


Abstract
A new approach to the surface engineering of superparamagnetic iron oxide nanoparticles (SPIONs) may encourage their development for clinical use. In this study, we demonstrated that nonpolymeric surface modification of SPIONs has the potential to be an advanced biocompatible contrast agent for biomedical applications, including diagnostic imaging in vivo. Methods: Adenosine triphosphate (ATP), which is an innate biomaterial derived from the body, was coated onto the surface of SPIONs. An in vivo degradation study of ATP-coated SPIONs (ATP@SPIONs) was performed for 28 d. To diminish phagocytosis, ATP@SPIONs were surface-modified with gluconic acid. We next studied the ability of the SPIONs to serve as a specific targeted contrast agent after conjugation of cMet-binding peptide. The SPIONs were conjugated with Cy5.5 and labeled with 125I for multimodality imaging. In vivo and in vitro tumor-targeted binding studies were performed on U87MG cells or a U87MG tumor model using animal SPECT/CT, an optical imaging system, and a 1.5-T clinical MR scanner. Results: ATP@SPIONs showed rapid degradation in vivo and in vitro, compared with ferumoxides. ATP@SPIONs modified with gluconic acid reduced phagocytic uptake, showed improved biodistribution, and provided good targetability in vivo. The gluconic acid–conjugated ATP@SPIONs, when conjugated with cMet-binding peptide, were successfully visualized on the U87MG tumors implanted in mice via multimodality imaging. Conclusion: We suggest that ATP@SPIONs can be used as a multiplatform to target a region of interest in molecular imaging. When we consider the biocompatibility of contrast agents in vivo, ATP@SPIONs are superior to polymeric surface–modified SPIONs.
Successful application of superparamagnetic iron oxide nanoparticles (SPIONs) in biomedicine is strongly dependent on their physicochemical characteristics and specifically a strong magnetic susceptibility and uniform shape, optimal particle size with a narrow size distribution, and colloidal stability in physiologic environments (1–4). In particular, it is essential to engineer the surface of SPIONs to prevent aggregation between magnetic particles by van der Waals attraction and to improve their colloidal stability in biologic solutions (5–7). Although various coating materials such as bovine serum albumin, polysaccharide-based materials (dextran, chitosan, and curdlan), polyethylene glycol, polyvinyl alcohol, and polyacrylamide have recently been used for the surface engineering of SPIONs, there are many problems associated with their use in biomedical applications (8–14). The fate of these polymeric materials in the body after intravenous injection has not yet been completely evaluated (15–17). It is important to consider that modification of SPIONs using polymeric materials may affect the biocompatibility, pharmacokinetic, biodegradation, and biodistribution profiles (18,19). Moreover, the polymeric materials need a fixation site affinity toward the surface of SPIONs and a coupling site to provide easy access for bioconjugation. Some of the polymeric materials that are used for surface modification may be the cause of unwanted high uptake in the liver and spleen, and they exert toxic effects in sites of accumulation (20). Therefore, the surface-coating material of SPIONs plays an important role in the biomedical application of SPIONs, and new approaches for surface modification of SPIONs with innate materials are needed to reduce unwanted uptake and toxic effects in molecular imaging. Adenosine triphosphate (ATP) consists of a purine base attached to a pentose sugar, and 3 phosphate groups are attached at the 5′ carbon atom of the pentose sugar. ATP is highly promising for surface engineering SPIONs because ATP has a part that has a strong affinity for anchoring onto the surface of SPIONs, and it also has a coupling part for conjugation with bioactive molecules (21). Moreover, ATP, which is an innate biomaterial derived from the body, has excellent biocompatibility compared with polymeric materials. If SPIONs coated with ATP have suitable chemicophysical properties for use in vivo, then they should be as safe as or safer than the polymer-coated SPIONs. Prolonged exposure to polymer-coated SPIONs due to delayed decomposition may have adverse effects in the body. Recently, some researchers reported that even at subclinical concentrations of SPIONs, SPIONs and static magnetic field exposure treatments showed synergistic cytotoxicity effects (22).
We report here the use of ATP as a surface-coating material for functionalizing SPIONs and multifunctional imaging. We designed and modified ATP-coated SPIONs (ATP@SPIONs) for tumor-targeted imaging as shown in Figure 1. ATP@SPIONs showed high colloidal stability and impressive biodegradation in vivo. When designed as an imaging agent with a tumor target ligand for diagnosing tumors, ATP@SPIONs were a successful multiplatform for molecular imaging. The goals of this study were to develop SPIONs using the innate material ATP for surface modification, to assess the biocompatibility and degradation of the SPIONs in vivo, and to investigate whether tumors can be enhanced by conjugation of tumor-specific ligands with the SPION.

