


Abstract
In vivo estimation of β2-nicotinic acetylcholine receptor availability with molecular neuroimaging is complicated by competition between the endogenous neurotransmitter acetylcholine and the radioligand 123I-3-[2(S)-2-azetidinylmethoxy]pyridine (123I-5-IA). We examined whether binding of 123I-5-IA is sensitive to increases in extracellular levels of acetylcholine in humans, as suggested in nonhuman primates. Methods: Six healthy subjects (31 ± 4 y) participated in a 123I-5-IA SPECT study. After baseline scans, physostigmine (1–1.5 mg) was administered intravenously over 60 min, and 9 additional scans were obtained. Results: We observed a significant reduction in the total volume of distribution after physostigmine administration (29% ± 17% in the cortex, 19% ± 15% in the thalamus, 19% ± 15% in the striatum, and 36% ± 30% in the cerebellum; P < 0.05). This reduction reflected a combination of a region-specific 7%–16% decrease in tissue concentration of tracer and a 9% increase in plasma parent concentration. Conclusion: These data suggest that increases in acetylcholine compete with 123I-5-IA for binding to β2-nicotinic acetylcholine receptor. Additional validation of this paradigm is warranted, but it may be used to interrogate changes in extracellular acetylcholine.
In vivo molecular imaging studies of muscarinic and nicotinic acetylcholine receptors have provided substantial contributions to our understanding of disorders related to cholinergic dysfunction but are limited by the lack of a suitable method for measuring fluctuations in brain acetylcholine levels. For example, our evaluation of the β2-nicotinic acetylcholine receptor (β2*-nAChR) demonstrated significantly lower receptor availability in unmedicated individuals with major depressive disorder than in control subjects (1); however, quantitation of total β2*-nAChR binding sites in postmortem brain revealed no differences between major depressive disorder and control samples, suggesting that increased acetylcholine levels in vivo may have resulted in lower β2*-nAChR availability and apparent lower receptor density.
In vivo imaging of β2*-nAChRs is possible with the high-affinity radioligand 123I-3-[2(S)-2-azetidinylmethoxy]pyridine (123I-5-IA) (2) and SPECT (3). 123I-5-IA has slow dissociation from the receptor–ligand complex, a good specific-to-nonspecific binding ratio, and high selectivity for β2*-nAChRs (3,4). 123I-5-IA has been used to measure β2*-nAChR availability in animals (4–6) and humans (7,8); however, there are no published studies demonstrating in vivo measurements of brain levels of acetylcholine in human subjects. Studies on nonhuman primates suggest that competition between acetylcholine and radioligand may be detectable (9,10), and microdialysis studies on rodents suggest an at least 10- to 30-fold increase (depending on dose) in brain acetylcholine levels after acetylcholinesterase inhibitor administration (11). In one study, 123I-5-IA binding in the thalamus was decreased (15%) after a challenge with a 0.067 mg/kg dose of physostigmine and was consistently decreased (14%–17%) after a challenge with a 0.2 mg/kg dose (10).
Here, we used 123I-5-IA SPECT to determine whether physostigmine-induced increases in extracellular acetylcholine in the brain compete with 123I-5-IA binding in vivo in humans. We hypothesized that physostigmine-induced increases in extracellular acetylcholine would significantly reduce 123I-5-IA binding.
MATERIALS AND METHODS
Six healthy control subjects (3 men and 3 women; mean age ± SD, 31 ± 4 y) gave written informed consent and completed the study, which was approved by the institutional review boards of Yale University School of Medicine, Veterans Affairs Health Care System, and the University of Toronto. Eligibility was evaluated via a structured interview, behavioral assessments, physical examination, laboratory blood tests, urine drug screening, and an electrocardiogram. Subjects had never smoked; had no life-time psychiatric, neurologic, or medical history; and had no contraindications for undergoing an MRI scan and a 123I-5-IA SPECT scan on the same day.
Mood symptoms were measured with the Center for Epidemiological Studies Depression Scale (12) and the Beck Depression Inventory (13). State and trait anxiety symptoms were measured with the Spielberger State-Trait Anxiety Inventory (14). All tests were administered at intake and on the scanning day (before and after physostigmine administration).
