


Abstract
6-18F-fluoro-l-dopa (18F-FDOPA) has proven to be a useful radiopharmaceutical for the evaluation of presynaptic dopaminergic function using PET. In comparison to electrophilic synthesis, the no-carrier-added (NCA) nucleophilic method has several advantages. These include much higher available activity and specific activity. Recently, we have described an NCA enantioselective synthesis using a chiral phase-transfer catalyst. However, some chemicals were difficult to implement into a commercially available synthesizer, restricting access to this radiopharmaceutical to only a few PET centers. Methods: In this paper, 2 important chemical improvements are proposed to simplify production of 18F-FDOPA, resulting in straightforward automation of the synthesis in a commercially available module. Results: First, a fast, simple, and reliable synthesis of 2-18F-fluoro-4,5-dimethoxybenzyl iodide on a solid-phase support was developed. Second, a phase-transfer catalyst alkylation of a glycine derivative at room temperature was used to enable enantioselective carbon–carbon bond formation. After hydrolysis and high-performance liquid chromatography purification, a high enantiomeric excess of 18F-FDOPA (∼97%) was obtained using a chiral catalyst available from a biphenyl 3 substrate. The total synthesis time was 63 min, and the decay-corrected radiochemical yield was 36% ± 3% (n = 8). Conclusion: By exploiting the advantages of this NCA approach, using a starting activity of 185 GBq of NCA 18F-fluoride, high activities of 18F-FDOPA (>45 GBq) with high specific activity (≥753 GBq/μmol) are now available at the end of synthesis for use in clinical investigations.
The radiopharmaceutical 6-18F-fluoro-l-dopa (18F-FDOPA), an analogue of 3,4-dihydroxy-l-phenylalanine (l-dopa), has proven to be useful for human brain studies of the dopaminergic system using PET (1–4). PET studies using this important 18F-fluorinated radiopharmaceutical allow visualization of the presynaptic distribution of the neurotransmitter dopamine and provide information on presynaptic dopamine metabolism. 18F-FDOPA is used in the evaluation of neuropsychiatric disorders such as schizophrenia (5) and Alzheimer disease (6) and in studies of cognitive function (e.g., in Parkinson disease) (4,7,8). More recently, 18F-FDOPA has also been applied in oncology for the investigation of neuroendocrine tumors (9).
18F-FDOPA is an aromatic amino acid with an asymmetric center. From a stereochemical point of view, this compound has 2 different 3-dimensional arrangements of atoms in space, nonsuperimposable on their mirror image, resulting in 2 enantiomers with d or (R) and l or (S) configuration. As the d isomer of 18F-FDOPA has a lower affinity for blood–brain barrier amino acid transport sites, enantiomeric purity is of great importance for PET imaging. Ideally, only the l form of 18F-FDOPA should be synthesized (10).
Since the beginning of the 1980s, as the specific activity of the amino acid was not a critical parameter for biologic applications, synthesis of 18F-FDOPA has generally been realized using an electrophilic approach. However, this synthesis was limited to PET centers with access to the 20Ne(d,α)18F nuclear reaction. So far, the most efficient electrophilic synthesis (radiochemical yield, ∼30%) has been based on a 2-step protocol with an 18F-fluorodestannylation reaction and subsequent deprotection of a protected tin l-derivative (11,12). Based on this chemical pathway, a fully automated synthesis module is available for the routine clinical preparation of 18F-FDOPA (13,14). However, this electrophilic strategy produces only low amounts (0.6–2.6 GBq) of 6-18F-fluoro-l-dopa even when using expensive posttarget-produced 18F-F2 (1.5 GBq) (15) or the “double-shoot” method (0.5 GBq) (16).
More recently, nucleophilic approaches have also been investigated and several enantioselective syntheses of 18F-FDOPA from NCA 18F-fluoride generated using the now-preferred 18O(p,n)18F reaction have been reported. Initially, NCA 18F-fluorination of the electron-rich aromatic ring of l-dopa was realized using a multistep approach (17–23). The most promising method requires a 3-step synthesis of a small 18F-fluoroaromatic compound that is used for phase-transfer catalyst enantioselective formation of a new carbon–carbon bond (21). As an alternative, an 18F-for-19F isotopic exchange reaction on a fully protected, masked, l-19F-fluoroamino acid has been described (24). However, this approach produced 18F-FDOPA with a low specific activity (2 GBq/μmol), after 105 min of synthesis and 2 subsequent transformation steps (Baeyer-Villiger oxidation and hydrolysis).
Recently, 2-step radiochemical syntheses (labeling and deprotection) of 18F-FDOPA based on the nucleophilic substitution of an iodonium ylide or iodyl group of a protected l-dopa derivative have also been proposed (25,26).
Presently, routine preparation of high specific activity (>37 GBq/μmol) 18F-FDOPA in our laboratory is achieved using a multistep phase-transfer catalyst strategy (21). However, some chemicals are difficult to implement in commercially available synthesizers. For example, synthesis of the 18F-fluorobenzyl bromide (7 with bromine replacing iodine) with high yields requires the use of a corrosive gas (HBr) that is arduous to handle and store. Moreover, the solid-phase support has to be kept “wet” with hexane before the halogenation step (21). Another limitation of this approach is the alkylation step, which necessitates a long precooling (10 min) of the electrophilic agent at 0°C before starting the enantioselective reaction (10 min, 0°C). As a result, the complexity of the synthesis makes the production challenging, increasing the chance of failure in routine production if the synthesis is performed by untrained chemists. For large-scale clinical investigations and wider use of this radiopharmaceutical, progress in the automation of this complex radiosynthesis is a necessity. In this paper, 2 significant improvements to the chemistry in the production of 18F-FDOPA are described, which have resulted in simplified automation of the synthesis in a FASTlab (GE Healthcare) synthesizer.
