


Abstract
Gastrin-releasing peptide receptor (GRPR) is overexpressed in human prostate cancer and is being used as a target for molecular imaging. In this study, we report on the direct comparison of 3 novel GRPR-targeted radiolabeled tracers: Al18F-JMV5132, 68Ga-JMV5132, and 68Ga-JMV4168 (JMV5132 is NODA-MPAA-βAla-βAla-[H-d-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2], JMV4168 is DOTA-βAla-βAla-[H-d-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2], and NODA-MPAA is 2-[4-(carboxymethyl)-7-{[4-(carboxymethyl)phenyl]methyl}-1,4,7-triazacyclononan-1-yl]acetic acid). Methods: The GRPR antagonist JMV594 (H-d-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2) was conjugated to NODA-MPAA for labeling with Al18F. JMV5132 was radiolabeled with 68Ga and 18F, and JMV4168 was labeled with 68Ga for comparison. The inhibitory concentration of 50% values for binding GRPR of JMV4168, JMV5132, natGa-JMV4168, and natGa-JMV5132 were determined in a competition-binding assay using GRPR-overexpressing PC-3 tumors. The tumor-targeting characteristics of the compounds were assessed in mice bearing subcutaneous PC-3 xenografts. Small-animal PET/CT images were acquired, and tracer biodistribution was determined by ex vivo measurements. Results: JMV5132 was labeled with 18F in a novel 1-pot, 1-step procedure within 20 min, without need for further purification and resulting in a specific activity of 35 MBq/nmol. Inhibitory concentration of 50% values (in nM) for GRPR binding of JMV5132, JMV4168, natGa-JMV5132, natGa-JMV4168, and AlnatF-JMV5132 were 6.8 (95% confidence intervals [CIs], 4.6–10.0), 13.2 (95% CIs, 5.9–29.3), 3.0 (95% CIs, 1.5–6.0), 3.2 (95% CIs, 1.8–5.9), and 10.0 (95% CIs, 6.3–16.0), respectively. In mice with subcutaneous PC-3 xenografts, all tracers cleared rapidly from the blood, exclusively via the kidneys for 68Ga-JMV4168 and partially hepatobiliary for 68Ga-JMV5132 and Al18F-JMV5132. Two hours after injection, the uptake of 68Ga-JMV4168, 68Ga-JMV5132, and Al18F-JMV5132 in PC-3 tumors was 5.96 ± 1.39, 5.24 ± 0.29, 5.30 ± 0.98 (percentage injected dose per gram), respectively. GRPR specificity was confirmed by significantly reduced tumor uptake of the 3 tracers after coinjection of a 100-fold excess of unlabeled JMV4168 or JMV5132. Small-animal PET/CT clearly visualized PC-3 tumors, with the highest resolution observed for Al18F-JMV5132. Conclusion: JMV5132 could be rapidly and efficiently labeled with 18F. Al18F-JMV5132, 68Ga-JMV5132, and 68Ga-JMV4168 all showed comparable high and specific accumulation in GRPR-positive PC-3 tumors. These new PET tracers are promising candidates for future clinical translation.
Prostate cancer (PCa) is the most frequently diagnosed cancer and the second leading cause of cancer death among men in the United States (1). There is a strong need for improved imaging techniques that provide accurate staging and monitoring of this disease, particularly at early stages. Conventional diagnostic techniques, such as ultrasound-guided biopsy, are limited by high false-negative rates (2). Emerging functional imaging techniques, including diffusion-weighted MR imaging, dynamic contrast-enhanced MR imaging, and PET, have shown improved sensitivity and staging accuracy for detecting primary prostate tumors and metastatic lymph nodes (3). Several PET radiotracers have shown promising clinical utility, such as the metabolic agents 18F-FDG, 11C-/18F-choline, and 11C-/18F-acetate for the assessment of distant metastasis and 18F-NaF for the detection of bone metastasis (4). However, their application is most valuable in late-stage, recurrent, or metastatic PCa. Increasing efforts are being made in developing PET imaging agents targeting specific biomarkers of PCa, such as gastrin-releasing peptide receptor (GRPR) (5) and prostate-specific membrane antigen (6).
The GRPR, or bombesin (BBN) receptor subtype II, has been shown to be overexpressed in several human tumors, including PCa (7). Overexpression of GRPR was found in 63%–100% of primary prostate tumors and more than 50% of lymph and bone metastases (8,9). The GRPR density was reported to be 26-fold higher in prostate carcinoma than prostatic hyperplasia (9). Because of the low expression in benign prostatic hyperplasia and inflammatory prostatic tissues, imaging of GRPR in localized PCa has potential advantages over choline- and acetate-based radiotracers (9,10).
A variety of radiolabeled BBN analogs have been developed for targeting GRPR-positive tumors and were evaluated in preclinical and clinical studies (5). Several recent reports have shown that GRPR antagonists show properties superior to GRPR agonists, affording higher tumor uptake and lower accumulation in physiologic GRPR-positive nontarget tissues (11,12). Moreover, GRPR agonists were shown to induce side effects in patients, mediated by virtue of their physiologic activity (13,14). Therefore, particular attention has been drawn to the development of GRPR antagonists for imaging and radionuclide therapy of PCa. Several GRPR antagonists have since been developed by the modification of C-terminal residues of GRPR agonists, including the statin-based BBN analog, JMV594 (H-d-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2) (15).
68Ga-labeled GRPR antagonists were developed for PET imaging, showing good targeting properties in preclinical studies (12,16–18) and recently also in clinical trials (19,20). Clinical evaluation of the 68Ga-labeled GRPR antagonist BAY86-7548 (68Ga-DOTA-4-amino-1-carboxymethylpiperidine-d-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2) has shown a specificity, sensitivity, and accuracy of 88%, 81%, and 83%, respectively, for the detection of primary PCa. In comparison, it was shown that 11C-choline was not able to discriminate PCa from benign prostatic hyperplasia, because maximum standardized uptake values in PCa were not significantly different from benign prostatic hyperplasia (21). Another study reported a specificity, sensitivity, and accuracy of 80%, 29%, and 71%, respectively, for the detection of PCa using 11C-choline (22). The detection of lymph node metastases with the 68Ga-labeled GRPR antagonist was suboptimal, partially due to the suboptimal physical characteristics of 68Ga, limiting the detection of small lesions (20). Therefore, the aim of the present study was to develop an 18F-labeled GRPR antagonist for high-resolution and sensitive PET imaging of primary, recurrent, and metastatic PCa and to compare the imaging properties of this tracer with those of 68Ga-labeled analogs. 18F has superior physical characteristics for PET imaging, such as a lower positron range and a higher positron yield, offering higher resolution and sensitivity (23). However, most methods for labeling peptides with 18F are laborious and require multistep procedures with moderate labeling yields. A good alternative is the Al18F-labeling method (24), allowing fast and facile labeling of peptides in a 1-step procedure. We designed a new GRPR antagonist conjugate (JMV5132 [NODA-MPAA-βAla-βAla-(H-d-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2); NODA-MPAA is 2-(4-(carboxymethyl)-7-{[4-(carboxymethyl)phenyl]methyl}-1,4,7-triazacyclononan-1-yl)acetic acid]), analogous to the previously described JMV4168 (DOTA-βAla-βAla-[H-d-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2]) (25), with a NODA-MPAA chelator for high-yield complexation of Al18F (26). Here, we report on the direct preclinical comparison of 3 novel radiolabeled tracers (Al18F-JMV5132, 68Ga-JMV4168, and 68Ga-JMV5132) for PET imaging of PCa. We determined the in vitro characteristics of the radiolabeled peptides and evaluated their tumor targeting properties in vivo in nude mice with subcutaneous human prostate tumors.
