


Abstract
Click chemistry has become a ubiquitous chemical tool with applications in nearly all areas of modern chemistry, including drug discovery, bioconjugation, and nanoscience. Radiochemistry is no exception, as the canonical Cu(I)-catalyzed azide-alkyne cycloaddition, strain-promoted azide-alkyne cycloaddition, inverse electron demand Diels-Alder reaction, and other types of bioorthogonal click ligations have had a significant impact on the synthesis and development of radiopharmaceuticals. This review will focus on recent applications of click chemistry ligations in the preparation of imaging agents for SPECT and PET, including small molecules, peptides, and proteins labeled with radionuclides such as 18F, 64Cu, 111In, and 99mTc.
The term click chemistry was first coined by K. Barry Sharpless to describe “a set of powerful, virtually 100% reliable, selective reactions for the rapid synthesis of new compounds.” Broadly defined, click chemistry ligations are a group of chemical reactions in which 2 molecular components can be joined under mild conditions in a manner that is high-yielding, rapid, modular, compatible with aqueous environments, and orthogonal to protecting and functional groups. In the years since its emergence, click chemistry has been used in nearly all disciplines of modern chemical science, including drug discovery, bioconjugation, materials science, and nanoscience (1).
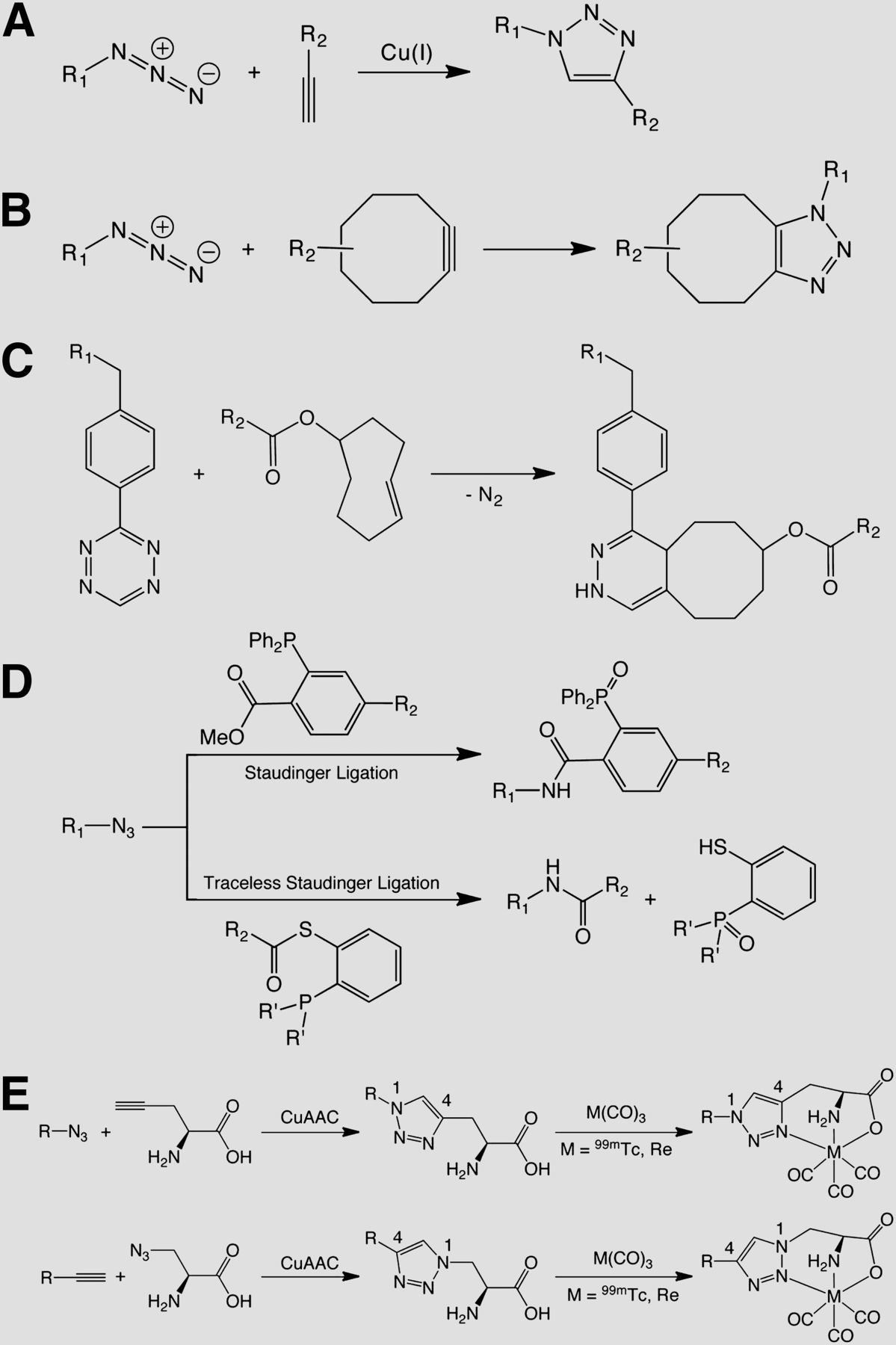
Without question, the most often used variant of click chemistry—and the reaction most closely associated with the term—is the Cu(I)-catalyzed 1,3-dipolar cycloaddition between azides and alkynes (CuAAC). Under certain circumstances, however, the requirement for a metal catalyst can be a complication. Therefore, significant effort has been dedicated to the development of catalyst-free click reactions, including strain-promoted azide-alkyne cycloadditions, inverse electron-demand Diels-Alder reactions, and Staudinger ligations (Fig. 1) (1).

Synthetic schemes of click reactions: CuAAC (A), SPAAC (B), Diels-Alder reaction (C), and Staudinger ligation (D), and 2 variations on click-to-chelate radiolabeling methodology using CuAAC and 99mTc(CO)3 (E).