MATERIALS AND METHODS
General
6-fluoroveratraldehyde and other chemicals were purchased from standard commercial sources (ABCR, Acros, Apollo Scientific, and Aldrich). Kryptofix 222 (4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosan), KOH, K2CO3, and hydroiodic acid (HI, 57%) were obtained from Merck. N-(diphenylmethylene)glycine tert-butyl ester 8 and cesium hydroxide monohydrate were purchased from Acros Organics. Before use, the hydriodic acid was distilled over red phosphorus and stored at 0°C after filtration. With the exception of this reagent, all commercially available chemicals were used without further purification. 18O-enriched water (H218O > 95%) was obtained from Rotem Industries. QMA Sep-Pak Light (130 mg) and tC18 (400 mg) Sep-Pak solid-phase extraction (SPE) cartridges were obtained from Waters. 2-formyl-4,5-dimethoxy-N,N,N-trimethylbenzenaminium trifluoromethanesulfonate 4 was synthesized according to procedures described in the literature (18) and characterized by standard methods. The 19F-fluoro reference compounds were also prepared according to reported methods (21,27). Chiral phase-transfer catalysts 2a–2e and 3 (Fig. 1) were prepared according to described methods (21,28–31). Catalyst 1, a chiral spiro-type (R,R)-3,4,5-trifluorophenyl-NAS bromide, also known as the Maruoka catalyst (32), was initially provided by the Maruoka group (33) and was later obtained from a commercial source (Wako Pure Chemical Industries).
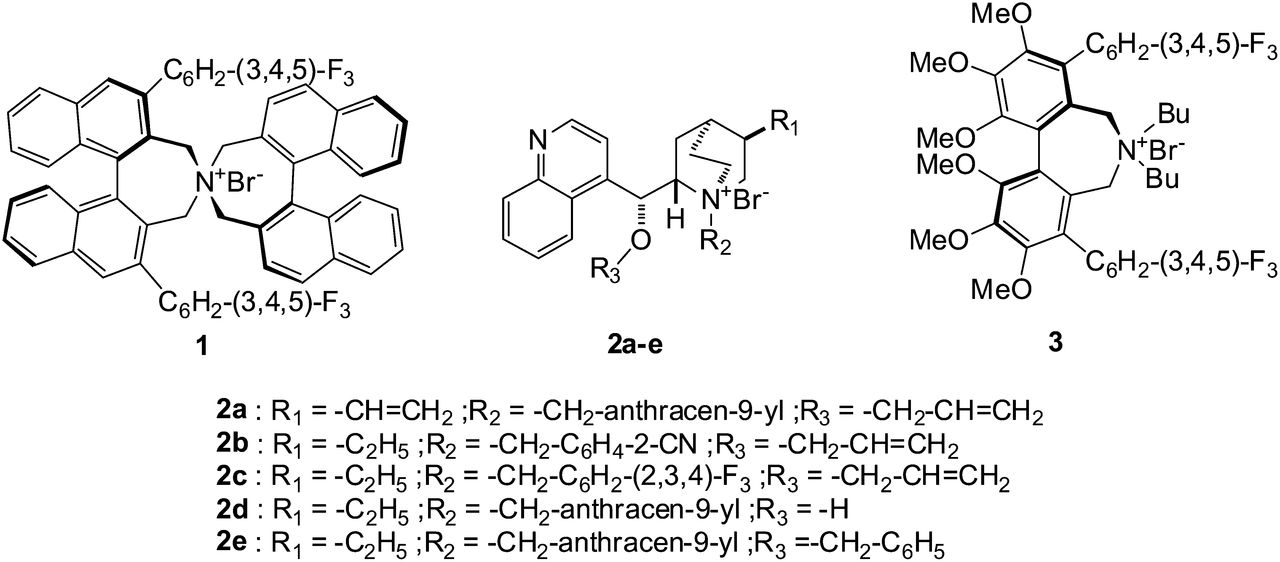
Chemical structure of 7 different chiral catalysts (1–3).
Thin-layer chromatography (TLC) was performed on Macherey Nagel Sil G/UV254 silica plates with dichloromethane/EtOAc (90/10) or dichloromethane (100%). The radioactive spots were quantitatively analyzed on a Bioscan TLC scanner (model AR 2000). Radioactivity was measured in a dose calibrator, and all radiochemical yields were decay-corrected. The nonradioactive fluoro-compounds were used as reference on the same TLC plates and the ultraviolet spot detected at 254 nm.
The TLC identity of the 18F-labeled compounds was confirmed by high-performance liquid chromatography (HPLC) analysis after injection and coinjection on the same analytic HPLC system of the corresponding 19F-fluorinated compounds (aldehyde 5, benzyl alcohol 6, benzyl iodide 7, and fluoroalkylated products 9). Analytic HPLC was performed with a Waters 600E pump, a manual Rheodyne injector (20-μL loop), a 996 Waters photodiode array detector, and the Empower chromatography manager software from Waters. For radioactivity detection, a NaI detector from Eberline was used. A symmetry Xterra RP18 column (3.5 μm, 4.6 × 150 mm; Waters) was eluted with a mixture consisting of CH3CN/water (70/30) at a flow rate of 1 mL/min.
Synthesis of 2-18F-Fluoro-4,5-Dimethoxybenzyl Iodide 7
NCA 18F-fluoride was produced by proton irradiation (18 MeV) of enriched 18O-water in a niobium or silver target (2.2-mL internal volume) using an IBA compact cyclotron 18/9. The 18F-fluoride was then transferred through Teflon (DuPont) tubing (60 m) to the chemistry laboratory and was trapped on a QMA Sep-Pak light cartridge. The 18F-fluoride was then eluted from the solid support into the reaction vessel with a small volume (500 μL) of a solution containing an equal volume of potassium carbonate in water (7 mg/250 μL) and cryptand in acetonitrile (22 mg/250 μL). The 18F-fluoride was dried under a stream of nitrogen for 5 min on an aluminum heating block at 120°C. At the end of the water/CH3CN evaporation, a small additional volume of acetonitrile was added (100 μL) and the mixture was concentrated to dryness. The azeotropic evaporation step was repeated twice (2 × 100 μL), and finally a dry fluorinating agent was obtained in the form of an 18F-fluoride potassium Kryptofix complex ([K/222]+18F−). The ammonium substrate 4 in dimethyl sulfoxide (800 μL, 20 mg/mL) was added to 0.1–185 GBq of the dry residue of the [K/222]+ 18F− complex. The vial was closed and the 18F-labeling reaction was then conducted at 140°C for 2.5 min. After labeling, the reaction solution was cooled and diluted with water (30 mL). The resulting solution was passed through a tC18 Sep-Pak cartridge previously activated with 5 mL of acetonitrile and 10 mL of water. The cartridge was then washed with 10 mL of water and dried for 15 s with a gentle flow of nitrogen.