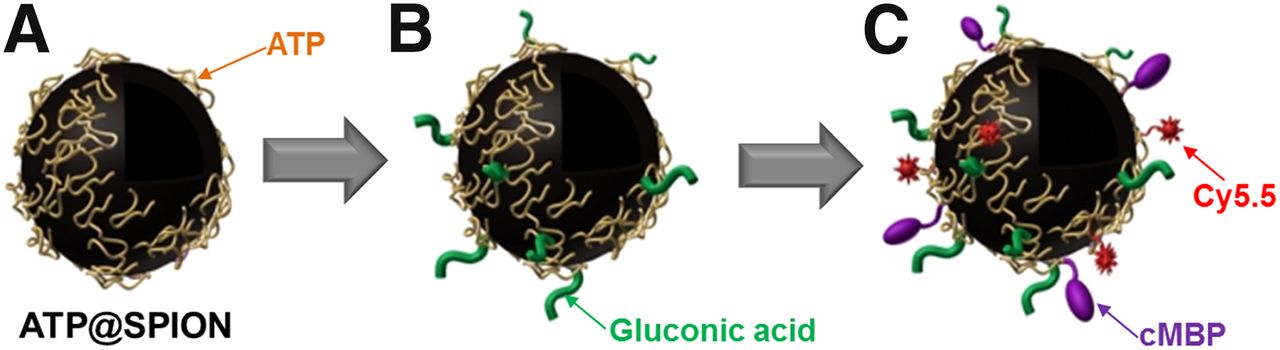
Schematic diagram for synthesis of ATP@SPIONs (A), GA-ATP@SPIONs (B), and Cy5.5-conjugated cMet binding peptide (cMBP)-GA-ATP@ SPIONs (C).
MATERIALS AND METHODS
Synthesis and Characterization of ATP@SPIONs
FeCl3·6H2O (250 mg) and FeCl2·4H2O (92 mg) were dissolved in deoxygenated water (15 mL) under N2 streaming. After the addition of ATP (200 mg), ammonium solution (32%, 7.5 mL) was injected into the ferrous solution and stirring was continued for 30 min. The resulting black solution was cooled and washed with distilled water 3 times. We characterized the ATP@SPIONs using analytic techniques including elemental analysis, dynamic light scattering (Jeonju Center, Korea Basic Science Institute), transmission electron microscopy, x-ray photoelectron spectroscopy, and superconducting quantum interference device analysis. MR phantom imaging was performed using a clinical 1.5-T MR scanner (Signa Exite Twin-speed; GE Healthcare).
Degradation of ATP@SPIONs
In vitro degradation of the ATP@SPIONs (1 mg/mL) was performed, as compared with that for ferumoxides (Feridex; Advanced Magnetics, Inc.) (1 mg/mL), in 20 mM citric acid (pH 3, 4, 5, 5.4, and 7.0) according to a previously described method (23). To evaluate in vivo degradation of ATP@SPIONs, mice were intravenously injected with 300 μg of ATP@SPIONs or Feridex and imaged using a 3-T clinical MR imaging instrument (Magnetom Tim Trio; Siemens Medical Solutions) with volume coil (RAPID Biomedical GmbH). The T2-weighted MR images were acquired over 21 d. A T2-weighted fast spin echo sequence with an echo train length of 10 was set as follows: repetition time/echo time, 3,000 ms/65 ms; flip angle, 150°; field of view, 4 cm; slice thickness, 1.2 mm; interslice gap, 0.12 mm (10% of slice thickness); and matrix, 256 × 210.
Synthesis and Characterization of GA-ATP@SPIONs
ATP@SPIONs were suspended in 2-(N-morpholino)ethanesulfonic acid buffer (10 mM, pH 5.5). Gluconic acid (GA) was added to the ATP@SPION solution at 0.1 mol/mol ATP followed by the dropwise addition of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide solution. The 1:1 molar ratio of this solution to GA was used. The reaction mixture was stirred for 2 h and then washed with distilled water 3 times. The GA-conjugated ATP@SPIONs (GA-ATP@SPIONs) were characterized by Fourier transform–infrared spectroscopy analysis. To investigate the antibiofouling effects of GA conjugation, Prussian blue staining was performed. RAW cells were placed on a 96-well plate at a density of 1 × 104 cells per well and were incubated overnight. After 1 h of adding 50 μg of ATP@SPIONs or GA-ATP@SPIONs, Prussian blue staining was performed and the blue dots bound on the cells were observed by a light microscope with a digital camera. ATP@SPIONs or GA-ATP@SPIONs were intravenously injected into mice at a dose of 13 mg/kg. The organs collected from the mice, such as liver, lung, spleen, and kidney, were stained at 1 h after injection. The organs were frozen-sectioned and stained by Prussian blue.
