just before and vildagliptin 1 h before intravenous injection of 10 pmol of the radiotracer. One hour later, the mice were sacrificed. Finally, radioactivity uptake in organs and tumors was calculated as percentage injected activity (%IA)/g. Biodistribution studies were conducted as described in detail in the supplemental data.
Small-Animal PET Studies
The mice were divided into 2 groups (n = 2/group). The mice of the first group were injected with 100 pmol/0.6–0.8 MBq/100 μL of 68Ga-EG4, and dynamic imaging was performed with the following acquisition frames: 30 × 10, 10 × 30, 6 × 600, 10 × 60, 10 × 120, and 4 × 300 s, amounting to a total imaging time of 60 min. Twenty-four hours later, the same mice were injected with 100 μL of Gelofusine (40 mg/mL) and 100 pmol/0.6–0.8 MBq/100 μL of 68Ga-EG4, and dynamic imaging was performed as described above. Immediately after the end of the dynamic scans, the mice were sacrificed and underwent nephrectomy, and static imaging was performed for 30 min. For the second group, on day 1 the mice were administered 150 μL of vildagliptin by oral gavage (10 μmol/kg). One hour later, intravenous injection of 100 pmol/0.6–0.8 MBq/100 μL of 68Ga-EG4 followed and dynamic imaging was performed as above. The day after, the same mice 1 h after oral administration of vildagliptin (10 μmol/kg) were injected with 100 μL of Gelofusine (40 mg/mL) and 100 pmol/0.6–0.8 MBq/100 μL of 68Ga-EG4, followed by dynamic imaging. Blocking experiments were performed as described above, and dynamic scans were obtained for 60 min following the same acquisition frames.
PET images were corrected for 68Ga decay and reconstructed with filtered backprojection. No correction was applied for attenuation. Images were generated using AMIDE software.
Statistical Analysis