Reduction of the 18F-fluorinated aldehyde 5 was realized directly on the tC18 SPE cartridge by passing an aqueous solution of NaBH4 (3 mL, 6 mg/mL) slowly through the cartridge (21). Before the subsequent halogenation step, the cartridge was then washed with 5 mL of water and flushed for 15 s with a flow of nitrogen. For the conversion of the benzyl alcohol 6 to the benzyl halide 7, the activity was kept trapped on the support and 0.8 mL of an aqueous solution of HI (57%) was slowly passed through the support (2 min). The reaction was conducted at room temperature. After reaction, the tC18 Sep-Pak was flushed with nitrogen for 30 s and the 2-18F-fluoro-4,5-dimethoxybenzyl iodide 7 was eluted with toluene (3 mL) (dichloromethane and ether were also considered, as described in the “Results” section). The excess of acid and water was removed by passing the solution through a custom-made online potassium carbonate cartridge (1.5 g). The 18F-fluorobenzyl iodide 7, ready for subsequent alkylation reaction, was then recovered in about 2.5 mL of toluene.
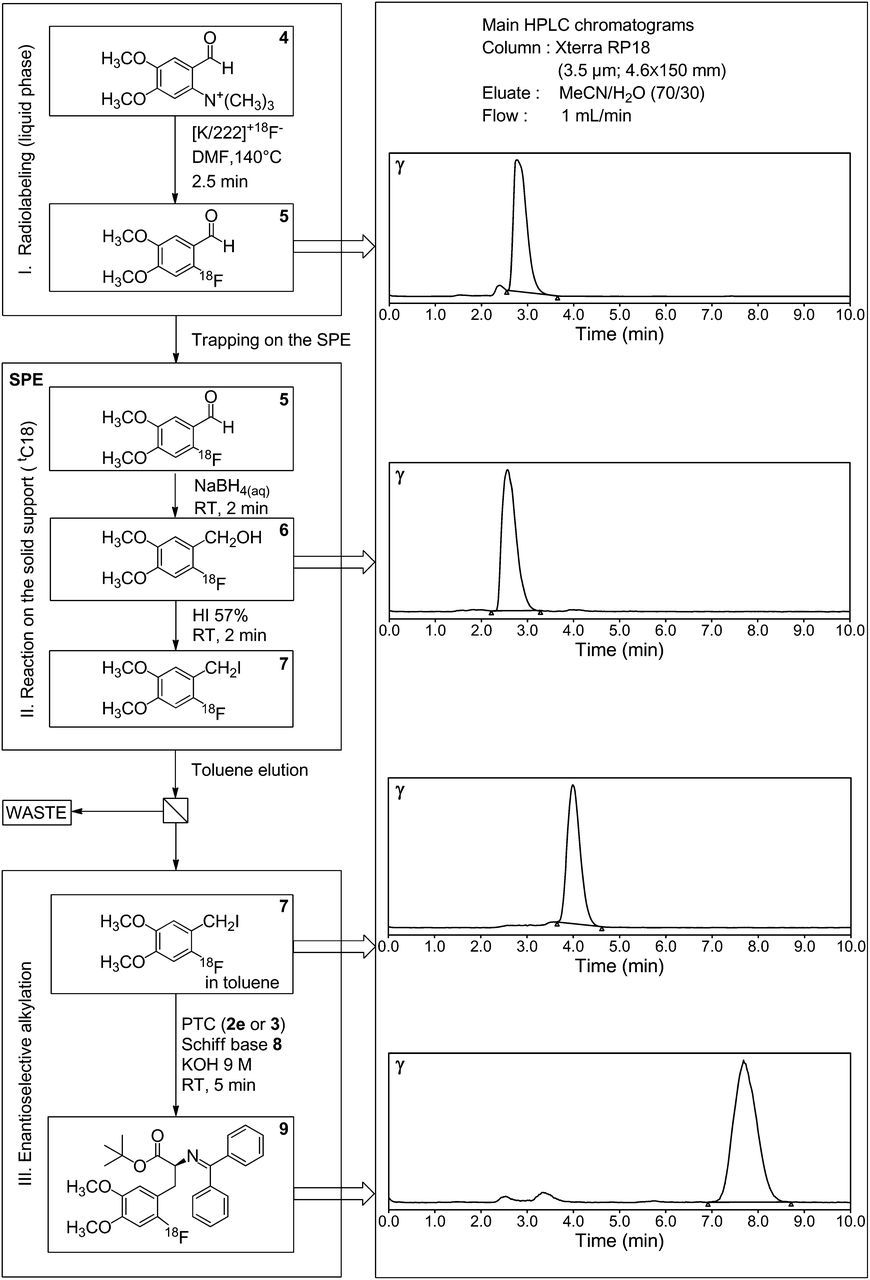
The radiochemical yield and purity of the different products were determined by TLC and HPLC analyses after elution of the tC18 cartridge with CH3CN. The retention times of the 6-18F-fluoroveratraldehyde 5, 2-18F-fluoro-4,5-dimethoxybenzyl alcohol 6, and iodide 7 were 2.8 min, 2.6 min, and 4.0 min, respectively (Fig. 2). (TLC: 90/10 dichloromethane/AcOEt; Rf CHO 5: 0.65; Rf CH2OH 6: 0.3; Rf CH2I 7: 0.74.)