Cell Cytotoxicity Tests for GA-ATP@SPIONs
The cytotoxicity of GA-ATP@SPIONs was investigated with an XTT kit. The CHO (hamster ovary cell line) and MDA-MB231 cells (human breast cancer cell line) were placed on a 96-well plate at a density of 1 × 104 cells per well and were incubated overnight. GA-ATP@SPIONs were added to the wells. After incubation for 24 h, XTT analysis was performed. The viability of the cells was determined by measuring the ultraviolet absorption at 450 nm on a plate reader (VersaMax; Molecular Devices Corp.).
Cellular Binding Studies of cMBP Conjugation to GA-ATP@SPIONs
To label the GA-ATP@SPIONs with Cy5.5, they were suspended in phosphate-buffered saline (PBS) (10 mM, pH 7.4). After Cy5.5-NHS in dimethyl sulfoxide was added, the reaction solution was stirred for 6 h in the dark. The nonreacted Cy5.5 was removed by dialysis (molecular-weight cutoff of 12 kDa; Spectrum Laboratories). Cy5.5-conjugated GA-ATP@SPIONs were suspended in PBS (pH 7.4) with ethylenediamine tetraacetic acid, and 200 μg of N-succinimidyl 3-(2-pyridyldithio) propionate (Pierce) dissolved in dimethyl sulfoxide were added to the Cy5.5-GA-ATP@SPION solution. After reaction for 3 h, the mixture was filtered and washed using a centrifugal tube (cutoff, 50 kDa; Falcon). cMet binding peptide (cMBP, NH2-Lys1-Ser2-Leu3-Ser4-Arg5-His6-Asp7-His8-Ile9-His10-His11-His12-Gly13-Gly14-Gly15-Cys16-Ac) was added to the N-succinimidyl 3-(2-pyridyldithio) propionate–modified Cy5.5-conjugated GA-ATP@SPION solution, and then the reaction solution was incubated overnight in the dark. The final solution was separated from the nonreacted cMBP.
cMBP-conjugated GA-ATP@SPIONs (MG-ATP@SPIONs) were radiolabeled with Na125I for in vitro cellular binding analysis and in vivo γ imaging as follows: 200 μL of MG-ATP@SPIONs solution were added to a tube, and 7.4 MBq of Na125I and 25 μg of chloramine T in 10 μL of PBS (0.5 M, pH 7.5) were sequentially added to the tube. The reaction was terminated with sodium bisulfide (25 μg/10 μL) in PBS. Radiolabeling efficiency was analyzed using instant thin-layer chromatography–silica gel strips (Gelman Sciences). The eluted strips were scanned on an automatic thin-layer chromatography scanner (Bioscan).
U87MG cells (human glioblastoma, 1 × 106) were seeded into 6-cm dishes. After overnight incubation, the medium was removed and replaced with serum-free medium. The cells were treated with 30, 60, or 90 μg of Cy5.5-conjugated cMBP-GA-ATP@SPIONs only (CMG-ATP@SPIONs) or were cotreated with CMG-ATP@SPIONs (90 μg) and free cMBP (20 nmol) for blocking studies. After 2 h of incubation, the cells were washed and harvested using trypsin-ethylenediamine tetraacetic acid. The fluorescence intensity of the cells bound with CMG-ATP@SPIONs was determined using an IVIS optical imaging system (Caliper Life Science) with the analysis conditions as follows: f-stop, 1 s; binning, 8; field of view, 12.8; exposure time, 1 s. For confocal laser-scanning fluorescence microscopy, U87MG cells were seeded and incubated on a 12-mm microscope cover glass (Marlenfeld GmbH) in 4-well plates at 37°C. CMG-ATP@SPIONs (30, 60, or 90 μg) were added and incubated for 1 h at 37°C. After being washed twice with cold PBS, the cells were fixed with 4% (w/v) paraformaldehyde solution, and 4,6-diamino-2-phenylindole was added to stain the nuclei. The cells were mounted and viewed using an LSM 510 META confocal laser scanning microscope (Carl Zeiss Inc.). For blocking studies, the cells were cotreated with CMG-ATP@SPIONs (90 μg) and free cMBP (20 nmol).