All data are expressed as the mean ± SD. Prism 5 software (GraphPad Software) was used to determine statistical significance at the 95% confidence level, with a P value of less than 0.05 being considered significantly different.
RESULTS
Peptides and Radiolabeling
The EG1, EG2, and EG4 GIP ligands were custom-synthesized according to the solid-phase peptide synthesis method by Peptide Specialty Laboratories GmbH. The analytic data of the purified conjugates are reported in Table 1 and in the supplemental material.
Analytic Data Along with IC50 Values of Conjugates
The labeling yields for 111In-conjugates were more than 95% at a specific activity of 7–8 GBq/μmol. The GIP conjugates were labeled with 68Ga with a labeling yield of more than 98% and a specific activity ranging between 20 and 25 GBq/μmol.
Binding Affinities
Compared with the reference peptide GIP(1–30) (IC50, 1.3 ± 0.6 nM), EG1, EG2, and EG4 retained high affinity to human GIP receptor (IC50: 2.1 ± 1.0, 2.5 ± 0.8, and 1.5 ± 0.7 nM, respectively) (Table 1; Supplemental Fig. 1).
Saturation Binding and Internalization Studies
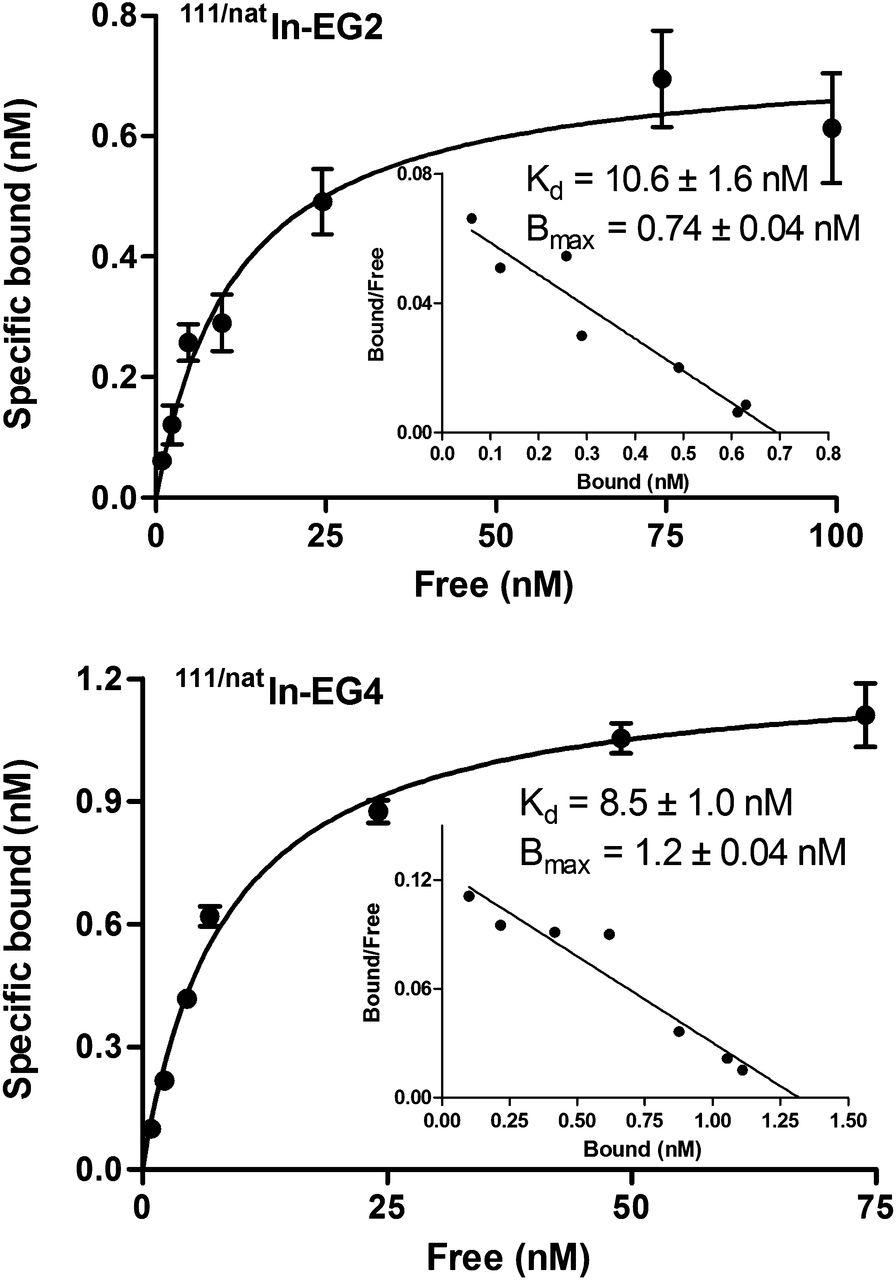
Saturation binding studies were performed at 4°C, to allow binding of the radioconjugates to the receptor but to avoid endocytosis. Both 111/natIn-EG2 and 111/natIn-EG4 exhibited similar affinity for the INR1G9-hGIP, with Kd values of 10.6 ± 1.6 nM for 111/natIn-EG2 and 8.5 ± 1.0 nM for 111/natIn-EG4 (Fig. 1). The Bmax values were also at the same level for both conjugates (0.7 ± 0.04 nM for 111/natIn-EG2 and 1.2 ± 0.04 nM for 111/natIn-EG4) and corresponded to approximately 6 × 105 receptors per cell (supplemental material).
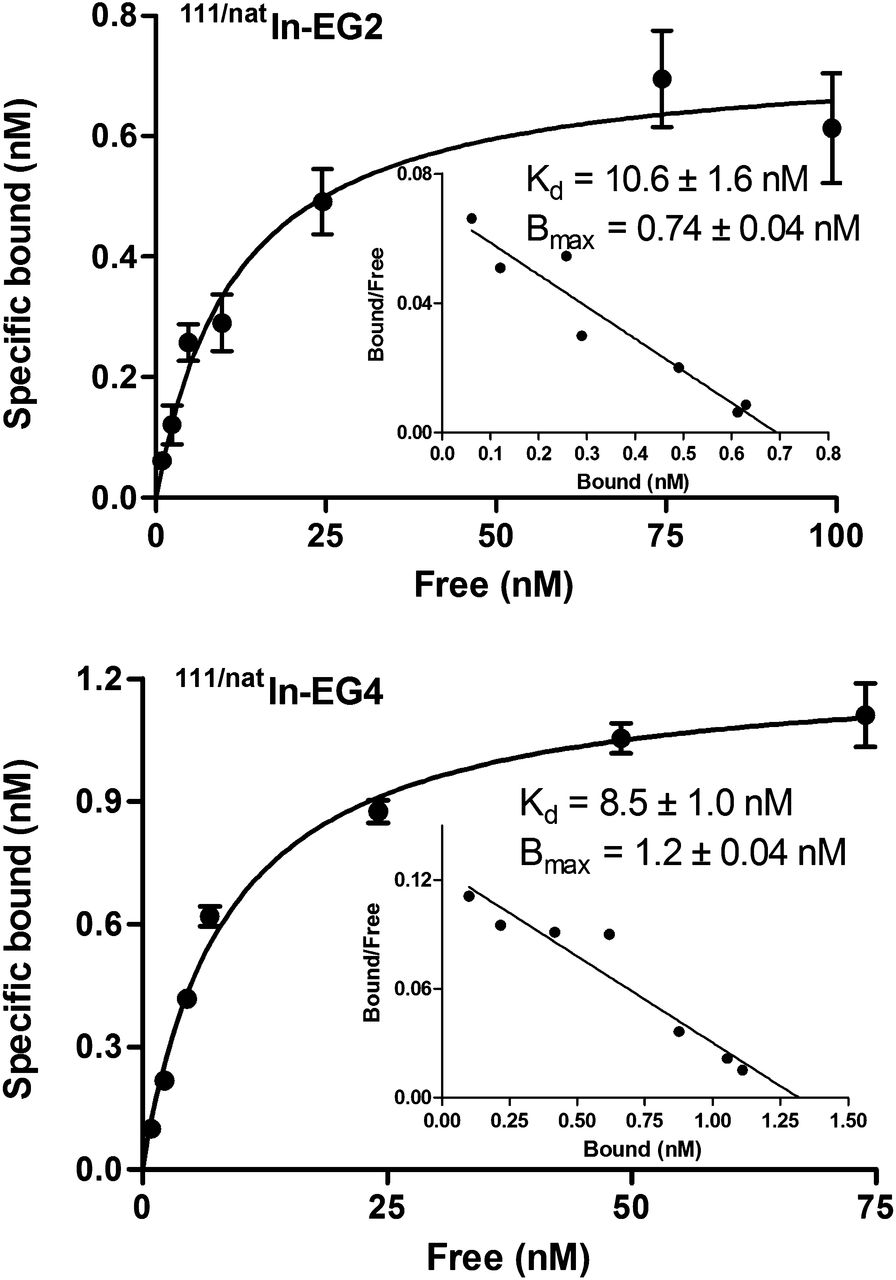
Saturation binding study on intact INR1G9-hGIPr cells, using increasing concentrations of 111/natIn-EG2 and 111/natIn-EG4, ranging from 0.1 to 1,000 nM. Kd and Bmax were calculated from nonlinear regression analysis using Prism 5 software.