The intrinsic characteristics of click chemistry—modularity, reliability, selectivity, rapidity, and efficiency—make it a singularly suitable strategy for the time-sensitive and impurity-conscious construction of radiopharmaceuticals. Over the past decade, click chemistry has been applied to the synthesis of radiolabeled imaging agents with increasing frequency and effectiveness. Several review articles have covered the broader applications of click chemistry to molecular imaging; therefore, in this short review, we will focus on the most recent applications of click chemistry to the development of radiopharmaceuticals (2).
APPLICATIONS OF CLICK CHEMISTRY TO RADIOCHEMISTRY
Cu(I)-Catalyzed Azide-Alkyne Cycloadditions (CuAAC)
Originally described by Huisgen, the [3 + 2] azide-alkyne cycloaddition, particularly the Cu(I)-catalyzed variant of the reaction reported independently in 2002 by Sharpless and Meldal, is the prototypical click reaction and has become a ubiquitous chemical tool in the years since its discovery. In radiochemistry, CuAAC has been used for labeling both peptidic and small-molecule radiotracers bearing the positron-emitting radiohalogen 18F (half-life, 109.8 min) using 18F-labeled alkyne- or azide-bearing building blocks. In 2008, Hausner et al. reported the first use of CuAAC for the 18F-labeling of peptides, comparing the in vivo behavior of an αvβ6-targeting radiotracer created using click chemistry with variants synthesized using traditional prosthetic groups (3). The authors clearly illustrated that CuAAC was an effective approach for the construction of the radiotracer. Yet perhaps equally important, this work illustrated how the introduction of a click reaction product (the triazole ring) can have a significant impact on the in vivo behavior of a radiotracer by altering its size, lipophilicity, hydrogen-bonding potential, or metabolism. In this case, the radiotracer synthesized via click chemistry displayed tumor uptake comparable to 2 analogous tracers created using traditional prosthetic groups yet also produced increased kidney and liver activity concentrations attributed to differences in metabolic fate between the click-derived radiopharmaceutical and the traditionally synthesized variants.