MRI was performed on a 3-T Magnetom Trio with Tim technology (Siemens) running Numaris/4 software (version: syngo MR B17) to guide placement of regions of interest for SPECT scans (series 1: 3-plane localizer; series 2: sagittal 3-dimensional turbo fast low-angle shot, 250-mm field of view, 1-mm-thick slices, 176 slices in total, echo time of 3.53 ms, repetition time of 2,500 ms, inversion time of 1,100 ms, flip angle of 7, 256 × 256 matrix, and 2 signal averages).
123I-5-IA was synthesized and administered for the duration of the study as described previously (7) using a bolus plus a constant infusion (7.3 ± 0.2 h) with a total injected dose (accounting for decay) of 390.2 ± 13.2 MBq. Six hours after injection of 123I-5-IA, a simultaneous transmission emission scan and 3 equilibrium 30-min emission scans (90 min total) were obtained on a Prism 3000 XP SPECT camera (Phillips). Subjects were administered glycopyrrolate (0.2 mg, intravenously) to minimize peripheral muscarinic side effects, followed by administration of physostigmine (a reversible acetylcholinesterase inhibitor that crosses the blood–brain barrier) over 1 h (1–1.5 mg, intravenously). Thereafter, up to 3 sets of 30-min emission scans were acquired (each set 90 min in duration, with a 20- to 30-min break between each set. In subject 5, the 123I-5-IA infusion was interrupted after the collection of 5 postphysostigmine scans; thus, data thereafter are not shown. In the other 5 subjects, all 9 postphysostigmine scans were collected). Venous blood samples were collected before and after physostigmine administration to correct for individual differences in radiotracer metabolism and protein binding. Pulse and blood pressure were measured before and after injection of 123I-5-IA and before and after physostigmine administration.
SPECT images were analyzed as described previously (7). Regional 123I-5-IA uptake (β2*-nAChR availability) was calculated as total volume of distribution (VT/fp), where VT is brain regional activity divided by metabolite-corrected plasma activity and fp is the free fraction of parent in plasma. Plasma for fp calculation was collected at 4 time points and applied to calculate the corresponding VT/fp. The first was before 123I-5-IA administration (baseline), the second was immediately before physostigmine administration, the third was immediately after physostigmine administration, and the fourth was at the end of the last set of postdrug scans. Specifically, fp values from time points 1 and 2 were averaged and applied to baseline VT to estimate baseline VT/fp; fp from time point 3 (immediately after physostigmine administration) was applied to VT from the first and second postphysostigmine scanning sessions to estimate VT/fp for those time points; and fp from time point 4 was applied to the last scanning session. The regions studied were frontal, parietal, anterior cingulate, temporal, and occipital cortices (averaged to obtained a single cortical value); striatum; thalamus; and cerebellum. The change in radioligand binding to β2*-nAChRs was calculated as the percentage difference between VT/fp before and VT/fp after physostigmine administration for each postdrug SPECT scan. To demonstrate the use of previously calculated nondisplaceable binding (VND/fP) (15) and estimate specific radioligand binding (BPf), we subtracted a fixed value (19.4 mL·cm−3) of VND/fP from VT/fp for each brain region and scan.
All statistical analyses were performed using SPSS, version 19.0 (IBM). Statistical significance was set at a P value of 0.05 or less, 2-tailed. Repeated measures of ANOVA (df = 5) were used to assess within-subject differences in pharmacokinetic parameters and mood variables before and after physostigmine administration. SD was calculated for all outcome variables (Table 1) and is represented as error bars in Figure 1.