MATERIALS AND METHODS
Synthesis of JMV4168 and JMV5132
JMV4168 was synthesized using Fmoc-based solid-phase peptide synthesis as described previously (25). JMV5132 was synthesized in the same manner as JMV4168 but was coupled to tert-butyl (tBu)–protected NODA-MPAA instead of tBu-protected DOTA. NODA-MPAA was prepared as previously described using NO2AtBu (Chematech) (26). The chemical structures of JMV4168 and JMV5132 are shown in Figure 1.

Chemical structures of DOTA-βAla-βAla-[H-d-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2] (JMV4168) (A) and NODA-MPAA-βAla-βAla-[H-d-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2] (JMV5132) (B).
Radiolabeling
Radiolabeling of JMV5132 with Al18F
18F− solution in enriched water (BV Cyclotron, VU) was purified from metal impurities and concentrated before use, as described in the supplemental materials (available at http://jnm.snmjournals.org). Labeling was performed by mixing 18F− solution (15–20 μL, 700–900 MBq), sodium acetate (NaOAc) (0.5 μL of 1 M solution, pH 4.1), Al3+ stock solution (20 nmol, 10 μL of 2 mM AlCl3⋅6H2O in 0.1 M NaOAc, pH 4.2), acetonitrile (67% v/v), quenchers (2.5 μL of 50 mM methionine, gentisic acid, and ascorbic acid), and finally JMV5132 (20 nmol, 3.26 μL of solution [10 μg/μL] in 2 mM NaOAc, pH 4.1). The reaction mixture was heated for 15 min at 105°C. To allow injection into mice, the peptide was diluted to less than 0.5% (v/v) acetonitrile with 0.5% (w/v) bovine serum albumin, 0.5% (w/v) polyoxyethylene (20) sorbitan monolaurate solution (polysorbate-20), and quenchers (1 mM methionine, gentisic acid, and ascorbic acid) in phosphate-buffered saline (PBS), pH 7.4. Bovine serum albumin and polysorbate-20 were added to reduce binding of radiolabeled peptide to plastic disposables, whereas quenchers (methionine, gentisic acid, and ascorbic acid) were added to prevent radiolysis of radiolabeled peptides.
Radiolabeling of JMV4168 and JMV5132 with 68Ga
Elution and purification of 68Ga from a 68Ga/68Ge generator (IGG-100; Eckert & Ziegler Europe) was performed using the sodium chloride–based procedure described earlier (27). A volume of 375 μL of 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (1 M, pH 3.6) was slowly added to 300 μL of purified 68Ga eluate, followed by addition of quenchers (methionine, gentisic acid, and ascorbic acid, 1.25 mM) and peptide (2 nmol). The reaction mixture was heated for 10 min at 95°C. After reaction, ethylenediaminetetraacetic acid (50 mM) was added to a final concentration of 5 mM to complex free 68Ga. For animal experiments, the labeled product was purified by reversed-phase high-performance liquid chromatography (RP-HPLC) using the gradient described in the “Quality Control” section of the supplemental materials and concentrated by evaporation. To allow injection into mice, the radiolabeled peptide was diluted with 0.5% (w/v) bovine serum albumin, 0.5% (w/v) polysorbate-20, and quenchers (1 mM methionine, gentisic acid, and ascorbic acid) in PBS and neutralized with sodium carbonate (NaHCO3) buffer (1 M, pH 8.5).
Cold Labeling of JMV4168 and JMV5132 with natGa and natF
The labeling methods of JMV4168 and JMV5132 with natGa and natF are described in the supplemental materials.
Quality Control
Quality control methods for peptide synthesis and radiolabeling are described in detail in the supplemental materials.
Lipophilicity
The octanol/PBS partition coefficients of 68Ga-JMV4168, 68Ga-JMV5132, and Al18F-JMV5132 were determined as described previously (28).
Cell Culture and Competitive Cell Binding Assay
The human PCa cell line PC-3 was cultured in Ham F-12K (Kaighn’s) medium (Life Technologies) supplemented with 10% fetal calf serum, penicillin (100 units/mL), and streptomycin (100 μg/mL). Cells were grown in tissue culture flasks at 37°C in a humidified atmosphere containing 5% CO2.
The binding affinities of JMV4168, JMV5132, natGa-JMV4168, natGa-JMV5132, and AlnatF-JMV5132 toward the GRPR were determined in a competitive binding assay using frozen cryostat sections (10-μm thick) of PC-3 xenografts and [125I-Tyr4]-BBN as a tracer. Tyr4-BBN (Sigma Aldrich) was iodinated as described earlier (29). The competitive binding assay protocol is described in detail in the supplemental materials.
Small-Animal PET/CT and Biodistribution Studies
Male nude BALB/c mice (age, 6–8 wk) were injected subcutaneously near the right shoulder with a PC-3 cell suspension (5 × 106 cells, 200 μL, 66% RPMI, 33% Matrigel [BD Biosciences]). Two to 3 wk after inoculation, when tumor size averaged 200 mm3, mice were injected intravenously with 5–10 MBq of radiolabeled peptide (200 pmol, 200 μL). To confirm the receptor specificity of the radiolabeled peptides, additional animals were coinjected with an excess (20 nmol) of unlabeled peptide. Mice were euthanized 1 or 2 h after injection by CO2/O2 asphyxiation. Mice were first scanned prone on a small-animal PET/CT scanner (Inveon; Siemens Preclinical Solutions). PET emission scans were acquired for 30–60 min, followed by a CT scan. After scanning, blood, tumor, and relevant organs and tissues were collected, weighed, and counted in a γ counter. Scanning and reconstruction parameters, as well as counting parameters, are described in detail in the supplemental materials. The percentage injected dose per gram (%ID/g) was determined for each tissue sample. All animal experiments were approved by local authorities and were in compliance with the institutions guidelines.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism (version 5.01; GraphPad Software) and described in the supplemental materials.