Typical radiochemical HPLC profiles of main steps of synthesis. DMF = N,N-dimethylformamide; PTC = phase-transfer catalyst; RT = room temperature.
Alkylation, Hydrolysis, HPLC Purification, and Formulation
2-18F-fluoro-4,5-dimethoxybenzyl iodide 7 in toluene was added to a vial containing the N-(diphenylmethylene)glycine tert-butyl ester 8 (25 mg, 85 μmol), the chiral phase-transfer catalyst (2–3 mg), CsOH⋅H2O (150 mg, 0.9 mmol), or KOH (9 M, 200 μL). The reaction mixture was stirred vigorously at room temperature for 5 min. A few microliters of the solution were then withdrawn and diluted with CH3CN, and the (S)-N-(diphenylmethylene)-2-(2′-18F-fluoro-4′,5′-dimethoxylbenzyl)-glycine tert-butyl ester 9 was analyzed by TLC and HPLC (Rt: 7.7 min; Rf: 0.15, 100% dichloromethane; Rf: 0.70, 90/10 dichloromethane/AcOEt).
After the alkylation reaction, HI (57%, 1 mL) was added and the toluene evaporated at 180°C (3 min). The reactor vial was then capped and the hydrolysis step conducted at this temperature for 15 min. The reaction mixture was cooled to approximately 50°C and diluted with 4 mL of the semipreparative HPLC eluent.
The crude diluted hydrolysis mixture was loaded into a manual Rheodyne injector (6-mL loop). HPLC purification was performed using an Econosphere C18 column (10 μm, 250 × 10 mm; Grace) with a Waters 600 pump and a 996 Waters photodiode array detector (280 nm) controlled by the Empower software. The radioactive elution profile was monitored with a custom-made Geiger-Müller radioactivity detector. The flow rate was 5 mL/min, and the mobile phase was aqueous acetic acid (50 mM) at pH 4 with ascorbic acid (0.57 mM) and ethylenediaminetetraacetic acid (1 mM). The retention time of the purified 6-18F-fluoro-l-dopa 10 was 9.3 min. The fraction containing 18F-FDOPA was collected into a small vial containing NaCl 0.9% (30 mL), NaCl 10% (0.5 mL), and ascorbic acid (30 mg).
For clinical use, the resulting solution was transferred to a pharmaceutical class A hot cell located inside the dispensing area, where microbiologic parameters are strictly controlled. The solution was filtered in a closed vial through 2 sterilizing 0.22-μm membrane filters (Millex-GS; Millipore).
Automation
Except for the HPLC purification, all the steps of the synthesis were automated in a FASTlab module. This module was equipped with a second reactor heater that can be rapidly cooled if necessary. A new sequence, based on the chemistry improvements reported in this paper, was developed and the entire chemical process was performed with a single-use cassette. The fully automated radiosynthesis, with the exception of the HPLC purification, was performed in 52 min from the end of activity recovery from the cyclotron.
Quality Control
The enantiomeric purity of 18F-FDOPA was determined after HPLC analysis on a CrownPak CR(+) column (5 μm, 150 × 4 mm) from Daicel Chemical Industries. The mobile phase was an aqueous solution of HClO4 (pH 2.0), and the flow rate was 0.8 mL/min (Rt: 5.0 min [d] and 6.4 min [l]; Fig. 3B). Specific activity was determined by ultra-performance liquid chromatography (UPLC) quantification of an aliquot of the final sample of 18F-FDOPA (ultraviolet and radioactive detectors). An ultraviolet calibration curve (280 nm) generated from solutions of known concentration was used to calculate the amount of cold 19F-FDOPA (Research Biochemical International) in the radioactive sample. UPLC assay (ultraviolet set at 280 nm and radioactive detection) was performed at 50°C on a Acquity UPLC BEH shield RP18 column (1.7 μm, 2.1 × 100 mm; Waters) eluted at a flow rate of 0.8 mL/min with water at pH 4 (100%). The retention time was 0.51 min.

Quality chromatogram of final 18F-FDOPA. Typical UPLC (A) and HPLC (B) profiles (radiochemical [γ] and ultraviolet [UV], 280 nm). (A) Acquity BEH shield RP18 (1.7 μm, 2.1 × 100 mm); mobile phase: H2O, 0.8 mL/min. (B) Crownpack CR(+) (5 μm, 150 × 4 mm); mobile phase: HClO4 (aqueous) (pH 2), 0.8 mL/min.