More recently, Doss et al. reported a first-in-human study of the biodistribution and radiation dosimetry of an integrin-targeted agent, 18F-RGD-K5, prepared using a similar CuAAC approach (Fig. 2A) (4). An analysis of maximum-intensity projections of 18F-RGD-K5 injected into a female volunteer revealed rapid clearance of activity from the kidneys, liver, and urinary bladder in addition to accumulation in the gallbladder, which peaked at 75 min and decreased thereafter (Fig. 2B). Importantly, compared with 18F-FDG, 18F-RGD-K5 produced higher absorbed doses in the bladder, liver, and kidneys but lower doses in the heart, brain, and breasts, information used to determine that the tracer can be applied safely for imaging αvβ3 expression in humans (4).
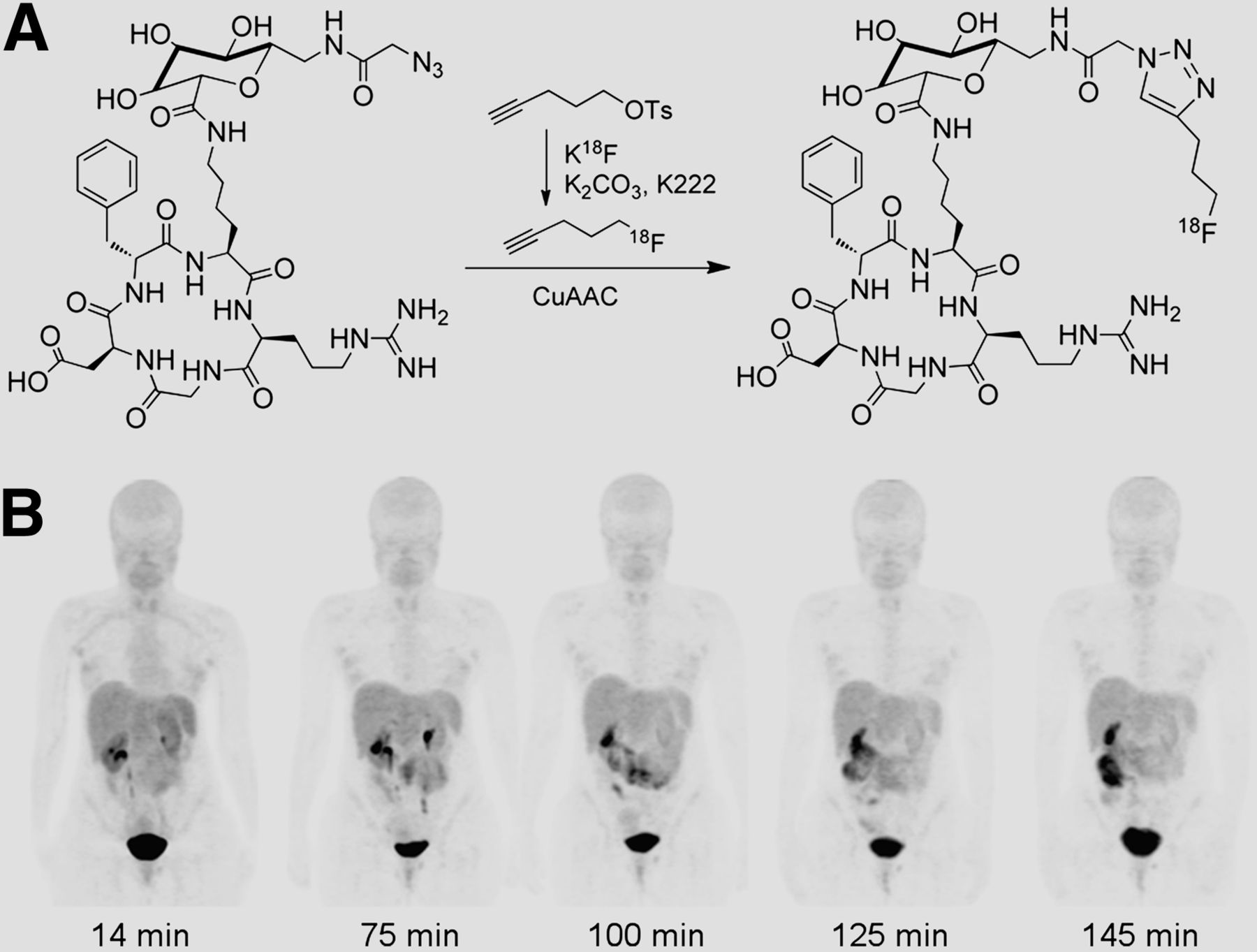
(A) Synthesis of 18F-RGD-K5. (B) Decay-corrected anterior maximum-intensity PET projections of female volunteer injected with 18F-RGD-K5.