Both 111In-labeled GIP ligands were well associated with the INR1G9-hGIPr cells within the 6-h incubation (Fig. 2). Continued exposure of the cells to the radioligands resulted in a gradual increase in the total cell-associated uptake from 30 min to 6 h, which was slightly higher for 111In-EG4 (19.7% ± 0.8%) than for 111In-EG2 (14.4% ± 1.4%) at 6 h (P = 0.0035). The internalized fraction increased with time from 30 min to 6 h, and at 6 h the internalization rate was somewhat higher for 111In-EG4 (18.3% ± 1.1%) than for 111In-EG2 (13.5% ± 1.1%) (P = 0.0054). Almost the entire cell-associated activity was internalized in both cases, supporting their agonist potency. Blocking experiments performed with an excess of GIP(1–30) showed negligible nonspecific binding on the cell surface, whereas less than 0.3% of total added radioactivity was found to be internalized (data not shown), demonstrating the high specificity of the GIP conjugates toward INR1G9-hGIPr cells.

Specific cell uptake and internalization rate after incubation of INR1G9-hGIPr cells with 111In-EG2 and 111In-EG4 for 6 h at 37°C. (A) Cell uptake calculated as cell surface–bound and internalized fraction. (B) Receptor-specific internalization expressed as percentage of applied radioactivity. Nonspecific binding was determined in presence of 1 μM GIP(1–30).
Biodistribution and Renal Uptake Reduction Studies
Both 111In-EG4 and 68Ga-EG4 showed fast blood clearance, with 0.04 %IA/g remaining in the blood at 4 h after injection of 111In-EG4 and 0.03 %IA/g at 2 h after injection of 68Ga-EG4, resulting in high tumor-to-blood ratios. 111In-EG4 and 68Ga-EG4 specifically targeted INR1G9-hGIPr xenografts. The tumor had the highest uptake—10.4 ± 2.2 and 17.0 ± 4.4 %IA/g, respectively—1 h after injection, and uptake dropped to 2.0 ± 0.2 and 1.3 ± 0.3 %IA/g 1 h after injection when a 2,000-times excess of GIP(1–30) was preinjected (Tables 2 and 3).