In Vivo Optical Imaging Studies
U87MG cells (5 × 106) were subcutaneously injected in the right flank of the mice. When the tumor implants reached 0.7 cm in diameter, optical imaging was performed using the IVIS optical imaging system. The imaging was performed at 1, 3, and 6 h after injection of 200 μL of CMG-ATP@SPIONs into the mice via a tail vein at a dose of 13 mg/kg. A Cy5.5 filter set (640 nm for excitation, 700 nm for emission) and a subtraction filter set (600–570 nm for excitation) were used for acquiring the in vivo images. The obtained images were analyzed using Living Image Software (Caliper Life Science) (24). All animal experiments were performed in accordance with the guidelines suggested by the Chonbuk National University Medical School Ethics Committee.
In Vivo SPECT/CT Imaging Studies
125I-labeled MG-ATP@SPIONs (5.5 MBq/200 μL) were injected into the U87MG xenograft mice via the tail vein at a dosage of 13 mg/kg. The static images were obtained for 10 min at an acquisition time of 1, 2, and 3 h after the injection of the radiolabeled SPIONs using a small-animal imaging system (X-SPECT/CT system; GE Healthcare) with pinhole collimation (aperture diameter, 1 mm; focal length, 9 cm) and a 15- to 45-keV photopeak energy window. After small-animal CT imaging, the whole-body small-animal SPECT images were acquired 4 h after injection using 32 projections over 360° (radius of rotation = 7.6 cm, 30 s/projection). The reconstructed data of SPECT and CT were visualized and coregistered using AMIRA, version 3.1 (Visualization Sciences Group).
In Vivo MR Imaging Studies
CMG-ATP@SPIONs and GA-ATP@SPIONs were intravenously injected via the tail vein at a dosage of 13 mg/kg with 200 μL. The mice were scanned using a 1.5-T clinical MR scanner (Signa Exite Twin-speed) with an animal coil (4.3-cm Quadrature volume coil, Nova Medical Systems). The T2-weighted MR images were acquired at 1, 3, and 5 h after injection of the SPIONs. A fast spin echo imaging method was used, and the imaging parameters were a repetition time/echo time of 4,200 ms/102 ms, a flip angle of 90°, an echo train length of 10, a field of view of 5 cm, a section thickness of 2 mm, an intersection gap of 0.2 mm, and a 256 × 160 matrix. The relative signal enhancement of tumors compared with that of normal muscle before (pre) and after (post) injection of the contrast agent was calculated using the following formula: [(signal intensity of post – signal intensity of pre)/signal intensity of pre] × 100.
Histology of Tumor Tissues
The tumor tissue of the U87MG mouse model was fixed with 4% formalin solution and then embedded in paraffin. Coronal sections (4 μm thick) of the tumor paraffin block were obtained, and the tumor tissue sections were identified by Prussian blue staining. All micrographs were taken using a light microscope with a digital camera.
Biodistribution Studies
125I-labeled MG-ATP@SPIONs (3.7 MBq/mL, 0.1 mL) were administrated into the tail vein in the U87MG-bearing mice. The mice were sacrificed and dissected at 3 h after injection. The main organs and tissues were collected, and the radioactivity was measured using a γ counter (Packard). The results were expressed as the percentage of the injected dose per gram of tissue. The radioactivity in the tissue samples was calibrated against a known quantity of the injected dose. Values are expressed as mean ± SD (n = 4).
Statistics
Quantitative data were expressed as mean ± SD. Means were compared by use of an independent-samples t test. P values of less than 0.05 were considered statistically significant.