RESULTS
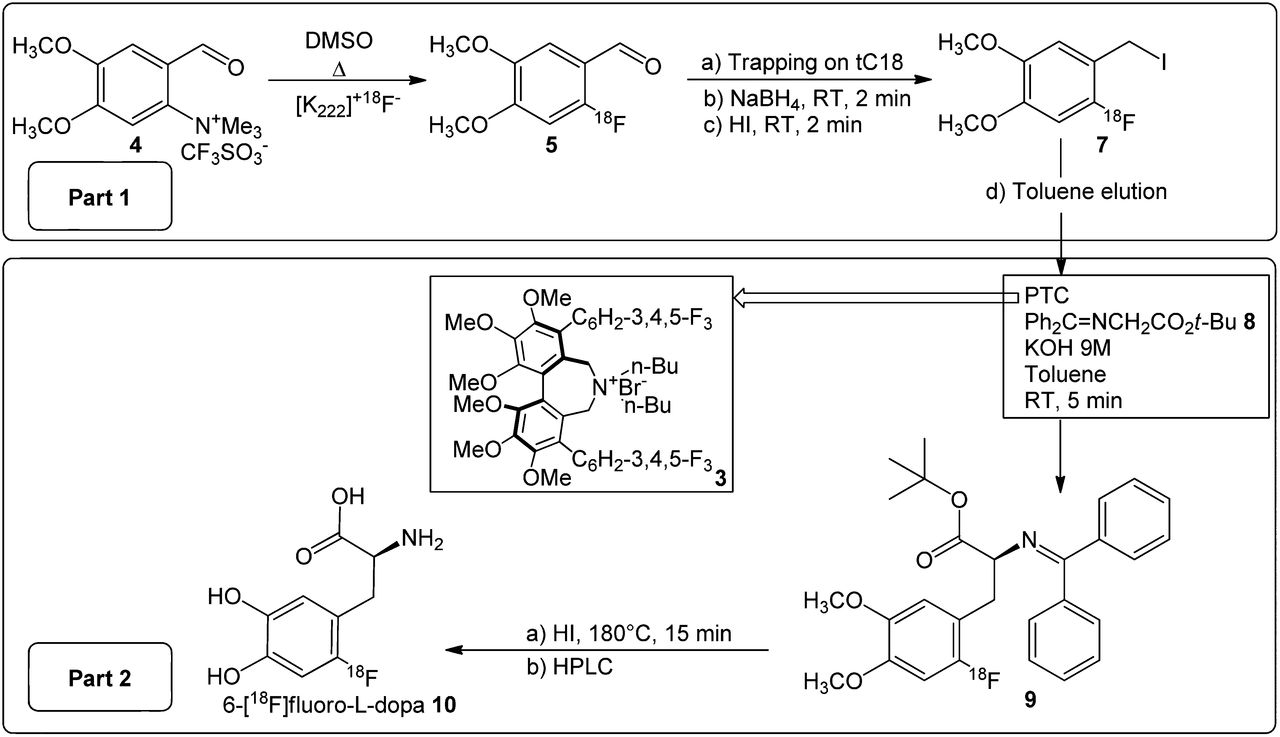
The approach followed for the NCA radiochemical synthesis of 6-18F-fluoro-l-dopa 10 (Fig. 4) is based on a similar pathway previously developed in our laboratory (21,34).
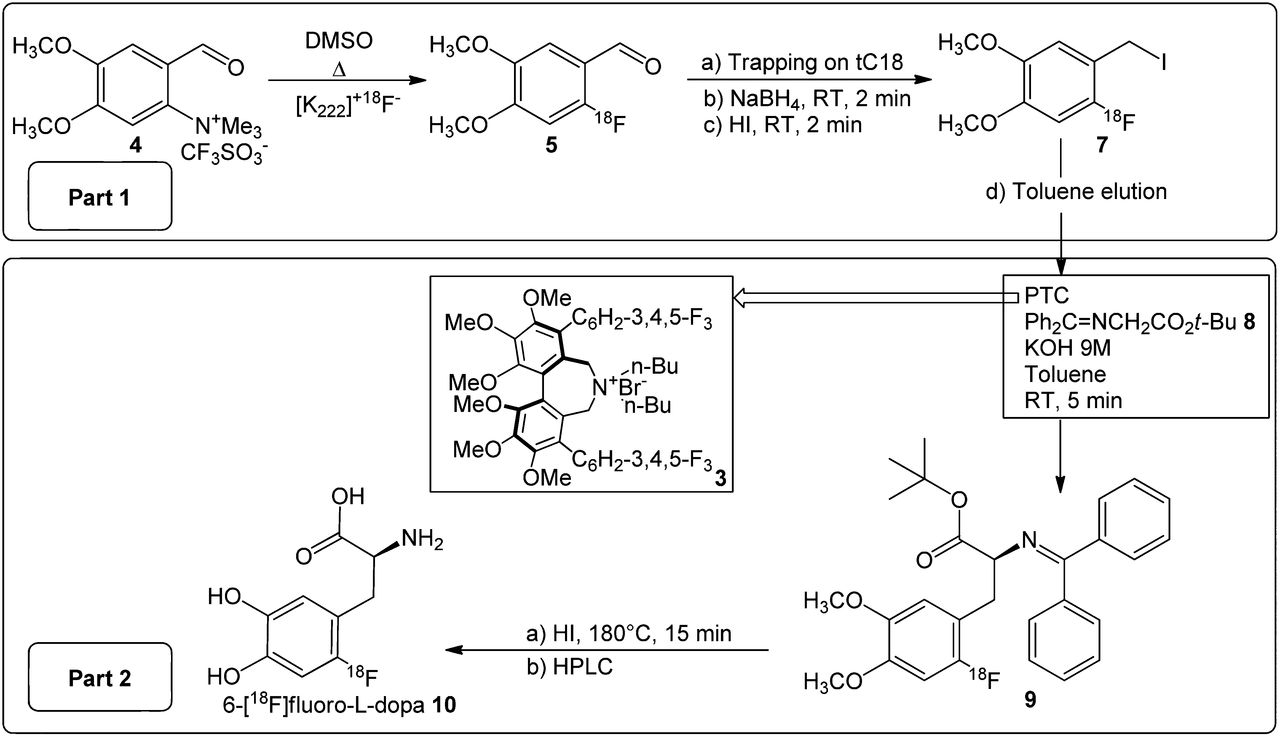
General phase-transfer catalyst strategy for radiochemical synthesis of 6-18F-fluoro-l-dopa. DMSO = dimethyl sulfoxide; PTC = phase-transfer catalyst; RT = room temperature.
The first part of the 18F-FDOPA synthesis consisted of the preparation of a 2-18F-fluoro-4,5-dimethoxybenzyl iodide 7 alkylating agent (part 1, Fig. 4). Briefly, fluorination of a trimethylammonium salt 4 was realized in dimethyl sulfoxide with 18F-fluoride in the presence of the aminopolyether Kryptofix 222 and potassium carbonate. A radiochemical yield of 50% was obtained at 140°C. After labeling, the aldehyde 4 was trapped on the SPE cartridge and reduced to the corresponding alcohol derivative 6 with a solution of NaBH4 in water. The 2-18F-fluoro-4,5-dimethoxybenzyl alcohol 6 was converted to the iodide derivative 7 by passing an aqueous solution of hydroiodic acid (57%) through the cartridge. The 2-18F-fluoro-4,5-dimethoxybenzyl iodide 7 was recovered from the SPE cartridge with a volume of toluene that did not exceed 2.5 mL. NCA preparation of 2-18F-fluoro-4,5-dimethoxybenzyl iodide 7 from the ammonium precursor 4 produces radiochemical yields of 42%–47% and high radiochemical purity (≥97%) (Fig. 2).