RESULTS
Synthesis of JMV4168 and JMV5132
JMV4168 and JMV5132 (Fig. 1) were obtained with an average yield of approximately 40% and a purity greater than 97% as confirmed by RP-HPLC. Conjugates were characterized by electrospray ionization mass spectroscopy (mass/charge, [M+2H]2+/2: JMV4168, calculated: 815.9414, found: 815.9412; JMV5132, calculated: 821.4416, found: 821.4433).
Radiolabeling and Stability Studies
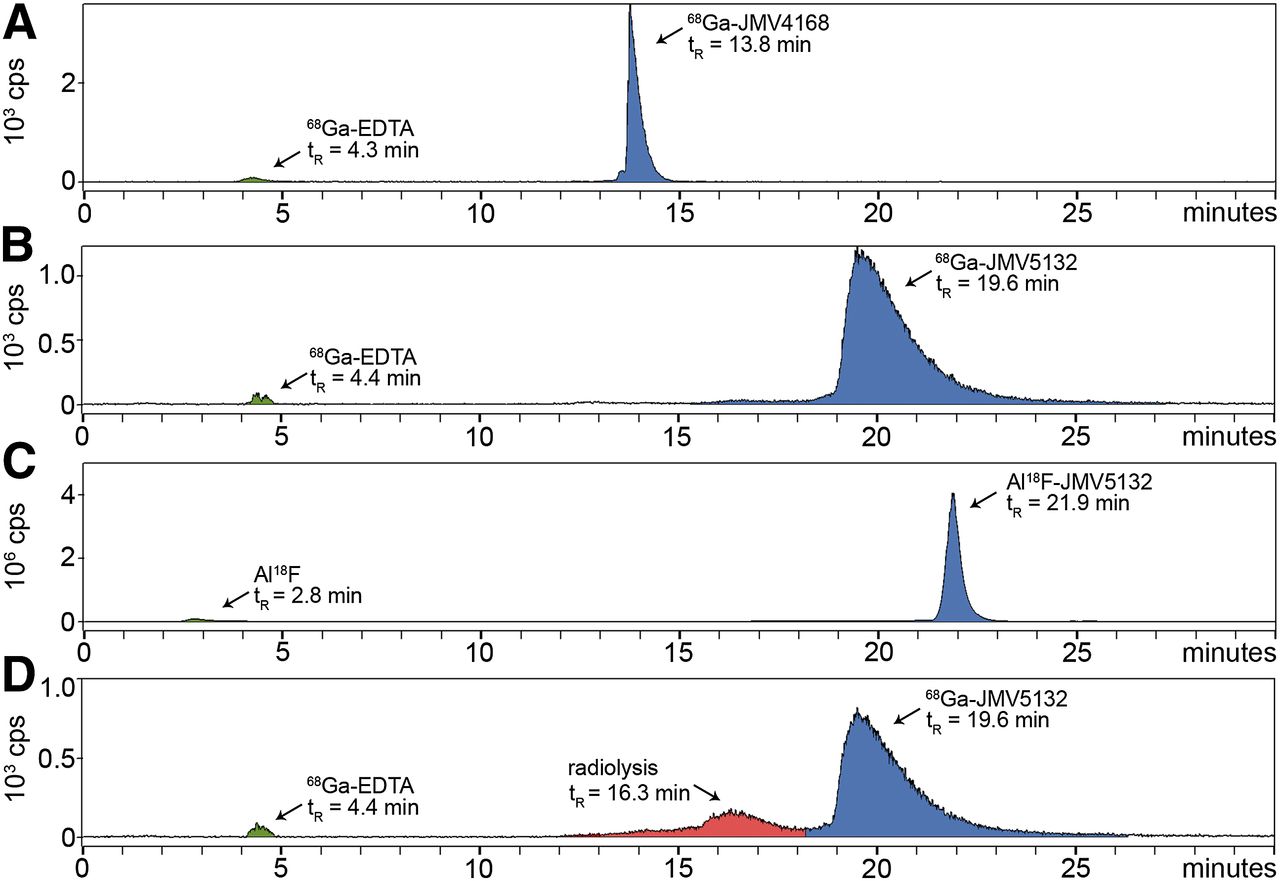
Al18F-JMV5132 was obtained with a specific activity of 40 ± 4 MBq/nmol (88% non–decay-corrected yield) and 68Ga-JMV4168 and 68Ga-JMV5132 with a specific activity of 47 ± 2 and 47 ± 4 MBq/nmol after purification, respectively. RP-HPLC analysis indicated that the radiochemical purity of the Al18F- or 68Ga-labeled peptide preparations used in in vitro and in vivo experiments always exceeded 95%. Radio-HPLC elution profiles of Al18F- and 68Ga-labeled peptides are shown in Figure 2A. 68Ga-JMV4168, 68Ga-JMV5132, and Al18F-JMV5132 had retention times of 13.8, 19.6, and 21.9 min, respectively. The addition of quenchers (methionine, gentisic acid, and ascorbic acid) prevented oxidation of the radiolabeled peptides, as shown in Figure 2B.