RESULTS
Characterization of ATP@SPIONs
ATP@SPIONs were strongly water-soluble, without any precipitation over a month’s time. The amount of ATP determined on the SPIONs was 0.319 mg/1 mg of the total particle weight. The x-ray photoelectron spectroscopy peaks for the iron in the ATP@SPIONs were observed at a binding energy of 715 eV (2p3) and 727 eV (2p1) (Supplemental Fig. 1). A single N peak for the adenine rings and a P 2p peak for the phosphate groups in the ATP@SPIONs were obtained at 398.3 eV and 132.7 eV, respectively. The O 1s peaks were observed at binding energies of 529.5, 530.7, 531.5, and 532.7 eV (C–O–C). These peaks match the main peaks of ATP and the iron particles, allowing us to confirm the preparation of the ATP@SPIONs. The shape and size of the ATP@SPIONs were observed and determined by transmission electron microscopy and dynamic light scattering, respectively (Fig. 2). As shown in Figures 2A and 2B, the ATP@SPIONs were spheric and had a mean diameter of 22.7 nm, with a narrow distribution. To evaluate the magnetic properties of the ATP@SPIONs, MR phantom studies and superconducting quantum interference device analysis were performed. The magnetic measurements by superconducting quantum interference device revealed the superparamagnetic behavior of the ATP@SPIONs (Fig. 2C). The T2-weighted MR phantom images of the ATP@SPION solution were obtained using a clinical 1.5-T MR scanner (Fig. 2D). The T2 signal intensity (darkness) was increased in an ATP@SPION concentration–dependent manner.

Characterization of ATP@SPIONs: transmission electron microscopy image of ATP@SPIONs (scale bar corresponds to 50 nm) (A), size distribution (B), magnetization curve (C), and phantom images of water and iron nanoparticles in water acquired from T2-weighted MR imaging (D). CHAN = channel; Oe = oersted.
Degradation of ATP@SPIONs
The dark brown color of the ATP@SPIONs turned rapidly to the bright yellow color of free iron ions at low pHs, except pH 7, whereas there was no color change of Feridex at any pH for 10 h (Supplemental Fig. 2). After injection into the tail vein of mice with an excess, Feridex showed the highest MR enhancement only at 3 h, and the saturated signal drop in the liver persisted without any recovery during experiment times (Fig. 3A). Despite an excess, ATP@SPIONs showed the degradation pattern (Fig. 3B). The liver MR enhancement was highest at 1 d after injection and started regaining from 6 d and recovered to 81% of normal within 3 wk after the MR signal of the liver had dropped (Fig. 3C).

In vivo degradation of ATP@SPIONs. (A and B) In vivo T2-weighted MR images of mouse liver before and after intravenous injection of ATP@SPIONs (A) and Feridex (B). (C) Changes in T2-weighted MR signal enhancements of liver after injection of ATP@SPIONs and Feridex.
Effects of GA Conjugation on ATP@SPIONs
The Fourier transform-infrared spectroscopy spectra of the SPIONs, ATP@SPIONs, and GA-ATP@SPIONs are shown in Supplemental Fig. 3. After GA conjugation, the specific peaks were observed at 870–906 cm−1 (CH2CH3), 1,064 cm−1 (C–N), 1,469 cm−1 (N–H), 1,563 cm−1 (C=O), and 2,797 cm−1 (C–H), respectively. We first examined whether GA-ATP@SPIONs have any cytotoxic biologic properties. The cytotoxicity studies of our nanoparticles were performed on 2 different cell lines (CHO and MDA-MB231). GA-ATP@SPIONs were reasonably nontoxic up to the particle concentration of 200 μg/mL (Supplemental Fig. 4). To investigate the antibiofouling effect by GA conjugation, the uptake by macrophage cells in vitro and the accumulation in major organs such as liver and lung after intravenous injection into mice were evaluated. Prussian blue staining was performed after 2 h of incubating the SPIONs with the RAW cells (Supplemental Fig. 5). Compared with that of the ATP@SPIONs, the uptake of GA-ATP@SPIONs into the cells was significantly reduced. When ATP@SPIONs were injected into mice via a tail vein, the particles accumulated mostly in the phagocytic organs such as liver and spleen, whereas the GA-ATP@SPIONs showed significantly lower accumulation in the liver and spleen (Supplemental Fig. 6).