The 18F-fluorobenzyl iodide 7 recovered from the SPE and K2CO3 cartridges was directly used for the formation of the new carbon-α carbon-β bond. The reaction was conducted at room temperature for 5 min in toluene with the prochiral Schiff base 8 in the presence of cesium hydroxide or potassium hydroxide (part 2, Fig. 4) and one of each of the chiral phase-transfer catalysts presented in Figure 1.
The radiochemical yield of the reaction was determined by HPLC and TLC (“Materials and Methods”). From these analyses, it appears that conversion of the benzyl iodide 7 into the 18F-fluorobenzylated product 9 was nearly quantitative with all the catalysts investigated (>80%–90%) (Fig. 2). However, to obtain a short reaction time, effective and adequate mixing was crucial.
In all cases, the chiral efficiency of these phase-transfer catalysts and the enantiomeric excess of the 18F-fluoro amino acid 10 (l isomer) were established after hydrolysis of the 18F-fluorobenzylated product 9 and HPLC purification. The alkylation reaction was stopped by addition of HI (57%, 1 mL), and the organic solvent was evaporated. After hydrolysis (15 min, 180°C), the crude mixture was purified on a semipreparative HPLC column. Enantiomeric excesses were determined on an analytic HPLC chiral column (“Materials and Methods,” Fig. 3B). The results obtained with catalysts 1–3 are summarized in Table 1.
Enantiomeric Excess of 18F-FDOPA with Different Phase-Transfer Catalysts (1-3, Fig. 1)
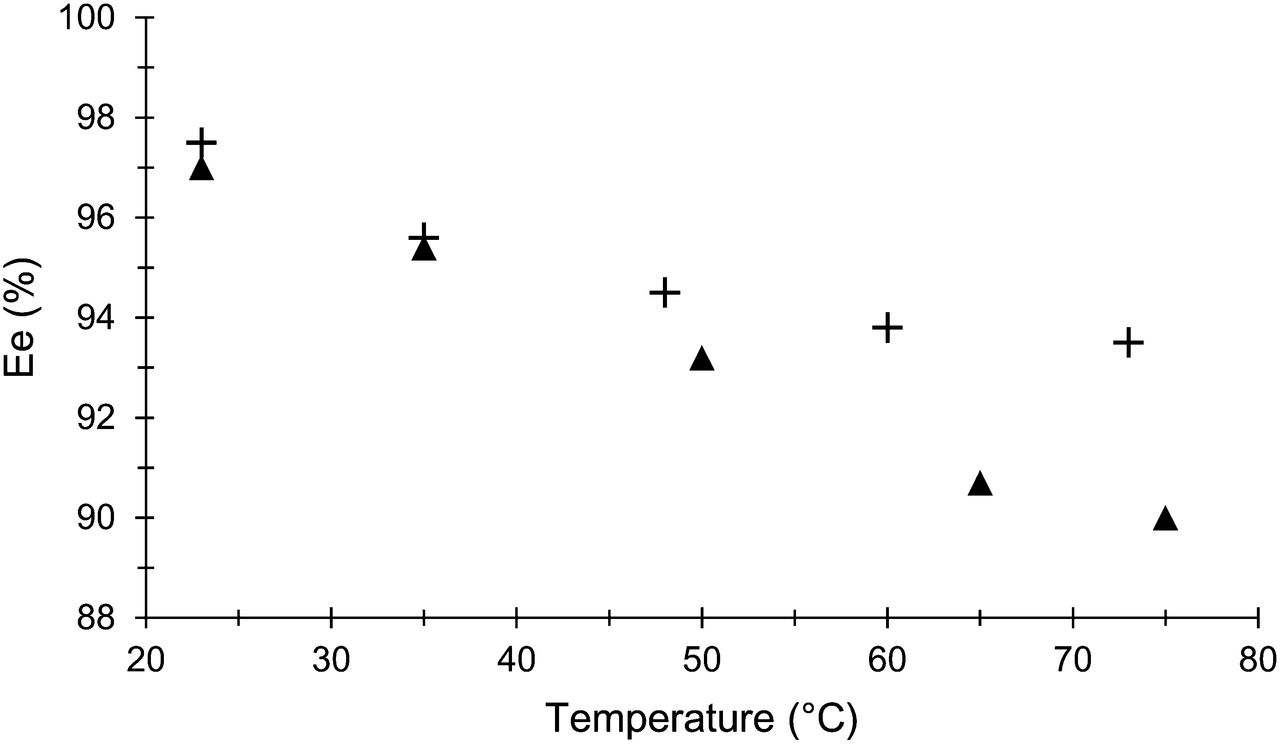
The effect of the temperature on the enantioselectivity of the reaction was evaluated between 25°C and 75°C with catalysts 2e and 3 (Fig. 5). These values were obtained after hydrolysis and HPLC purification.

Effect of temperature on enantiomeric excess (Ee) of 18F-FDOPA with catalysts 2e (▲) and 3 (+).
The last step of the synthesis, before HPLC purification, requires evaporation of the toluene and hydrolysis of the protective groups. To facilitate automation of the evaporation step, the possibility of replacing this solvent with another of lower boiling point was evaluated. These data were acquired with the biphenyl catalyst 3 (Table 2).
Effect of Solvent on Enantiomeric Excess of 18F-FDOPA
For preliminary automation in a FASTlab, the synthesis was performed using the 2-18F-fluoro-4,5-dimethoxybenzyl iodide 7 obtained from the ammonium salt 4, and the alkylation step was realized in toluene with catalyst 3, at room temperature, for 5 min. After hydrolysis and HPLC purification, the collected peak of 18F-FDOPA was formulated to a final solution of 35 mL with 0.9% NaCl, 10% NaCl, and ascorbic acid.