The CuAAC reaction has also been used to synthesize 18F-labeled small-molecule probes. In 2009, for example, Nguyen et al. reported the in vivo evaluation of 18F-ICMT-11, a caspase 3/7–specific radiotracer synthesized using CuAAC. The authors found that mice treated with cyclophosphamide chemotherapeutics exhibited elevated levels of tumoral tracer uptake compared with mice treated with vehicle alone (5). Recently, 18F-ICMT-11 finished its phase 1 clinical trial and demonstrated significant potential for the assessment of tumor apoptosis and response to therapy (6).
Moving beyond 18F, Schibli et al. have developed an elegant application of the CuAAC reaction for radiometal chemistry: the click-to-chelate methodology (Fig. 1E). In this system, the alkyne-azide cycloaddition reaction does not simply serve to link a metal chelator to a peptide; rather, the ligation facilitates the formation of a tridentate scaffold for the coordination of M(CO)3 cores in which the 1,2,3-triazole actually participates in chelation. With this click-to-chelate approach, substrates ranging from d-galactose to peptides have been successfully radiolabeled with 99mTc(CO)3, and further investigations have illustrated that these click-based constructs show improved in vivo stability compared with their traditionally labeled cousins (7).
Strain-Promoted Azide-Alkyne Cycloaddition (SPAAC)
Despite the manifold advantages offered by CuAAC, the presence of the Cu(I) catalyst can be problematic in certain circumstances, particularly syntheses involving radiometals. Significant effort has therefore been dedicated to the development of a catalyst-free variant of azide-alkyne click chemistry: SPAAC. In this reaction, the release of ring strain in a cycloalkyne precursor lowers the activation energy for the cycloaddition process, enabling the ligation to be performed in high efficiency without a catalyst. Initially hampered by the synthetic complexity of its cyclooctyne precursors, SPAAC has been used increasingly in radiochemistry as more easily synthesized cycloalkynes have emerged and as other variants, for example, dibenzocyclooctyne, have become commercially available.
Zeng et al. have recently used SPAAC in the construction of 64Cu-labeled, micellar shell-crosslinked nanoparticles (8). The authors first radiolabeled DOTA-modified dibenzocyclooctyne with 64Cu and then purified the construct in high specific activity (318 GBq [8,600 mCi]/μmol end of bombardment [EOV]). Subsequently, the purified 64Cu-DOTA-dibenzocyclooctyne was clicked to nanoparticles containing azide groups in their core and alkyne moieties on their surface. Compared with traditional CuAAC, SPAAC offered several advantages in this case, most importantly the elimination of competition between 64Cu and the Cu(I) catalyst. Using this strategy, the radiolabeled nanoparticles were obtained with a specific activity of 164 TBq (4,420 Ci)/ μmol (EOB), a 500-fold amplification over that of the 64Cu-DOTA-dibenzocyclooctyne construct. This result represents significant progress toward overcoming low specific activity as a limitation to the clinical application of nanoparticle-based PET imaging agents.
Soon thereafter, Chen et al. used a similar dibenzocyclooctyne-SPAAC approach to prepare a 64Cu-labeled cyclo(RGD) variant by clicking a 64Cu-DiAmSar-dibenzocyclooctyne construct to an azide-modified cyclo(RGD) peptide (9). Subsequently, Nedrow-Byers et al. reported two 99mTc-labeled prostate-specific membrane–targeted SPECT agents using an inverted pathway: clicking dibenzocyclooctyne-modified agents to azide-bearing 99mTc-DTPA constructs. The in vivo performance of these radiotracers was shown to be similar to that of analogous tracers constructed in traditional fashions (10).