Biodistribution Data and Tissue Radioactivity Ratios of 111In-EG4 in INR1G9-hGIPr Xenografts
Biodistribution Data and Tissue Radioactivity Ratios of 68Ga-EG4 in INR1G9-hGIPr Xenografts
Kidney uptake was high and persistent for both 111In-EG4 and 68Ga-EG4, reaching values greater than 200 %IA/g. In separated groups of mice (n = 4/group), 100 μL (40 mg/mL) of Gelofusine or 150 μL (10 μmol/kg) of vildagliptin were administered before the injection of the radiopeptides followed by biodistribution studies at 1 h after injection (Tables 2 and 3). The administration of Gelofusine reduced kidney uptake by about 40% (32% for 68Ga-EG4 [P = 0.035] and 42% for 111In-EG4 [P = 0.034]) without significantly affecting the overall biodistribution profile. Similarly, when vildagliptin was administered, kidney uptake was reduced by a factor of about 2, leading to a tumor-to-kidney ratio of 0.09. The combination of Gelofusine and vildagliptin did not cause a further reduction of kidney uptake (P < 0.05) (Supplemental Table 2).
Small-Animal PET Studies
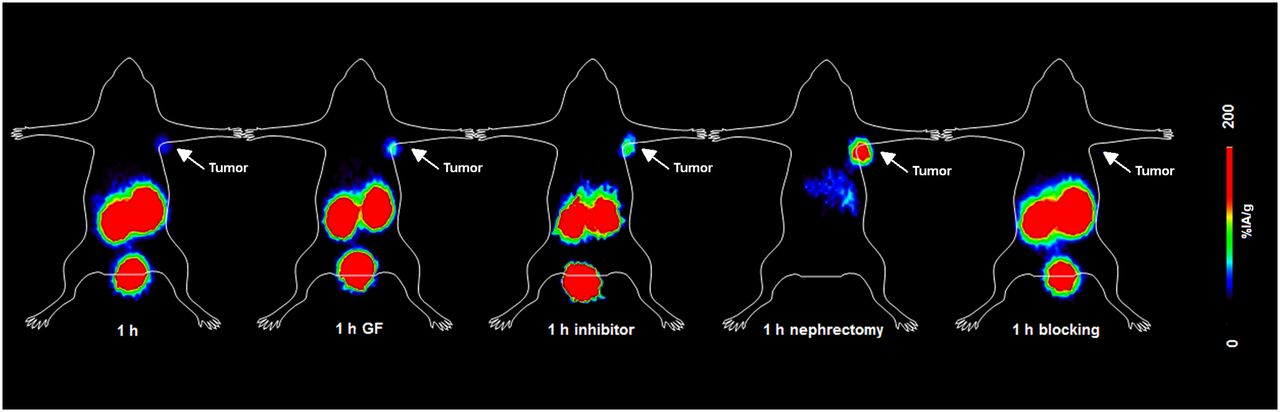
Representative PET images of INR1G9-hGIPr mice on injection of 68Ga-EG4 were obtained (Fig. 3). Specific tumor uptake was visualized within the 60-min time frame; the initial background uptake of 68Ga-EG4 in the excretion organs (kidneys) rapidly increased over time. These images well represented the results from the biodistribution studies and particularly the reduction of kidney uptake after administration of Gelofusine or vildagliptin. Furthermore, the nephrectomy allowed better visualization of the abdominal organs that were overlapped by the high kidney uptake. Urinary excretion of the tracer was represented by the high focal activity concentration in the urinary bladder at all time points.