In Vitro Tumor Targeting
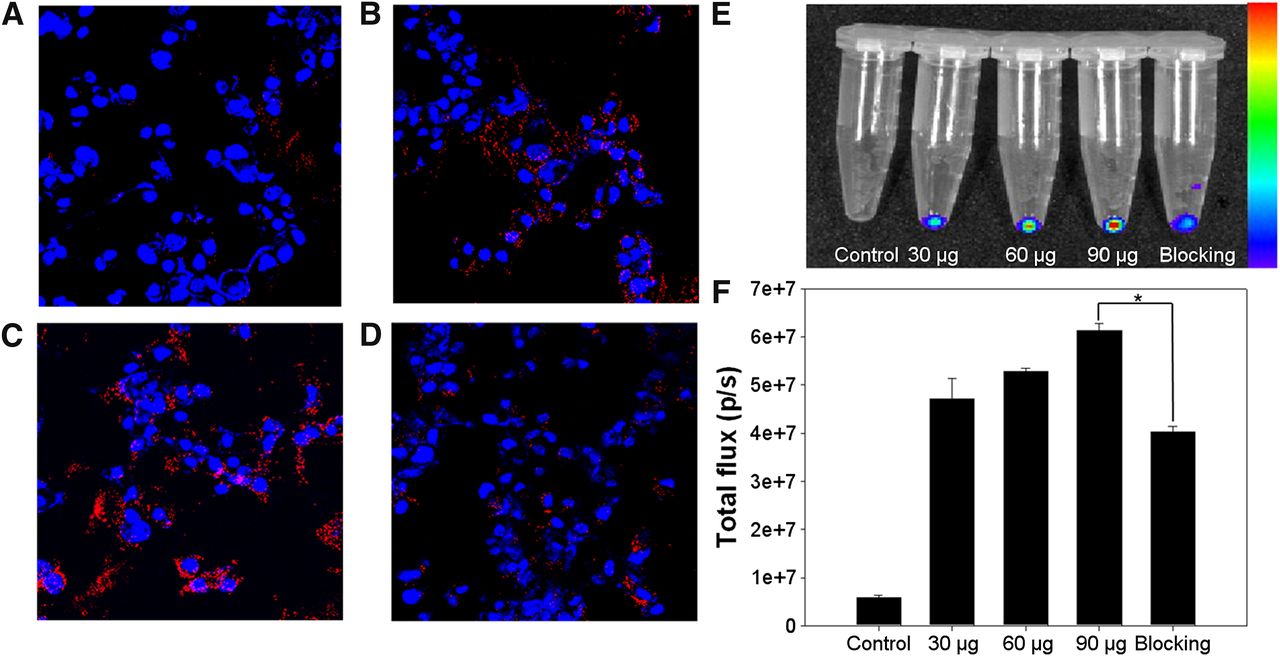
The cellular binding of MCG-ATP@SPIONs with U87MG cells was visualized by confocal fluorescent laser scanning microscopy (Fig. 4A–4D). As shown in Figure 4, the MCG-ATP@SPIONs showed higher binding ability as the amount the SPIONs was increased. The cellular binding of MCG-ATP@SPIONs was significantly reduced by blocking the receptors with free cMBP. In quantification analysis, the fluorescence signal intensity of the cellular binding of the MCG-ATP@SPIONs gradually increased with the particle concentration and clearly was decreased by blocking the receptor with cMBP (Figs. 4E and 4F). The quantification results show that the total flux values of the cellular binding of MCG-ATP@SPIONs were dependent on the concentration of the particles, and blocking with cMBP led to a significant decrease in the values for the binding of MCG-ATP@SPIONs (P < 0.001). Further, the specific cellular binding of cMBP-GA-ATP@SPIONs was confirmed through radiolabeling 125I (P < 0.001, Supplemental Fig. 7).

Cellular binding of cMBP-GA-ATP@SPIONs. (A–C) Confocal laser scanning fluorescence images of U878MG cells. Images of U87MG cells were obtained after incubation for 1 h in presence of 30 μg (A), 60 μg (B), or 90 μg (C) of cMBP-GA-ATP@SPIONs. (D) U87MG cell image was obtained after incubation for 1 h in presence of 90 μg of cMBP-GA-ATP@SPIONs with cMBP. (E) Near-infrared fluorescent imaging for quantification of cellular binding. (F) Quantitative analyses of cellular binding by calculation of total flux values of region of interest using IVIS imaging software. Cellular binding of cMBP-GA-ATP@SPIONs was significantly reduced by cMBP (*P < 0.001). Nuclei of cells were stained using 4,6-diamino-2-phenylindole (blue). Data represent mean ± SD (n = 4). p/s = photons per second.
In Vivo Multimodal Tumor Imaging
Figure 5 shows near-infrared fluorescent optical images obtained for 4 h after injection of the MCG-GA-ATP@SPIONs. A strong fluorescence signal in the tumor was clearly detected, and the ratio of the fluorescence signal intensity of the region of interest of the tumor to that of normal muscle was 5.3 ± 1.7. Figure 5B shows the γ-imaging series in which the data were obtained over the course of 3 h after the injection of 125I-labeled cMBP-GA-ATP@SPIONs. The tumor area was visualized with high contrast relative to the contralateral normal muscle. Tumor accumulation at 3 h after the injection of 125I-labeled cMBP-GA-ATP@SPIONs was 2.96% ± 0.74% of the injected dose per gram (Supplemental Fig. 8). The SPECT/CT fusion image of the mouse is presented in Figure 5C. Tumor uptake was more clearly visualized on the reconstructed coregistered transaxial SPECT/CT image.