Using this approach, the radiochemical yield of NCA 18F-FDOPA was around 24% (not corrected for decay) after a little more than 1 h of synthesis (63 min). The enantiomeric excess was always greater than 96%. The specific activity determined at the end of the synthesis on the sample without ascorbic acid was 0.75–1.3 TBq/μmol (Fig. 3A).
DISCUSSION
The multistep NCA nucleophilic approach described in this paper consists of 2 main parts. The first consists of the synthesis of a 18F-fluoro–labeled benzyl iodide 7 (part 1, Fig. 4), and the second, of the enantioselective alkylation of a prochiral protected derivative 8 by using a phase-transfer catalyst and a chiral catalyst, followed by hydrolysis and HPLC purification (part 2, Fig. 4).
The complexity of the synthesis makes automation challenging. To automate this synthesis on a FASTlab module, improvements to the 2 synthesis parts were necessary (Fig. 4).
The first part in the synthesis (part 1, Fig. 4) implies a 3-step preparation of a 2-18F-fluoro-4,5-dimethoxybenzyl iodide 7. According to previous results (27), its synthesis is feasible from either ammonium or nitro precursors with similar radiochemical labeling yields (∼50%). However, the shorter reaction time of the ammonium salt 4 (2 min) than of the corresponding nitro compound (15 min) can be considered a significant advantage in terms of available activity at the end of the process. In contrast to the nitro precursor, another advantage of this ammonium salt 4 results from its solubility in water. This solubility facilitates purification of the 18F-fluorinated aldehyde 5 during the tC18 SPE trapping, allowing excess starting precursor to easily be removed.
The labeled aldehyde 5 was trapped on a tC18 SPE cartridge before being then quantitatively reduced into the corresponding alcohol 6 with an aqueous solution of NaBH4 (Fig. 2). We have recently shown that for the subsequent conversion of the benzyl alcohol 6 into the corresponding halogenated derivative, gaseous hydrogen bromide, which is difficult to handle, could be substituted by a concentrated aqueous solution of hydroiodic acid (27). This advantageous approach was then used for the preparation of the 2-18F-fluoro-4,5-dimethoxybenzyl iodide 7. According to typical HPLC profiles obtained during the synthesis, the reduction and iodination steps are nearly quantitative (Fig. 2).
In summary, this strategy greatly simplifies the synthesis and facilitates automation of the process by introducing fast reduction and halogenation steps at room temperature, on the same solid support, with easy-to-handle reagents (NaBH4 [aqueous] and HI [57%]).
The second part of the modified 18F-FDOPA synthesis described here entailed phase-transfer catalyst enantioselective construction of a new carbon–carbon single bond (part 2, Fig. 4) and the use of a chiral phase-transfer catalyst. To obtain high enantioselectivity, the 2-18F-fluoro-4,5-dimethoxybenzyl halide 7 in toluene must previously be cooled for at least 10 min at 0°C and the alkylation reaction conducted at this same temperature for 10 additional minutes (21). However, an alkylation reaction at this temperature is difficult to automate, because of the requirement for additional equipment such as a stirrer and a cooling system. To increase the overall radiochemical yield of the synthesis and to simplify automation, this alkylation should be performed at room temperature rather than at 0°C—a temperature that should also improve synthesis time. The results of this second improvement are presented below.
Fortunately, in recent years, a great number of new chiral phase-transfer catalysts have been described (30,35,36). A few of them with a potential high enantioselectivity at room temperature were evaluated. Their structures are illustrated in Figure 1.
All of these compounds are quaternary ammonium salts. Five of these new catalysts are cinchonidine derivatives with several substituents, including ethyl, anthracenyl, benzyl, allyl, or vinyl functions 2a–2e. Another has a conformationally fixed biphenyl core 3, and the last, which was commercially available at the beginning of this work, is a C2-symmetric chiral catalyst 1 that can be obtained after 11 synthesis steps from (S)-binaphthol.
With the exception of the Maruoka catalyst 1 (32), all these chiral catalysts have been synthesized in our laboratory. The synthesis of the cinchona alkaloid-derived phase-transfer catalysts is generally fast and requires only a limited number of steps (2 or 3) from the low-cost cinchonidine precursor available from the inexpensive chiral pool.
Catalysts 2a–2e have a common structure involving a sterically hindered tertiary amino group in a quinuclidine moiety, one alcohol function, and a quinoline structure. Depending of the stereo-structure and type of functional groups, 4 generations of these catalysts are now described in the literature (37).
Catalyst 3 is certainly the most difficult to prepare. Its expensive preparation requires 6 steps from the resolved (R)-4,5,6,4′,5′,6′-hexamethoxybiphenyl-2,2′-dicarboxylic acid, previously synthesized from commercially available 3,4,5-trimethoxybenzoic acid (31,38).
All radiochemical alkylation reactions were conducted with similar amounts of catalyst (2–3 mg/3–5 μmol). As shown in entries 1 and 2 of Table 1, summarizing enantiomeric excess for 18F-FDOPA, reaction temperature has a high effect on the enantioselectivity of the reaction with catalyst 1. An increase in temperature from 0°C to room temperature decreases the enantiomeric excess from 96% to 38%. Under the same conditions with catalyst 2a, temperature has only a slight effect on enantiomeric excess (entries 3 and 4, Table 1).
With catalysts derived from cinchonidine 2a–2e (entries 3–9, Table 1), enantiomeric excesses of 52%–97% are obtained. A reaction performed using 9N potassium hydroxide in place of CsOH⋅H2O produces a similar asymmetric induction at room temperature (entries 4 and 5, Table 1). Thus, to facilitate automation, all other experiments were conducted with KOH. An enantiomeric excess of only 93% was observed with catalyst 2a at room temperature in toluene. This value is in accordance with the enantiomeric excess previously observed in our laboratory with dichloromethane and a greater amount of catalyst (15 mg/25 μmol) (21,39). However, under the same conditions, enantiomeric excesses close to 100% have been reported (19,40). In comparison, catalysts 2c (entry 7, Table 1) and 2d (entry 8, Table 1) produce relatively moderate enantioselectivity at room temperature. In this cinchonidine family, the highest enantioselectivity is obtained when cinchona is substituted by an ethyl, anthracenylmethyl, and benzyl function (entry 9, Table 1, catalyst 2e) (37).