In addition to radiometal-based applications, several 18F-labeled radiotracers have been prepared using SPAAC. In 2011, Campbell-Verduyn et al. reported the synthesis of an 18F-labeled bombesin analog that maintained high affinity for the gastrin-releasing peptide receptor in vitro in human prostate cancer cells (11). Almost simultaneously, Bouvet et al. reported another successful application of SPAAC in the preparation of an 18F-labeled variant of the complex natural product geldanamycin (12).
Diels-Alder Click Chemistry
Without question, the 2 azide-alkyne cycloaddition reactions are the variants of click chemistry most often used in the synthesis of radiopharmaceuticals. Yet these methodologies are not without their limitations. The requirement of a Cu(I) catalyst in CuAAC can prove problematic, especially when used in conjunction with radiometals. In contrast, the hydrophobicity and somewhat cumbersome synthesis of the cyclooctyne precursors in the SPAAC systems have proven limiting to their widespread application.
In response to these limitations, another promising click ligation has garnered increasing interest among radiochemists over the past 5 y: the inverse electron demand Diels-Alder cycloaddition between 1,2,4,5-tetrazines and strained alkene dienophiles (13). Like other click reactions, the ligation is selective, clean, efficient, and bioorthogonal, but unlike many Diels-Alder reactions, the coupling is irreversible, forming a stable pyridazine product after a retro-Diels-Alder reaction driven by the release of dinitrogen from the reaction intermediate. The rapidity of the reaction is governed by the identity of the tetrazine and dienophile. The earliest work used norbornene derivatives as the dienophile, with rate constants around 1–2 M−1s−1 in water at 37°C. However, norbornenes quickly gave way to transcyclooctene-based dienophiles, which accelerate the reaction by over 3 orders of magnitude. Several different 1,2,4,5-tetrazines have also been tested for their kinetics, with rate constants with transcyclooctene ranging from 210 M−1s−1 to almost 30,000 M−1s−1. Not surprisingly, stability and reaction rate were observed to be inversely proportional: the more stable tetrazines generally reacted less rapidly, whereas the less stable compounds generally reacted more rapidly (14).
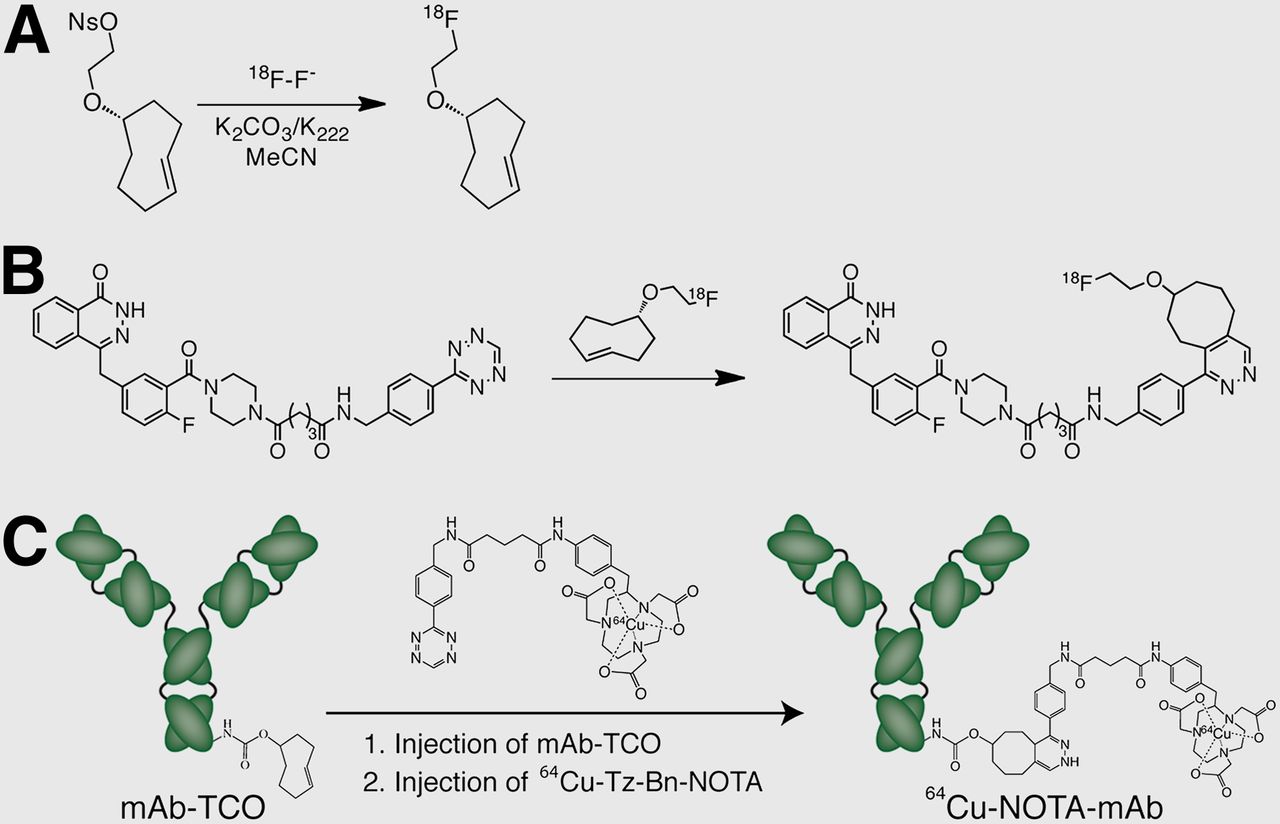