Multimodality tumor imaging using cMBP-GA-ATP@SPIONs. (A) In vivo noninvasive near-infrared fluorescent images of mice with U87MG tumors. Images were taken at 1, 2, 3, and 4 h after injection of CMG-ATP@SPIONs. (B) In vivo static planar γ images of mice with U87MG tumors. Images were taken at 1, 2, and 3 h after injection of 125I-cMBP-GA-ATP@SPIONs. (C) Reconstructed coregistered transverse SPECT/CT image of same mouse 4 h after injection. In vivo T2-weighted MR images of mice with U87MG tumors were obtained before (D) and at 3 h after (E) injection of cMBP-GA-ATP@SPIONs. (F) Prussian blue staining on tumor sections. Blue dots, representing iron contents, were widely found across tumor, with inhomogeneous pattern. PI = after injection.
Figures 5D and 5E show the T2-weighted MR images of the U87MG-bearing mice before and after injection of cMBP-GA-ATP@SPIONs. The tumor appears as a hyperintense area on the T2-weighted MR images before injection of the cMBP-GA-ATP@SPIONs. The signal intensity of the T2-weighted images of the tumor clearly dropped after injection of the iron oxide particles. The relative signal enhancement of the tumor area on the MR image was 22.1% compared with before injection. On the other hand, after GA-ATP-SPIONs, the MR images for the tumor area showed feeble signal enhancement, compared with the enhancement after injection of the cMBP-GA-ATP@SPIONs (Supplemental Fig. 9). As shown in Figure 5F, the presence of the iron particles in the tumor was readily indicated by blue dots on Prussian blue staining.
DISCUSSION
The goal of this study was to develop SPIONs with a new approach using an innate material, ATP, to assess their biocompatibility and degradation in vivo and to investigate whether tumors can be enhanced by conjugation to tumor-specific ligands on the SPION. ATP@SPIONs were prepared simply by an alkali-mediated coprecipitation method as described previously (14). It is well recognized that phosphate molecules are stably anchored on the surface of SPIONs (25,26). Among various candidates for the surface modification of SPIONs, ATP is a good stabilizer of SPIONs because it has 3 phosphate groups, is produced in the body, and so is a naturally occurring compound in the body itself. The kinetic properties of iron oxide nanoparticles for their degradation in the body strongly depend on their surface coating (27,28). The transformation and degradation take longer for iron oxide nanoparticles with a polymeric surface coating than for iron oxide nanoparticles with a noncoated surface because of the limited availability and affinity of the iron chelating agents produced intracellularly (23,29). The iron ions from the transformed and degraded nanoparticles may allow bonding with ferritin proteins, leading to minimum or no toxicity. ATP@SPIONs rapidly degraded to iron ions at a slightly lower pH, such as that in endosomes or lysosomes, which exist in the intracellular environment, but Feridex maintains the iron particle formulation without transformation to ions (Supplemental Fig. 2). To confirm whether ATP@SPIONs exhibit rapid degradation compared with Feridex in vivo, as they do in vitro, we performed sequential MR studies after intravenous injection of ATP@SPIONs or Feridex into mice (Fig. 3). It is well known that Feridex shows the degradation profiles in the liver within 7–10 d after injection with approximately 1 mg of iron per kilogram (30–32). An excess (15 mg/kg) of ATP@SPIONs or Feridex was injected for degradation studies in vivo to maximize differences in degradation between the 2 agents. Feridex showed the saturated signal drop in the liver only at 3 h, and the drop persisted, without any recovery. On the other hand, despite an excess, ATP@SPIONs were degraded in the liver like the usual dose of Feridex was, with the result that liver MR enhancement started regaining at 6 d and recovered to 81% of normal within 3 wk. These data mean that compared with Feridex, fewer ATP@SPIONs are taken up into Kupffer cells, and ATP@SPIONs show prolonged circulation in the blood and are degraded gradually after phagocytosis in vivo. Considering the degradation of iron particles into the body, we expect that ATP@SPIONs will show good kinetics for degradation and elimination, compared with polymer-coated SPIONs. We found that ATP@SPIONs were nontoxic in cell viability studies. Considering the degradation of iron oxide after injection into the body and the noncytotoxicity of ATP@SPIONs, we expect that they will show good kinetics for degradation and elimination, compared with polymer-coated SPIONs.