Outcome Values for Each Subject at Baseline and 2–4 Hours After Physostigmine Injection

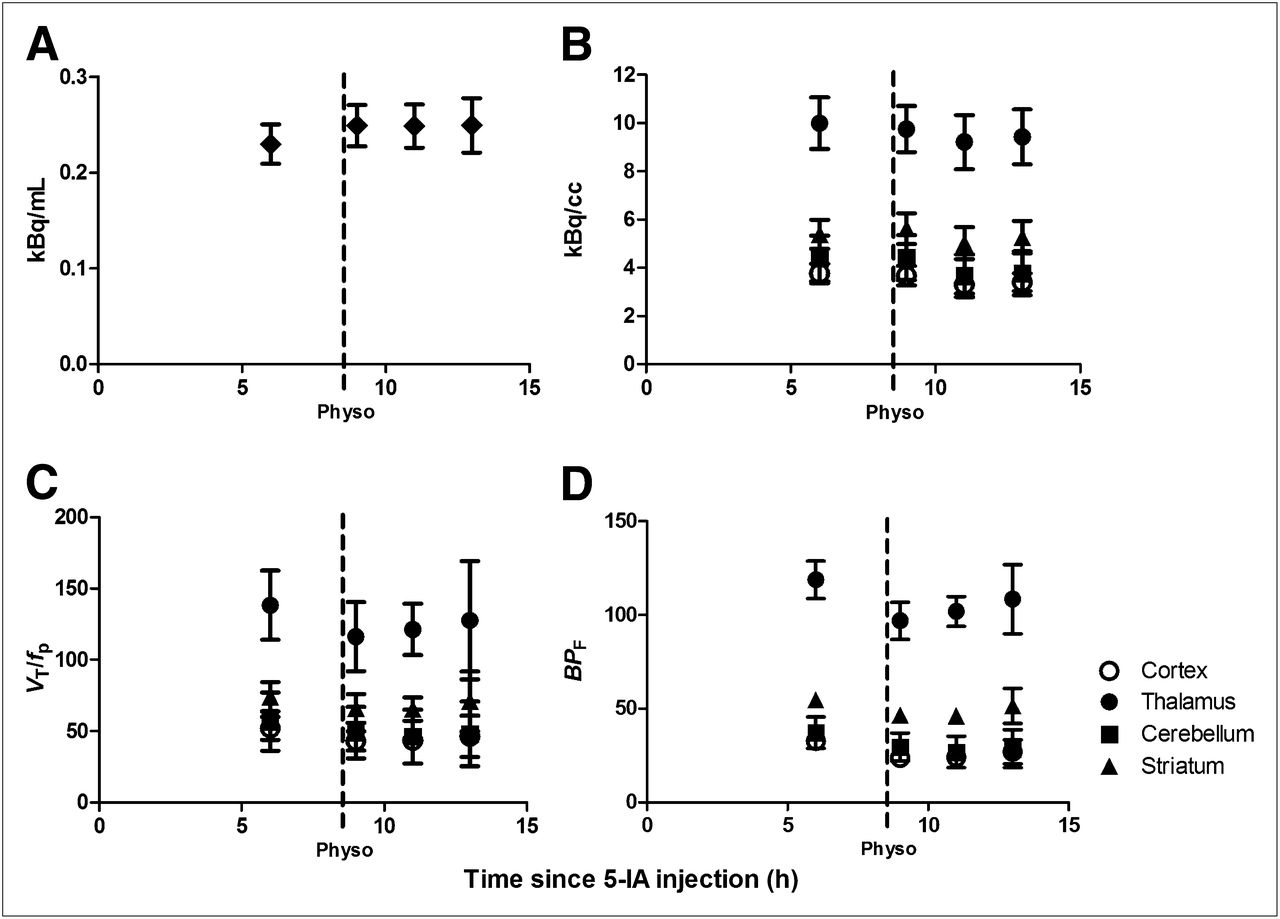
First point in each graph represents baseline data obtained starting 6 h after beginning of tracer infusion, when state of equilibrium was achieved, and provided baseline specific binding. After completion of baseline scans, physostigmine was administered intravenously (1.0–1.5 mg over 1 h). At onset of physostigmine infusion, scanning was resumed for up to 9 h. Bars represent SEM. (A) Plasma 123I-5-IA concentration (kBq/mL) (total parent) measured during 123I-5-IA constant infusion in healthy volunteers. After physostigmine administration, there was significant 9% increase in mean plasma 123I-5-IA concentration as compared with before physostigmine administration. (B) Tissue 123I-5-IA concentration (kBq/cm3) in thalamus, striatum, cortex, and cerebellum measured during 123I-5-IA constant infusion. We observed 7%–16% region-specific decrease in 123I-5-IA tissue concentration after physostigmine challenge. (C) 123I-5-IA VT/fp in thalamus, striatum, cortex, and cerebellum measured during 123I-5-IA constant infusion. VT/fp values measured after physostigmine infusion were significantly reduced (14%–18% region-specific), compared with baseline values. (D) 123I-5-IA BPf in thalamus, striatum, cortex, and cerebellum measured during 123I-5-IA constant infusion. BPf values measured after physostigmine infusion were significantly reduced (19%–36% region-specific), compared with baseline values.
RESULTS
Nonsmoking status was verified by negligible urine cotinine (0 ηg/mL), plasma cotinine (<2ηg/mL), and nicotine (<1.0 ηg/mL) levels and exhaled carbon monoxide (0.7 ± 0.8 ppm). There were no significant differences in subjects’ mood or anxiety before and after physostigmine administration (P > 0.2).