A high enantioselectivity is also observed at room temperature with catalyst 3 (entry 10, Table 1). Under these conditions, 6-18F-fluoro-l-dopa 10 is recovered with 97% enantiomeric excess. This value means that at least 98.5% of the l isomer and only 1.5% of the d are generated during the process. The enantiomeric excesses obtained with catalysts 3 and 2e (>95%) are similar.
In the literature, 3 is described as a catalyst with a very high catalytic activity, and very small amounts of 3 (0.01 mol%) are required for the benzylation of a Schiff base without decreasing the enantioselectivity of the reaction (35). Synthesis with cinchonidine derivatives requires generally 10 mol%. However, under our radiochemical conditions, both of these catalysts are always in large excess compared with the amount of NCA 18F-fluorobenzyl iodide 7 used. This makes it difficult to evaluate the benefits of 3 compared with 2e. At this NCA scale, the ratio of the different reagents (i.e., catalyst and Schiff base amount) used during the alkylation step can change the enantiomeric excess (21). Although this effect has been previously described, no attempts to optimize this reaction parameter were made in this study.
Enantiomeric excesses with catalysts 2e and 3 are similar at room temperature (97%) and at 35°C (95.5%). However, increasing the reaction temperature to 75°C leads to a slight decrease in these values (90% and 93.5% enantiomeric excess, respectively) (Fig. 5). Compared with the first chiral catalysts described in the literature, these results are impressive.
As clearly shown in Table 2, chlorinated solvents are best avoided with catalyst 3 as they result in a low enantiomeric excess (maximum, 51%). On the other hand, toluene (boiling point, 110°C) could be replaced by an ethereal solvent of lower boiling point providing an enantiomeric excess of 96% or more (diethyl ether, diisopropyl ether, and methyl tert-butyl ether). However, the first automation with toluene in the FASTlab showed that this solvent could be evaporated in less than 5 min, and thus replacing it is not of great importance.
Using this approach (Fig. 4) and a FASTlab module, automated synthesis of 18F-FDOPA was realized in less than 65 min with a decay-corrected radiochemical yield of 36% ± 3% (n = 8). Nevertheless, the final decay-corrected radiochemical yield of this synthesis depends on the chemical purity of the starting ammonium salt 4, as this purity has a direct influence on the yield of the labeling step.
This approach included preparation of the 2-18F-fluoro-4,5-dimethoxybenzyl iodide 7 (42%–47%, 25 min), phase-transfer catalyst enantioselective alkylation with catalyst 3, hydrolysis, and HPLC purification and afforded 18F-FDOPA with a high specific activity at the end of the synthesis (0.75–1.3 TBq/μmol).
The injectable solution of 18F-FDOPA contains ascorbic acid. This acts as a radical scavenger and contributes to the stability of the final solution. All quality control steps performed on the 18F-FDOPA preparation met the basic criteria of the European Pharmacopeia. These included the appearance test, osmolarity (300 mOsm), pH (3.6), residual solvent (acetonitrile < 410 ppm; toluene < 890 ppm; dimethyl sulfoxide < 5,000 ppm; acetic acid < 5,000 ppm), enantiomeric purity (Fig. 3B), specific activity (Fig. 3A), half-life, and radionuclidic and radiochemical purities (Fig. 3A).
CONCLUSION
After application of the improvements to the chemistry described in this paper, the NCA synthesis of 18F-FDOPA was greatly simplified. This is highlighted in, for example, the aldehyde 5 conversion into the corresponding alcohol 6 and iodide 7 derivatives, which is easily feasible on an SPE cartridge. After elution with a low volume of toluene, the 18F-fluorobenzyl iodide 7 is ready to use in the subsequent alkylation step. This step, resulting in the enantioselective formation of a new carbon–carbon bond, proceeds readily at room temperature by phase-transfer catalyst in the presence of a chiral phase-transfer catalyst. This catalyst is readily available from a cinchonidine (2e) or biphenyl (3) substrate. In both cases, enantiomeric excesses were approximately 97%.
Because of its operational simplicity and mild reaction conditions, the phase-transfer catalyst synthesis was easily implemented in a FASTlab module equipped with a second reactor heater. After 100 min of bombardment (185 GBq) and 63 min of synthesis, more than 45 GBq (1.2 Ci) of 6-18F-fluoro-l-dopa (enantiomeric excess > 96%) with a specific activity in excess of 0.75 TBq/μmol are routinely produced with a reaction yield of 24%.
With such reliable production, 6-18F-fluoro-l-dopa is now available for clinical investigation.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This work was supported by the Université de Liège and a GE grant. Alain Plenevaux is a research associate of FNRS Belgium. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank the cyclotron operators Jean-Luc Genon and Patrick Hawotte for providing us with 18F and Geoffrey Warnock for useful discussions.
Footnotes
Published online May 8, 2013.
- © 2013 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication September 14, 2012.
- Accepted for publication January 22, 2013.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Dopamine D2 receptor upregulation in dorsal striatum in the LRRK2-R1441C rat model of early Parkinsons disease revealed by in vivo PET imaging
- In Vivo Biodistribution of No-Carrier-Added 6-18F-Fluoro-3,4-Dihydroxy-L-Phenylalanine (18F-DOPA), Produced by a New Nucleophilic Substitution Approach, Compared with Carrier-Added 18F-DOPA, Prepared by Conventional Electrophilic Substitution
- In Vivo PET/CT in a Human Glioblastoma Chicken Chorioallantoic Membrane Model: A New Tool for Oncology and Radiotracer Development