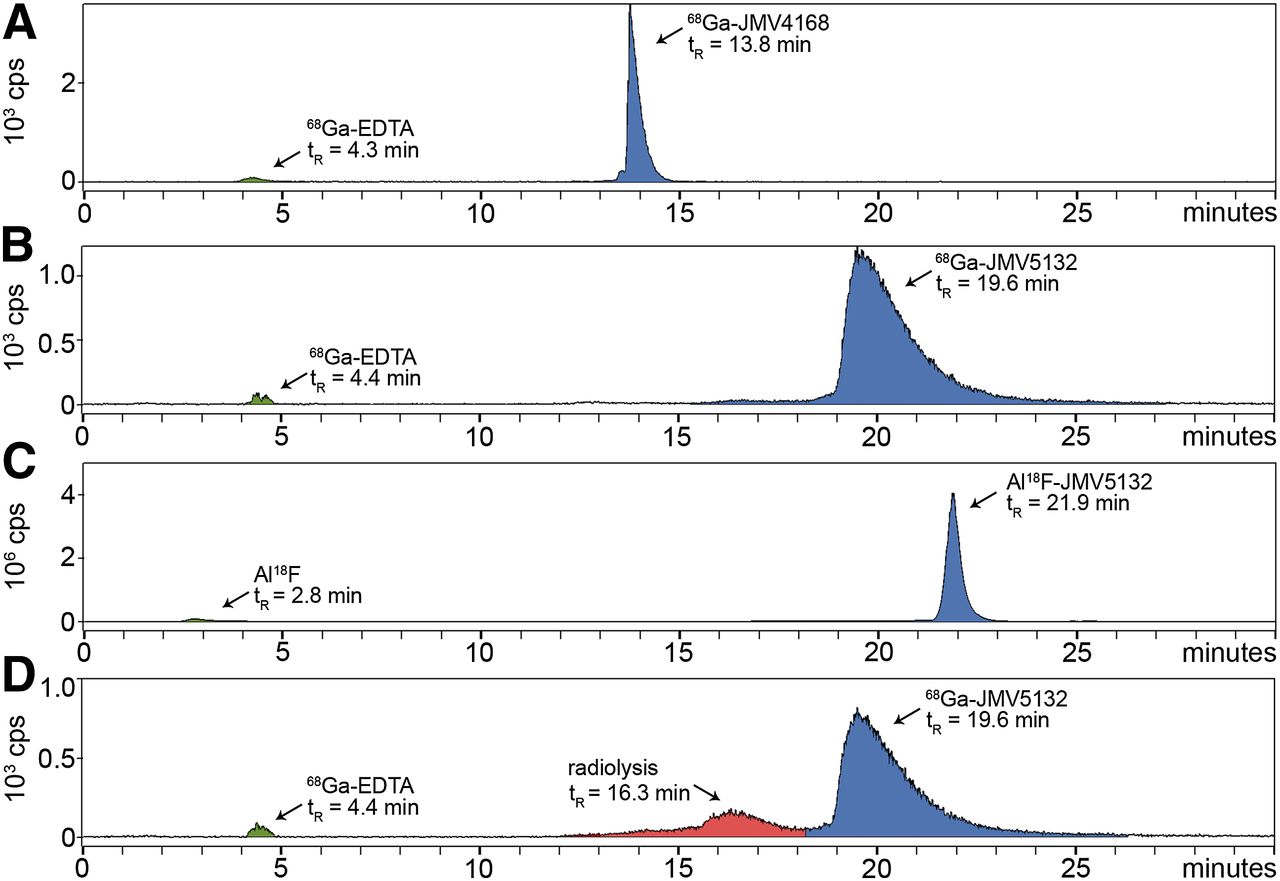
Radio–high-performance liquid chromatograms of 68Ga-JMV4168 (A), 68Ga-JMV5132 (B), and Al18F-JMV5132 (C). (D) Radio–high-performance liquid chromatogram of 68Ga-JMV5132 without added quenchers. y-axis = radioactivity in count per second (cps); x-axis = retention time in min. EDTA = ethylenediaminetetraacetic acid; tR = ▪▪▪retention time.
Lipophilicity
The octanol/PBS partition coefficients were determined to estimate the lipophilicity of the Al18F- or 68Ga-labeled peptides. The log Poctanol/PBS values for 68Ga-JMV4168, 68Ga-JMV5132, and Al18F-JMV5132 were −2.53 ± 0.04, −1.40 ± 0.01, and −1.56 ± 0.08, respectively. This result shows that the 68Ga-DOTA analog (JMV4168) was more hydrophilic than the 68Ga- and 18F-NODA-MPAA analogs (JMV5132).
Competitive Cell Binding Assay
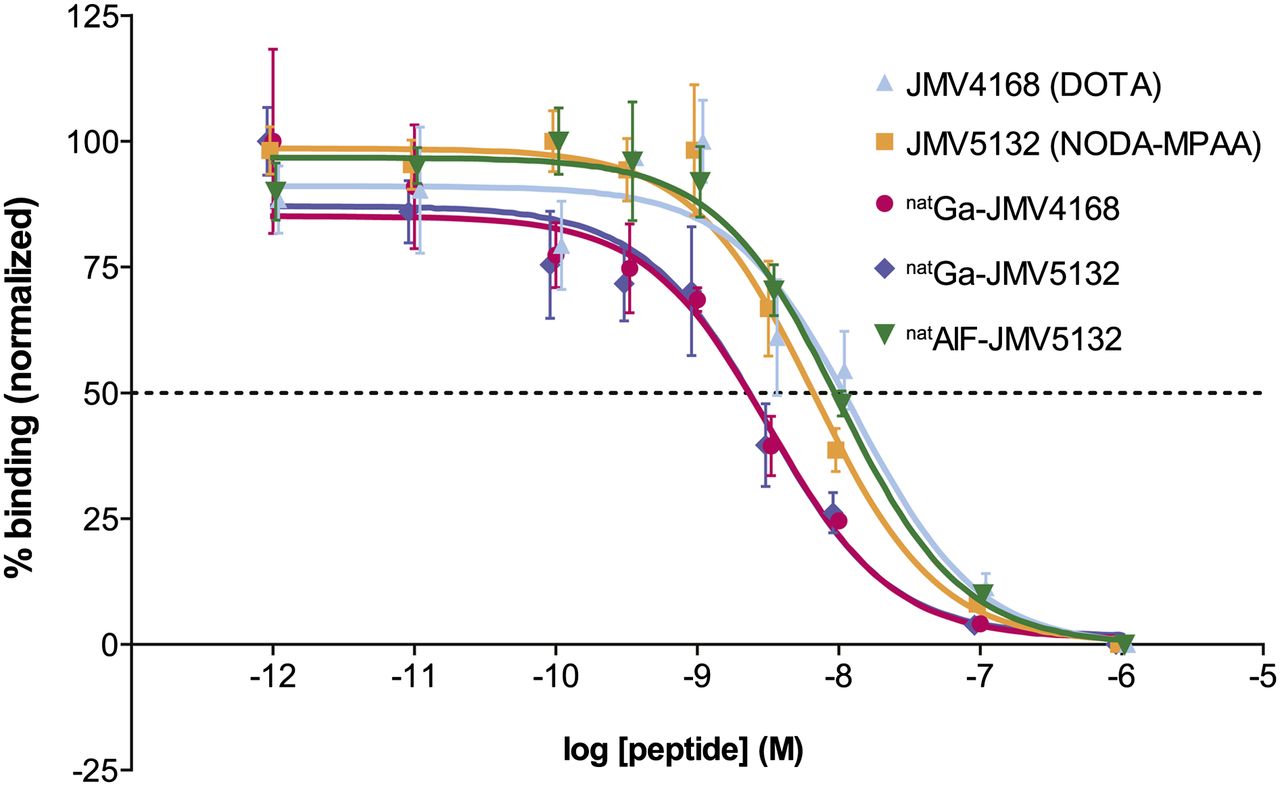
The affinity of JMV4168, JMV5132, natGa-JMV4168, natGa-JMV5132, and AlnatF-JMV5132 for the GRPR was determined in a competitive binding assay, using [125I-Tyr4]-BBN as radioligand. The displacement binding curves are shown in Figure 3. Inhibitory concentration of 50% (IC50) values (in nM) for binding to GRPR for JMV5132 (NODA-MPAA), JMV4168 (DOTA), and AlnatF-JMV5132 were not significantly different: 6.8 (95% confidence interval [CI], 4.6–10.0), 13.2 (95% CI, 5.9–29.3), and 10.0 nM (95% CI, 6.3–16.0), respectively. IC50 values for natGa-JMV5132 (3.0 [95% CI, 1.5–6.0]) and natGa-JMV4168 (3.2 [95% CI, 1.8–5.9]) were lower than their unlabeled counterpart, indicating a higher binding affinity for the GRPR.