Administration of physostigmine did not significantly alter free fraction (P > 0.2) but resulted in significantly increased 123I-5-IA total plasma parent activity for all subjects (9.1% ± 8.6%, t = −2.56, P = 0.05, 1 h after a challenge, stable thereafter; Fig. 1A) and significantly increased free parent activity (9.9% ± 7.7%, t = −3.1, P = 0.03) at 1 h after a challenge but not thereafter (t = −1.2, P = 0.30, 6 h after a challenge).
Equilibrium 123I-5-IA binding (<5% change in receptor availability/h), was reached 6–8 h after injection (average change across subjects: 2.7% ± 1.7%/h in the thalamus, 3.6% ± 2.2%/h in the striatum, 2.5% ± 2.5%/h in the cortex, and 1.7% ± 1.6%/h in the cerebellum). 123I-5-IA tissue concentration was reduced after physostigmine, with the peak reduction reached 2–4 h after a challenge (Fig. 1B), the same time point for the greatest decrease in 123I-5-IA binding after nicotine administration (15,16). A reduction in 123I-5-IA tissue concentration was observed in all brain regions 2–4 h after a challenge: thalamus, 7.8% ± 4.7%; striatum, 7.0% ± 0.9%; mean cortex, 12.7% ± 18.1%; and cerebellum, 16.5% ± 13.6%.
Administration of physostigmine significantly reduced the total volume of distribution of 123I-5-IA at 2–4 h after physostigmine administration. The peak average decrease in VT/fp was 18% ± 11% in cortical regions (F5 = 15.4, P = 0.01), 17% ± 12% in the thalamus (F5 = 11.3, P = 0.02), 14% ± 11% in the striatum (F5 = 11.0, P = 0.02), and 17% ± 10% in the cerebellum (F5 = 10.4, P = 0.02) (Figs. 1C and 2A).

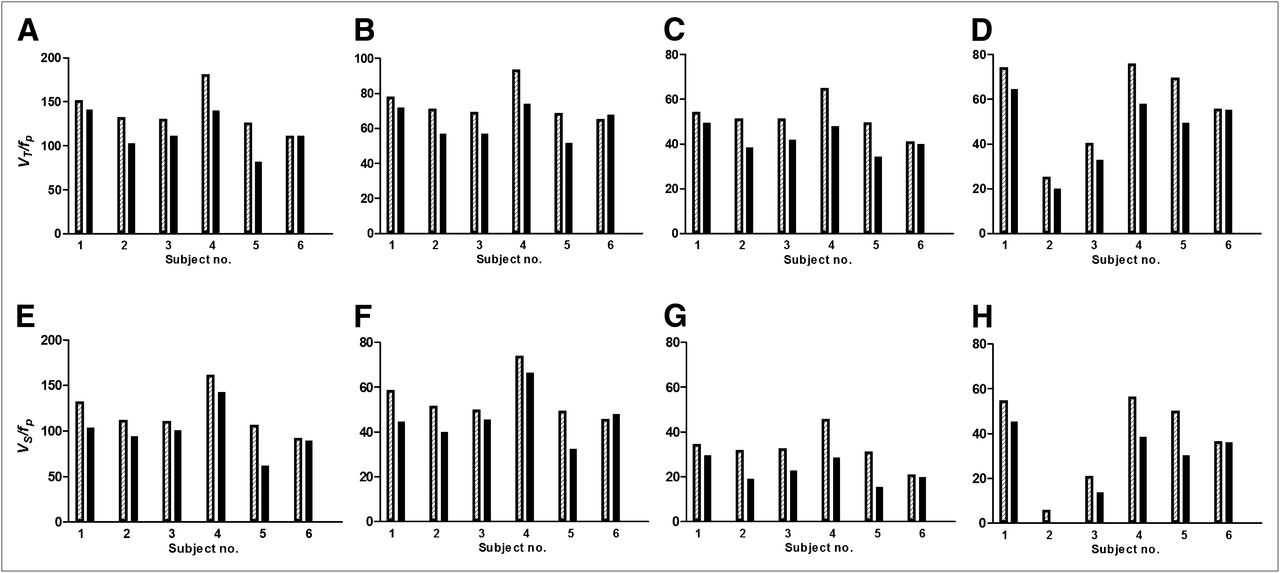
(A–D) β2*-nAChR availability (VT/fP) before (hatched bars) and after (black bars) physostigmine injection for each subject. For thalamus (A), percentage displacement of 5-IA for subjects 1–6 was −7%, −22%, −14%, −22%, −35%, and −0%. For striatum (B), percentage displacement was −8%, −20%, −18%, −20%, −25%, and −4%. For cortex (C), percentage displacement was −9%, −25%, −18%, −26%, −30%, and −2%. For cerebellum (D), percentage displacement was −12%, −20%, −18%, −23%, −28%, and 0%. (E–H) BPf before (hatched bars) and after (black bars) physostigmine injection for each subject. For thalamus (E), percentage displacement of 5-IA for subjects 1–6 was −8%, −25%, −17%, −25%, −41%, and 0%. For striatum (F), percentage displacement was −10%, −27%, −24%, −26%, −34%, and +5%. For cortex (G), percentage displacement was −13%, −40%, −29%, −36%, −50%, and −6%. For cerebellum (H), percentage displacement was −17%, −90%, −35%, −32%, −40%, and −1%.