SPIONs having amino groups on their surface are quickly captured by phagocytic cells such as macrophages in the liver and spleen (33,34). We used GA to modify the ATP@SPIONs so as to diminish phagocytosis by the reticuloendothelial system and adsorption of plasma proteins on the particle’s surface. GA is also not a polymer, and GA has a simple structure and is a biocompatible material like ATP. In a previous paper, we reported that uptake of the amino-Q dots in liver and lung could be reduced in vivo through GA conjugation to their surface (35). These results indicate that GA conjugation on the surface of ATP@SPIONs drastically minimizes hepatic and splenic accumulation through reducing the recognition by reticuloendothelial cells. It is essential to diminish phagocytic uptake of the SPIONs because they may cause oxidation stress in the organs and cytotoxicity and deprive the SPIONs of the opportunity to be taken up in the target site. Most of the recently reported SPIONs, including polymer-coated iron oxide nanoparticles, have not clearly shown their biodistribution in the whole body after intravenous injection. It is important to evaluate whether SPIONs accumulate in any organs and tissues and whether they efficiently reach the target sites, although they show a good paramagnetic effect in target sites.
We next studied the ability of the SPIONs as a specific targeted contrast agent. cMet, which is normally expressed by epithelial cells, is a tyrosine-kinase receptor for hepatocyte growth factor and scatter factor. cMet signaling stimulated by hepatocyte growth factor and scatter factor is essential for cell proliferation, migration, angiogenesis, embryogenesis, organogenesis, and tissue regeneration. Compared with normal cells, cMet is overexpressed and amplified in a variety of human tumors, including lung, breast, colorectal, prostate, pancreatic, head and neck, gastric, and hepatocellular tumors; glioma; melanoma; and several sarcomas (36–38). cMet has recently been considered as a promising target for diagnosing cancer and determining its therapy. In our previous studies, we suggested that the epitope-mimicking peptide (NH2-Lys1-Ser2-Leu3-Ser4-Arg5-His6-Asp7-His8-Ile9-His10-His11-His12-Gly13-Gly14-Gly15-Cys16-Ac) binding to cMet may be a good candidate for the detection of cancer (39,40). We evaluated whether cMBP-GA-ATP@SPIONs can specifically detect cMet-expressed cancer cells in vitro. cMBP-GA-ATP@SPIONs showed specific binding to the cells through receptor mediation (Fig. 4). The in vivo near-infrared fluorescent/MR/SPECT imaging studies indicated that cMBP-GA-ATP@SPIONs were clearly accumulated in the cMet-positive U87MG tumor. These results of 3 types of multimodality imaging show the potential benefits of cMBP-GA-ATP@SPIONs as a multifunctional imaging probe.
CONCLUSION
We have presented in this study the novel approach of using ATP in place of polymeric materials as the surface modification agent in the engineering of SPIONs for tumor-targeted multimodality imaging in vivo. ATP@SPIONs degraded spontaneously in a pH lower than 7 in vitro, and they caused T2-weighted MR images of the liver to be enhanced gradually, with enhancement restored to 81% of normal within 3 wk. The ATP@SPIONs were modified with GA to reduce phagocytic uptake after administration. MG-ATP@SPIONs could visualize the U87MG tumors using multimodality imaging, including near-infrared fluorescent optical imaging, MR imaging, and SPECT imaging in vivo. Successful accumulation of MG-ATP@SPIONs in tumors was confirmed using Prussian blue staining of the tumor tissues. Therefore, ATP@SPIONs not only biodegrade well in vivo but also are a multiplatform for targeting specific regions of interest in molecular imaging. We are planning to further investigate the abilities of ATP@SPIONs in targeted imaging and therapy in various tumor models as an extension of this study.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. The authors had final responsibility for the decision to submit for publication. This work was supported by a grant from the National R&D Program for Cancer Control, Ministry of Health, Welfare and Family Affairs, Republic of Korea (0620220). This work was also supported by National Research Foundation of Korea (NRF) grants funded by the Korean Government (MEST) (grants 2011-0028581, 2012M2A2A7035779, and 2012M2A2A7014020). No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Sep. 19, 2013.
- © 2013 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication February 27, 2013.
- Accepted for publication June 24, 2013.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}