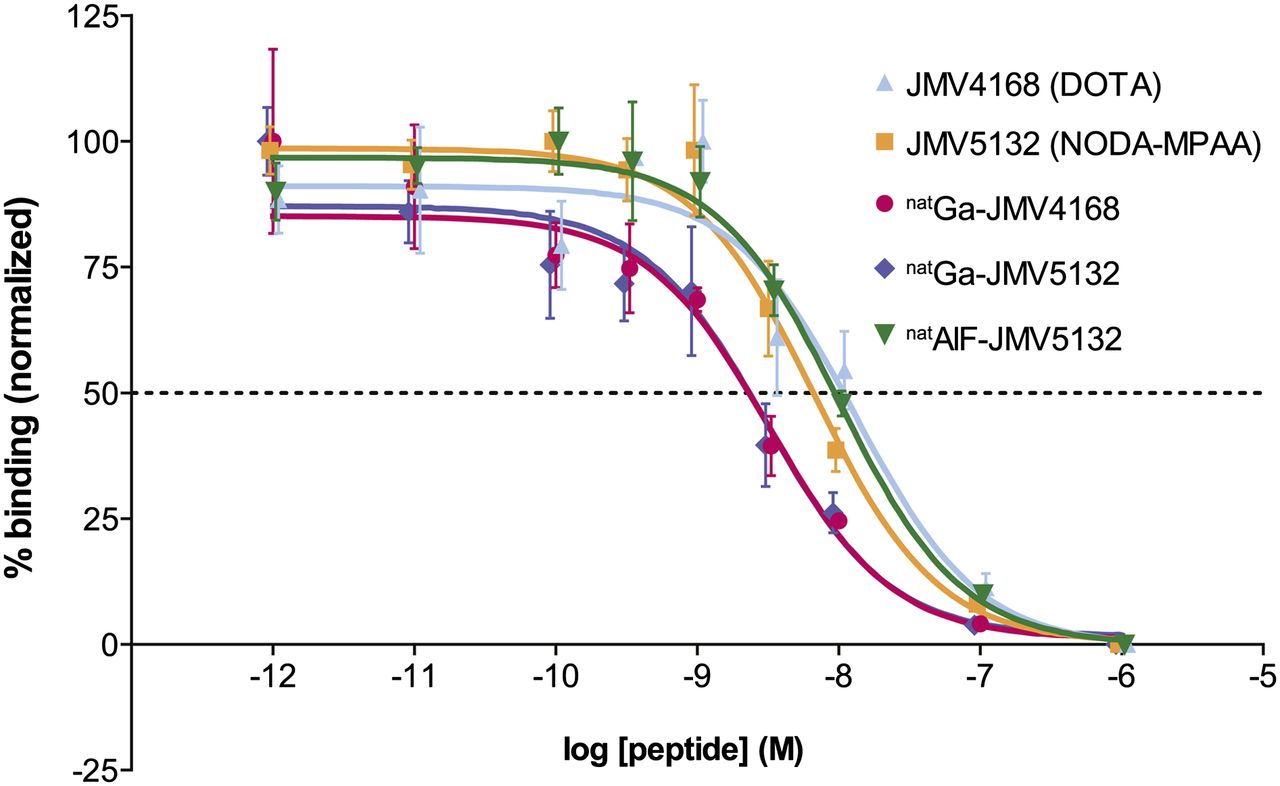
Competition binding curves. PC-3 frozen sections were incubated in presence of 5.10−10 M [125I-Tyr4]-BBN and increasing amounts of JMV4168, JMV5132, natGa-JMV4168, or natGa-JMV5132. IC50 values (with 95% CIs in parentheses) were 6.8 nM (4.6–10.0) for JMV5132, 13.2 nM (5.9–29.3) for JMV4168, 3.0 nM (1.5–6.0) for natGa-JMV5132, 3.2 nM (1.8–5.9) for natGa-JMV4168, and 10.0 nM (6.3–16.0) for AlnatF-JMV5132.
Small-Animal PET/CT and Biodistribution Studies
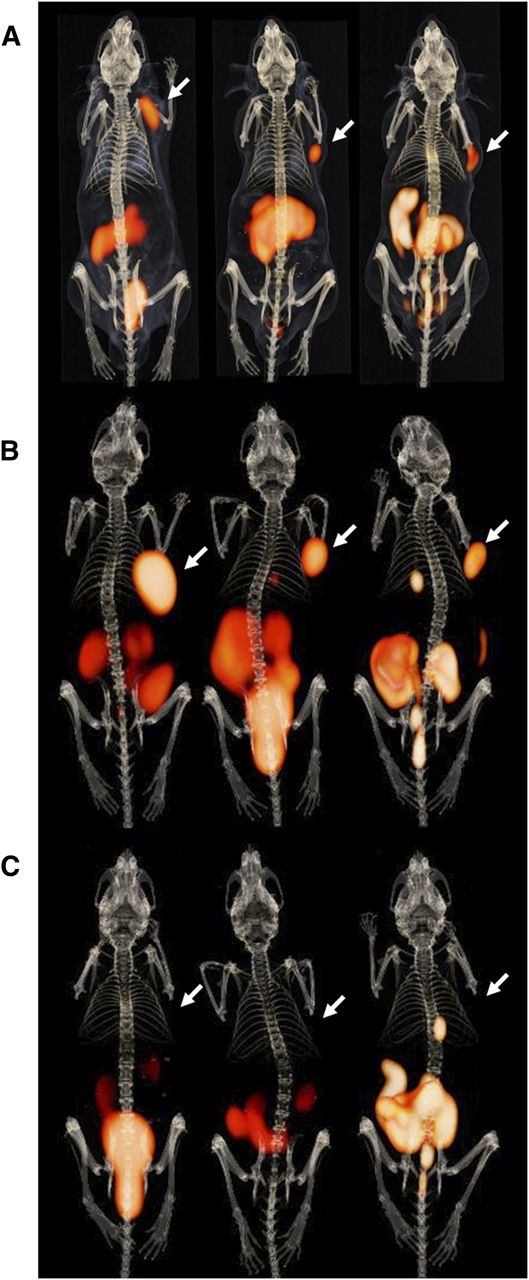
Fused PET and CT images obtained at 1 and 2 h after injection are shown in Figure 4. Maximum-intensity projections showed clear visualization of PC-3 tumors with very low background. Predominant renal excretion was observed for all 3 radiolabeled peptides. Partial hepatobiliary excretion was observed for Al18F-JMV5132 and 68Ga-JMV5132, as indicated by the nonspecific uptake in the gallbladder and intestines. PET images obtained at 2 h after injection showed partial clearance of radioactivity in nontarget tissues such as pancreas, kidney, and intestines as compared with the images obtained at 1 h after injection.