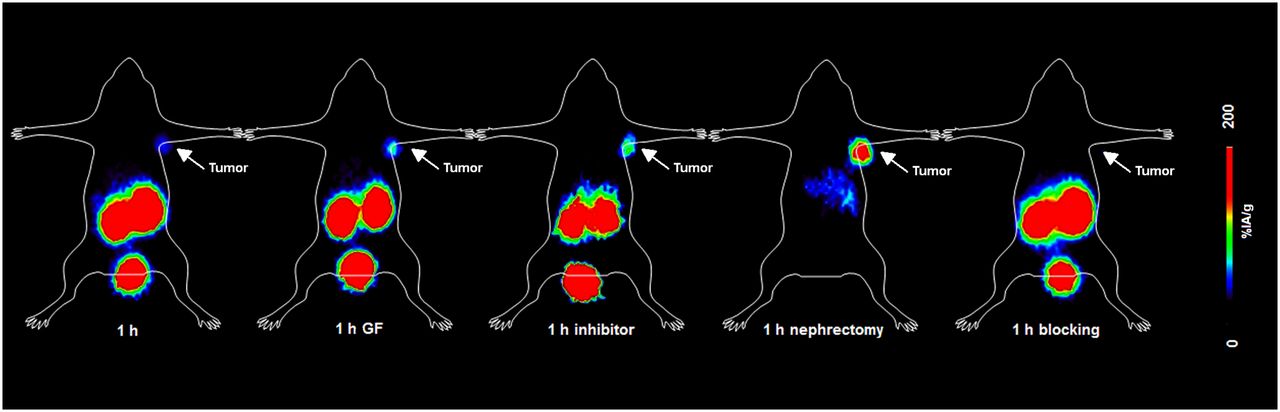
PET dynamic imaging of INR1G9-hGIPR tumor–bearing mice on injection of 68Ga-EG4 at 1 h after injection along with blocking studies. Images clearly visualized tumor and proved the specificity of the radiopeptide; kidney uptake was high. After administration of Gelofusine or vildagliptin, kidney uptake was lower, resulting in better delineation of tumor. After bilateral nephrectomy, 1 h after injection, uptake in abdomen was distinguishable from tumor with weak signal.
DISCUSSION
Despite almost 30 y of intense research on the development of radiopeptides, only the targeting of NETs has a real impact on patient care (7), because of the high expression of somatostatin and GLP-1 receptors (1). Unfortunately, the lack of somatostatin or GLP-1 receptor in specific NETs, such as insulinomas, demands the identification of a new family of receptors that qualifies for successful in vivo targeting of those tumors (20). In vitro autoradiography studies on tumor tissues from surgical specimens, evaluating the expression of GIP receptor versus somatostatin receptor, have shown that GIP receptor is overexpressed in a given number of samples originating from pancreatic NETs whereas only about 65% of the same samples were found to be positive for somatostatin receptor. Of 24 bronchial NETs, 22 were positive for GIP receptor but only 12 for somatostatin receptor (20). The fact that most of these tumors retain the GIP receptor even when they lack the somatostatin receptor paves the way for targeting a broader spectrum of NETs. Consequently, the importance of the overexpression of GIP receptor in NETs relies on 2 major advantages: first, the NETs that lack the somatostatin or GLP-1 receptor but retain the GIP receptor may be integrated into the successful peptide receptor targeting method. Furthermore, the simultaneous coexistence of GIP receptor with somatostatin or even GLP-1 receptor in a large number of NETs might argue for multiple receptor targeting using a cocktail of radioligands (8).
In this frame, we developed and evaluated a new class of radioligands with the potential to be used for the in vivo targeting of GIP receptor–positive tumors. Because the truncated peptide GIP(1–30) activates the GIP receptor as efficiently as the full-length peptide GIP(1–42) (25), the GIP(1–30) was chosen as the peptide motif of our study. The peptide was functionalized with the chelator DOTA via 6-Ahx coupled to the side chains of Lys16 and Lys30, respectively, to obtain EG1, EG2, and EG4. In EG4, the Met14 was substituted with Nle, as a widely used strategy for stabilizing peptides against oxidative damage and allowing easier handling. The GIP conjugates were labeled with 68Ga and 111In. Because of its availability from generator systems and the relative ease of labeling chemistry, the positron emitter 68Ga has gained increasing interest in the field of molecular imaging with PET (28). On the other hand, 111In is being used as an important SPECT label and as a surrogate of β emitters such as 90Y.
The coupling of Ahx-DOTA to Lys side chains of GIP(1–30) independently of the site of modification does not influence the binding affinity of the conjugates toward GIP receptor. The substitution of Met14 with Nle improved the affinity of EG4 about 1.5-fold compared with EG1. Furthermore, saturation binding studies exhibited high and comparable affinities for both 111/natIn-EG2 and 111/natIn-EG4, with Kd values in the low nanomolar range.
The further in vitro evaluation showed comparable cellular uptake of the 111In-labeled radiopeptides, with 111In-EG4 exhibiting a somewhat superior profile. In both cases, more than 90% of the total cell-associated activity was internalized into the cancer cells. The almost identical affinity of the DOTA conjugates in comparison to the parent agonist GIP(1–30) and the efficient receptor-mediated internalization led us to conclude that EGs are agonists (25,29).