The Diels-Alder cycloaddition reaction has been used in the construction of both 18F- and radiometal-based radiopharmaceuticals (Fig. 4). The first application was published by Li et al. in 2010, when they developed the synthesis and purification of an 18F-labeled transcyclooctene and tested its reactivity with 3,6-diaryl-s-tetrazines (Fig. 3A) (15). The authors reported radiochemical yields of up to approximately 70% for 18F-transcyclooctene under mild conditions in addition to rapid and clean reactions between the 18F-transcyclooctene and its tetrazine partner. In the same work, efforts to label a 1,2,4,5-tetrazine variant with 18F proved elusive, likely because of decomposition of the tetrazine during fluorination. The first targeted probes synthesized using 18F-labeled transcyclooctene emerged soon thereafter, including an 18F-labeled RGD peptide developed by the same group and an 18F-labeled poly[ADP-ribose]polymerase 1 (PARP1) inhibitor developed by Reiner et al. using an innovative wrinkle: an “orthogonal scavenger-assisted high-performance method” in which a transcyclooctene-modified magnetic resin is used to remove unreacted tetrazine-modified precursor from the click reaction mixture (Fig. 3B) (16,17).

(A) Synthetic route to 18F-transcyclooctene. (B) Synthesis of PARP1 inhibitor 18F-AAZD2281 using inverse electron-demand Diels-Alder click chemistry. (C) Schematic for methodology for pretargeted 64Cu-immunoPET imaging agent based on inverse electron Diels-Alder click chemistry. TCO = transcyclooctene.
To date, the sole PET radiometal-based application of the ligation has been a modular strategy for the construction of radiolabeled antibodies geared at eliminating the intrinsic variability in the synthesis of radioimmunoconjugates (18). In this system, norbornene is first conjugated to the antibody, and this single covalently modified construct can then be clicked with any tetrazine-modified chelator and subsequently radiolabeled. With a model system using trastuzumab, 64Cu, and 89Zr, it was found that for a given initial stoichiometry of norbornene to antibody, the final 64Cu-DOTA– and 89Zr-desferrioxamine–labeled constructs were effectively identical. For example, using an initial conjugate bearing 3 norbornene dienophiles per antibody, 89Zr-labeled and 64Cu-labeled radioimmunoconjugates were constructed that possessed identical numbers of chelates per antibody (2.3 ± 0.4 desferrioxamine/monoclonal antibody and 2.2 ± 0.3 DOTA/monoclonal antibody, respectively), specific activities (115 ± 7 MBq [3.1 ± 0.2 mCi]/mg and 107 ± 11 MBq [2.9 ± 0.3 mCi]/mg), immunoreactive fractions (0.95 ± 0.05 and 0.96 ± 0.05), and in vitro stabilities (96% ± 2% and 97% ± 2%) and were able to effectively and selectively delineate HER2-positive breast cancer xenografts in vivo.