PET/CT images of mice bearing subcutaneous PC-3 xenografts on right shoulder (arrow) injected with 68Ga-JMV4168 (left), 68Ga-JMV5132 (center), or Al18F-JMV5132 (right) at 1 h after injection (A), 2 h after injection (B), and 2 h after injection with coinjection of excess unlabeled peptide (C). Besides tumor (arrow), pancreas, intestines, and kidneys can be observed in abdominal region. Bladder can be distinguished by hot spot below abdomen. Gallbladder can be recognized as hot spot below rib cage.
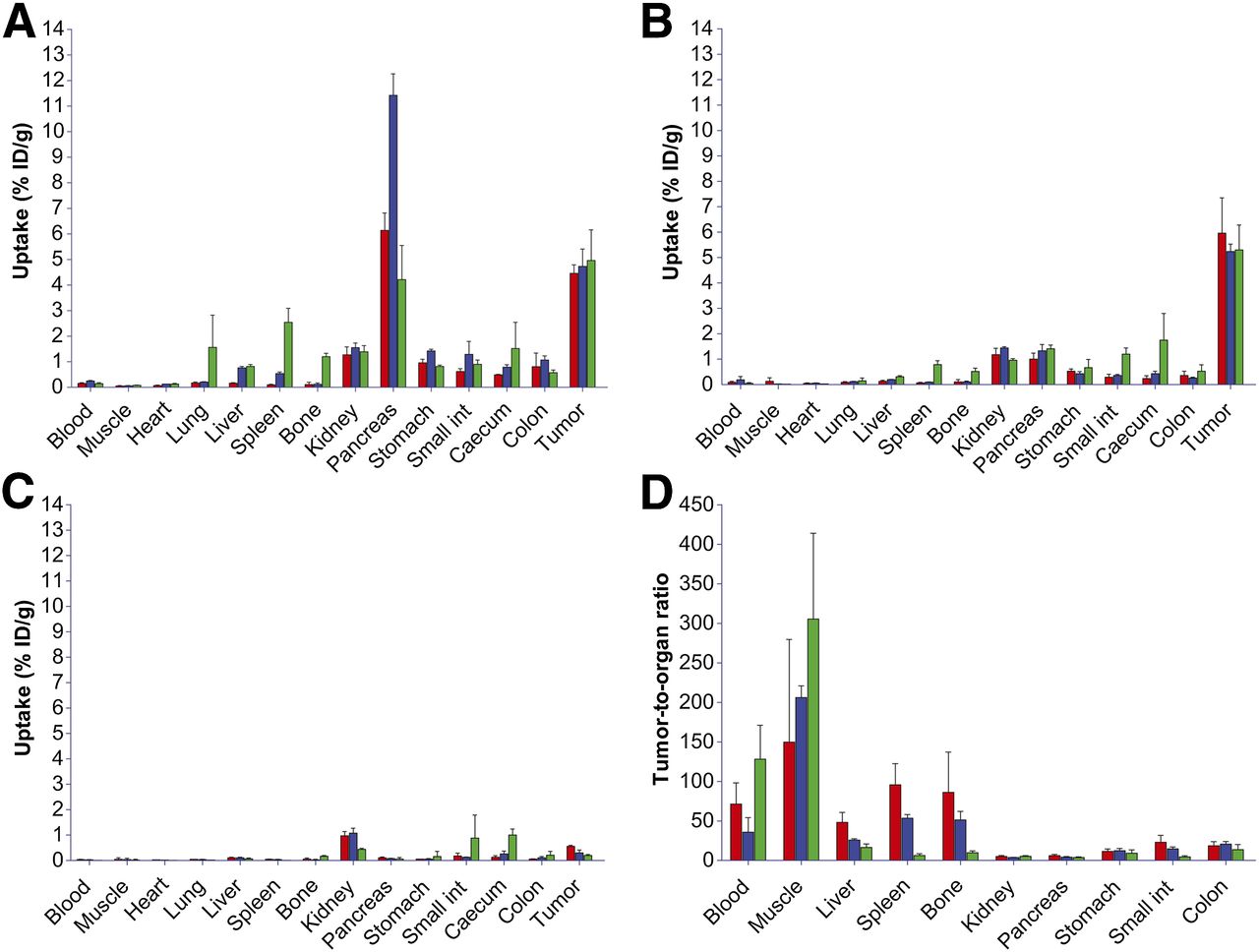
The results of the biodistribution studies of 18F- and 68Ga-labeled peptides are summarized in Figure 5. These pharmacokinetic data obtained at 1 and 2 h after injection were in line with the PET images. High and specific uptake of the tracer was observed in the PC-3 tumors. There were no significant differences for 68Ga-JMV4168, 68Ga-JMV5132, and Al18F-JMV5132, with uptake values (in %ID/g) of 4.46 ± 0.33, 4.73 ± 0.68, and 4.96 ± 1.20, respectively, at 1 h after injection and 5.96 ± 1.39, 5.24 ± 0.29, and 5.30 ± 0.98, respectively, at 2 h after injection. The uptake in GRPR-positive organs, such as tumor, pancreas, stomach, and intestines, was significantly decreased by coinjection of an excess of unlabeled peptide, indicating GRPR-specific targeting. All tracers displayed fast blood clearance, with 0.09 ± 0.04, 0.19 ± 0.13, and 0.05 ± 0.01 %ID/g remaining in blood at 2 h after injection for 68Ga-JMV4168, 68Ga-JMV5132, and Al18F-JMV5132, respectively. The 3 tracers cleared rapidly from the GRPR-positive pancreas between 1 h (6.14 ± 0.68, 11.42 ± 0.84, and 4.21 ± 1.34 %ID/g) and 2 h after injection (1.00 ± 0.24, 1.33 ± 0.25, and 1.41 ± 0.15 %ID/g) for 68Ga-JMV4168, 68Ga-JMV5132, and Al18F-JMV5132, respectively, whereas PC-3 tumor uptake was preserved. The uptake and retention of all tracers in blood, muscle, heart, lung, liver, and bone were relatively low as measured at 2 h after injection (all ≤0.5 %ID/g). Mice injected with Al18F-JMV5132 showed a (significantly) higher uptake in spleen and bone.