We further proceeded with the in vivo evaluation of the most promising candidate, EG4, aiming at the development of a proof-of-principle radioligand that leads to feasible imaging of GIP receptor–positive tumors. Biodistribution studies of 111In-EG4 and 68Ga-EG4 showed fast blood clearance and specific targeting of INR1G9-hGIPr xenografts. The washout from the tumor is not yet understood and is contrary to the GLP-1 receptor–targeting radiopeptides, which impress by a long tumor retention time (12,13). The kidneys showed the highest uptake, which remained high at all tested time points. Similar kidney uptake was reported for [Lys40(Ahx-DTPA-111In)NH2]exendin-4, [Lys40(Ahx-DOTA-111In)NH2]exendin-4, and [Lys40(DOTA)]exendin-3 labeled either with 111In or 68Ga (12–14). Uptake of radiolabeled peptides in kidneys is a major problem because of the nephrotoxicity caused by the accumulated activity. This problem is even more pronounced if therapeutic applications are planned (30). The rationale for using Gelofusine as a reuptake blocking agent relies on the fact that the metallated DOTA-conjugated peptides contain 3 Asp, 1 Glu, and 1 Lys, having 3 negative charges at physiologic pH, Gelofusine is a succinylated gelatin with a net negative surface charge. Indeed, the administration of Gelofusine led to a reduction of kidney uptake by about 40%. One additional major parameter that needs to be considered is the high GIP peptide cleavage rate by DPP-4. Natural or synthetic regulatory peptides that possess at the N-terminus penultimate position a proline or alanine residue have proved to be good substrates for DPP-4 (30). Especially, for GIP, the minimal N-terminal truncation by DPP-4 leads to full inactivation of the truncated peptide GIP(3–42) (25). DPP-4 can be detected in most organs, such as the kidneys, liver, pancreas, and intestine; high activity was found in the kidneys, in which the enzyme is mostly localized in the brush-border and microvillus fractions. Indeed, Deacon et al. have shown that DPP-4 inhibition significantly reduces renal clearance, most probably because GIP degradation by DPP-4 takes place in the renal capillary endothelium (31). On the basis of these findings and in an attempt to increase the metabolic stability that should lead to higher tumor uptake or longer retention, and to further reduce the accumulated activity in the kidneys, we investigated the influence of the clinically used DPP-4 inhibitor, vildagliptin (32). Biodistribution studies showed a 2-fold reduction in kidney uptake compared with the control group of mice, leading to an improved tumor-to-kidney ratio. Surprisingly, the administration of vildagliptin did not affect tumor uptake, but the lower kidney uptake led to higher PET contrast.
Despite the high kidney uptake, PET imaging studies proved that delineation of the tumor is feasible. Higher contrast was achieved after the administration of Gelofusine or vildagliptin. At a high threshold level after bilateral nephrectomy, uptake in the abdomen was distinguishable from tumor, with a weak signal.
CONCLUSION
The preclinical evaluation of 111In- and 68Ga-labeled EG4 as proof-of-principle radioligands indicated that diagnostic imaging of GIP receptor–positive tumors is feasible. Coupling of GIP(1–30) to a DOTA chelator leads to GIP receptor–specific ligands able to be used for PET or SPECT imaging. Despite kidney uptake, which could be reduced by approximately 40%–50% either by Gelofusine or vildagliptin, these results prompt us to further continue the development of this family of radioligands that allows the imaging of a broad spectrum of NETs.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This work was supported by the German Consortium for Translational Cancer Research (DKTK) and Région Midi-Pyrénées, France. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Roswitha Toennesmann and Yvonne Kiefer for technical assistance and Véronique Gigoux for help in INR1G9-hGIPr cloning.
Footnotes
Published online Apr. 17, 2014.
- © 2014 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication October 10, 2013.
- Accepted for publication February 5, 2014.





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Drug Occupancy Assessment at the Glucose-Dependent Insulinotropic Polypeptide Receptor by Positron Emission Tomography
- Approaches to Multireceptor Targeting: Hybrid Radioligands, Radioligand Cocktails, and Sequential Radioligand Applications
- Prognostic Impact of Novel Molecular Subtypes of Small Intestinal Neuroendocrine Tumor
- Triple-Peptide Receptor Targeting In Vitro Allows Detection of All Tested Gut and Bronchial NETs
- Targeting Neuropeptide Receptors for Cancer Imaging and Therapy: Perspectives with Bombesin, Neurotensin, and Neuropeptide-Y Receptors