The tremendous rapidity and bioorthogonal nature of the ligation have also been harnessed for in vivo pretargeted imaging, an area in which the inverse electron-demand Diels-Alder reaction offers the greatest advantage over SPAAC, for the somewhat sluggish kinetics of the strain-promoted click variant almost certainly preclude its use for in vivo pretargeting. The first study, reported by Rossin et al., was the development of a pretargeted SPECT imaging methodology based on a transcyclooctene-modified CC49 antibody and an 111In-DOTA–labeled tetrazine (19). More recently, 2 different pretargeted PET imaging methodologies have been published: one in which an 18F-labeled, tetrazine-modified macromolecular dextran serves as the radioligand and another in which a small-molecule 64Cu-NOTA–labeled tetrazine is used (Fig. 3C) (20,21). All 3 systems rely on the injection of a monoclonal antibody–transcyclooctene conjugate, a localization period during which the antibody accumulates in the tumor and clears from the blood, the injection of a radiolabeled tetrazine, and the in vivo ligation of the 2 components followed by the clearance of excess radioligand. In murine models of cancer, all strategies yielded high-quality images and proved effective at selectively delineating tumor from background organs, suggesting that click-based pretargeted methodologies may provide a safe and effective alternative to traditional antibody-based nuclear imaging.
Other Types of Click Chemistry
The catalog of reactions that can be categorized as click chemistry does not end with the three we have discussed thus far. If the requirement for bioorthogonality is relaxed, the list of reactions grows considerably to include ligations between thiols and maleimides, aldehydes and amines, isothiocyanates and amines, and epoxides and nucleophiles. Although we cannot address all these reactions here, many, if not all, have been used in radiopharmaceutical chemistry. Some, such as the reaction between vinyl sulfones and thiols, have been reported only a few times, whereas others, such as the reaction between maleimides and thiols, are widely used (2).
The final reaction that merits our attention here represents another bioorthogonal ligation: the traceless Staudinger reaction between an azide and a phosphane-substituted ester to create an amide bond. In an early example, Gaeta et al. used the ligation between 2-18F-fluoroethylazide and a diphenylphosphane-bearing precursor to produce a high-affinity, 18F-labeled subtype A γ-aminobutyric acid receptor ligand in 7% uncorrected yield over 2 steps and a specific activity of 0.9 GBq/μmol (22). In similar work, Carroll et al. reacted thioester-based phosphane precursors and 2-18F-fluoroethylazide to create the 18F-labeled products in greater than 95% radiochemical yield for the single ligation step (23). Pretze et al., in contrast, have moved the 18F to the phosphane, enabling the radiolabeling of an array of azide-functionalized targets (24). In their work, an 18F-fluoroacyl-functionalized phosphane was synthesized and reacted with azide-containing model compounds to produce 18F-labeled products in 30%–35% decay-corrected radiochemical yields in 1 pot over 2 steps. Finally, the Staudinger ligation has also recently been applied to pretargeted PET imaging by Vugts et al., who ultimately concluded, however, that this reaction is not suitable for in vivo pretargeting, likely because of the relatively slow kinetics of the ligation and the in vivo oxidation of the phosphane-based radioligand (25).
SUMMARY
Strategies using click chemistry have shown significant advantages over traditional synthetic techniques for the modular, rapid, clean, and efficient synthesis of radiopharmaceuticals, particularly biomolecular radiotracers. In this regard, click chemistry has already begun to revolutionize radiopharmaceutical chemistry. As new click chemistry tools are developed and existing methodologies are increasingly applied to radiotracer development, click techniques will facilitate the facile, safe, and efficient synthesis and development of an ever-widening range of radiolabeled agents for the detection, therapy, and treatment monitoring of disease.
DISCLOSURE
No potential conflict of interest relevant to this article was reported.
Footnotes
↵* Contributed equally to this work.
Published online Apr. 24, 2013.
- © 2013 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication January 23, 2013.
- Accepted for publication April 4, 2013.





{kind=link}
{kind=link}
{kind=link}