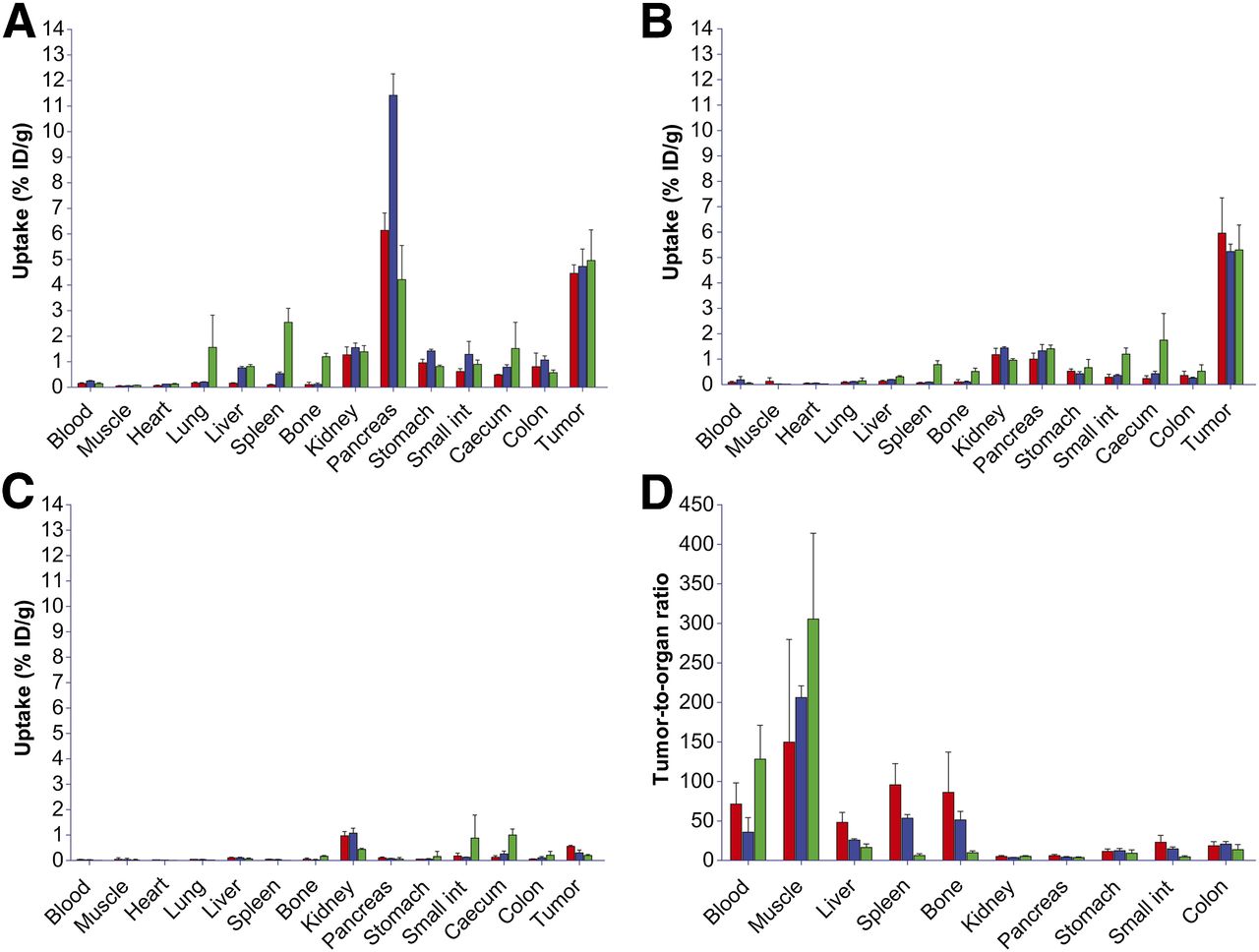
Biodistribution of 68Ga-JMV4168 (red), 68Ga-JMV5132 (blue), and Al18F-JMV5132 (green) in mice bearing PC-3 xenografts at 1 h after injection (A), 2 h after injection (B), and 2 h after injection with coinjection of excess unlabeled peptide (C) and tumor-to-organ ratios at 2 h after injection (D). Int = intestines.
DISCUSSION
The use of radiolabeled GRPR antagonists for targeting tumors in vivo has attracted considerable attention, starting with somatostatin receptor antagonists showing higher tumor uptake and targeting more receptor-binding sites than their agonists (30). This finding was also extended to GRPR antagonists, with the seminal work of Cescato et al. (11). Recently, more articles have appeared showing the promise of novel radiolabeled GRPR antagonists for GRPR-positive tumor imaging (12,16–18,25). The studies revealed favorable pharmacokinetics of radiolabeled antagonists, including high tumor uptake and fast clearance from nontargeted tissues. Several 64Cu- and 68Ga-labeled receptor antagonists developed for PET imaging of prostate tumors have shown pharmacokinetics superior to 64Cu- or 18F-labeled GRPR agonists described in earlier literature (12,16,17). Besides a favorable pharmacokinetic profile, the use of antagonists should reduce the occurrence of side effects. In a study in which BBN was infused intravenously in a dose of 15 ng/kg/min over a 90-min period, side effects—among which were nausea, hot flush, and sweating—were observed in 80% of the patients (13).
Here, we report on the development of a NODA-MPAA–conjugated GRPR antagonist (JMV5132) labeled with 18F for PET imaging of GRPR-positive tumors and the direct comparison with 68Ga-radiolabeled analogs. In our previous work, the statin-based GRPR antagonist JMV594 was linked to DOTA via a (βAla)2 linker and labeled with 111In. It showed good tumor targeting in PC-3 xenografts in mice (25). In the present study, we conjugated JMV594 to NODA-MPAA for radiolabeling with 18F. The NODA-MPAA-(βAla)2-JMV594 peptide (JMV5132) was labeled with 18F and 68Ga and compared with the 68Ga-labeled DOTA-(βAla)2-JMV594 peptide (JMV4168).
The radiolabeling of peptides via complexation of Al18F by a NOTA chelator was first described by McBride et al. (24). This novel technique has been successfully applied to several peptides, including a GRPR agonist (28) and recently to GRPR antagonists (31,32). Recently, McBride et al. reported the labeling of peptides with Al18F in a 1-pot, 1-step procedure using the NODA-MPAA chelator (26,33), leading to a kit formulation, after which the labeled peptide could be purified by solid-phase extraction.
In the present study, we further optimized the labeling conditions to achieve Al18F-labeled JMV5132 in less than 20 min with complete incorporation of 18F-fluoride, resulting in a high specific activity (35 MBq/nmol), without the need for purification by solid-phase extraction. In receptor-binding studies using PC-3 tumor sections, the in vitro affinities of JMV5132 and JMV4168 were comparable, as shown by the similar IC50 values, indicating that both chelators apparently affected receptor affinity in a similar way. The peptides labeled with natGa had slightly higher receptor affinities than their unlabeled counterpart, indicating that the presence of Ga3+ in the chelator enhanced the affinity of the peptides for the GRPR. The affinity of the peptide labeled with natAlF, on the other hand, was not significantly different from its unlabeled counterpart, indicating that the presence of AlF in the chelator did not affect the affinity of the peptide for the GRPR.
The PET images obtained with Al18F-JMV5132 showed higher spatial resolution than the images obtained with the 68Ga-labeled tracers, which is most likely due to the longer positron range of 68Ga (34) as compared with 18F.
The comparative biodistribution study showed GRPR-specific accumulation of all 3 radiolabeled GRPR antagonists in the tumor. 68Ga-JMV4168, 68Ga-JMV5132, and Al18F-JMV5132 tracers showed similar uptake in the GRPR-positive tumor and organs, including PC-3 tumor, pancreas, stomach, and colon. The uptake was receptor-mediated, as confirmed by the reduction of uptake in tumor and other receptor-positive organs after coinjection of excess unlabeled peptide. The washout from receptor-positive organs occurred at different rates. The pancreas uptake decreased from 1 to 2 h after injection by a factor of 6.1, 8.6, and 3.0 for 68Ga-JMV4168, 68Ga-JMV5132, and Al18F-JMV5132, respectively, whereas tumor uptake was increased by a factor of 1.3, 1.1, and 1.1 for 68Ga-JMV4168, 68Ga-JMV5132, and Al18F-JMV5132, respectively. This outcome indicates a higher retention of the tracers in the tumor than in the pancreas.
Despite their low internalization rate, the high and persistent tumor uptake of these radiolabeled antagonists was expected, as it was previously described for a few other radiolabeled antagonists (11,12,16,18). This might be explained by a higher number of binding sites for receptor antagonists than agonists, a higher metabolic stability of antagonists, or a strong interaction of the antagonist with the receptor (11,16). In previous studies using radiolabeled GRPR antagonists, a faster clearance from the pancreas and abdominal organs was already observed between 1 and 4 h after injection, in contrast with data concerning radiolabeled GRPR agonists, which showed more sustained retention of activity in the abdominal region. Several reasons for these differences in tissue clearance kinetics have been postulated, including species differences or more efficient perfusion in the pancreas and intestine (16). Possible metabolic degradation of the peptide by enzymes in the pancreas might also explain the faster washout from the pancreas.
Clearance from background tissues, such as blood, muscle, heart, lung, liver, and bone, was fast for all tracers tested, leading to high tumor-to-background ratios, which allowed clear visualization of the tumor. Overall, Al18F-JMV5132 showed improved imaging properties, compared with the previously reported Al18F-NOTA-8-Aoc-BBN(7-14)NH2 GRPR agonist (28), because that analog showed lower tumor uptake, much higher pancreatic uptake, and higher liver and intestinal uptake in the same animal model.
The slightly higher uptake of Al18F-JMV5132 in bone may be due to the presence of trace amounts (<1%) of uncomplexed Al18F or 18F-fluoride or partial defluorination of the tracer in vivo. The uptake of Al18F-JMV5132 in bone was relatively low (0.52 ± 0.13 %ID/g 2 h after injection), in comparison with the values reported for the Al18F-labeled RM1 derivative (1.58 %ID/g 2 h after injection) (31). The increased uptake of Al18F-JMV5132 and 68Ga-JMV5132 in the gallbladder, liver, and gastrointestinal excretions indicates partial hepatobiliary excretion of the tracers, because of their higher lipophilicity, which may be caused by the benzyl group. Considering the clinical application of the tracers, high signal intensity in the intestines may affect visualization of prostate-confined tumor or spread to lymph nodes. Nevertheless, considering the superior imaging characteristics of 18F, further development of Al18F-JMV5132 as a tracer for PCa diagnostic and therapy follow-up is certainly warranted.
CONCLUSION
Highly sensitive and receptor-specific imaging of PCa with PET/CT can be achieved using 68Ga- and 18F-labeled GRPR antagonists. In this study, labeling of JMV5132 with Al18F was completed within 20 min with high specific activity without a need for purification. The 68Ga-JMV4168 tracer showed the most favorable biodistribution, because of its lower hepatobiliary excretion, but the PET images showed a higher resolution with the 18F-JMV5132 tracer. These new PET tracers are promising candidates for future clinical translation.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This project is funded by the Erasmus MC grant “Novel Radio-Antagonists for PET/MRI Imaging and Therapy of Prostate Cancer.” Drs. McBride and Goldenberg have employment and stock ownership with Immunomedics, Inc., which has patented the Al18F labeling technology. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Bianca Lemmers-van de Weem, Kitty Lemmens-Hermans, Henk Arnts, and Iris Lamers-Elemans for their technical assistance during animal experiments and Linda Van der Graaf for her contribution to receptor-binding experiments.
Footnotes
Published online Nov. 20, 2014.
- © 2014 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication April 3, 2014.
- Accepted for publication September 5, 2014.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- 18F-AlF-Labeled Biomolecule Conjugates as Imaging Pharmaceuticals
- Bombesin-Targeted PET of Prostate Cancer
- 68Ga-NOTA-Aca-BBN(7-14) PET/CT in Healthy Volunteers and Glioma Patients
- Radiopeptides for Imaging and Therapy: A Radiant Future
- A Novel 111In-Labeled Anti-Prostate-Specific Membrane Antigen Nanobody for Targeted SPECT/CT Imaging of Prostate Cancer