Subtraction of VND/fp revealed a greater percentage reduction in specific binding of 123I-5-IA. The peak average decrease in BPf was 29% ± 17% in cortical regions (F5 = 15.4, P = 0.01), 19% ± 15% in the thalamus (F5 = 11.3, P = 0.02), 19% ± 15% in the striatum (F5 = 11.0, P = 0.02), and 36% ± 30% in the cerebellum (F5 = 10.4, P = 0.02) (Figs. 1D and 2).
There was a significant decrease in pulse rate after physostigmine administration (12.3% ± 13.3%; P = 0.08) but no significant changes in blood pressure (P > 0.7).
DISCUSSION
We evaluated whether increases in extracellular levels of the endogenous neurotransmitter acetylcholine compete with 123I-5-IA binding to β2*-nAChRs. The goal of this study was to establish a novel paradigm to interrogate the cholinergic system in vivo in humans and to provide a more comprehensive interpretation of our (1,17) and other’s (18) findings in populations with compromised cholinergic systems. Physostigmine administration resulted in a significant decrease in total 123I-5-IA binding, suggesting that 123I-5-IA is sensitive to extracellular acetylcholine levels. This reduction in 123I-5-IA binding is similar to that achieved after smoking a cigarette from which the nicotine has been removed ((19) and unpublished data from our group). Such cigarettes have only 0.05 mg of nicotine, equivalent to smoking about 1 puff from a regular cigarette. However, given that nicotine is a direct agonist at β2*-nAChRs and has high affinity for the receptor, and physostigmine-induced radioligand displacement has an indirect action, it is not surprising that administration of a high dose of physostigmine leads to displacement of 123I-5-IA comparable to that from administration of 0.05 mg of nicotine.
The degree of reduction of 123I-5-IA binding in the thalamus after a physostigmine challenge is in line with the findings of a previous study on nonhuman primates (10), although the amount of injected physostigmine was 10-fold less in the present human study. The similarity in the decrease of radioligand binding may be attributed to lower levels of acetylcholine release in anesthetized nonhuman primates than in conscious humans or to physiologic differences between species. The observed decrease in 123I-5-IA binding here and in the previous study (10) was due to a combination of increases in 123I-5-IA plasma concentration and a reduction in 123I-5-IA tissue concentration. Thus, administration of physostigmine appears to alter radioligand distribution throughout the body, affecting tracer levels in the blood. Specifically, the increase in acetylcholine due to administration of physostigmine occurs throughout the body, and thus the peripheral nAChR binding sites previously available for binding of 123I-5-IA are now also occupied by acetylcholine. The displacement of 123I-5-IA in the body likely causes more free radioligand to circulate in the plasma. Thus, the tissue concentration alone is not an accurate measure for the evaluation of physostigmine-induced acetylcholine displacement of 123I-5-IA, and total volume of distribution or specific ligand binding should be used in this paradigm.
There were several limitations to this study. First, physostigmine may have an allosteric effect on radioligand binding to β2*-nAChRs; however, physostigmine did not alter 123I-5-IA binding in rat in vitro studies and had a significantly lower affinity for β2*-nAChR than did 123I-5-IA (25,000 nM for physostigmine vs. 0.010 nM for 123I-5-IA) (4). Thus, competition between physostigmine and 123I-5-IA for binding to the receptor is not a likely explanation for the observed outcome of decreased 123I-5-IA binding after physostigmine administration. Second, the use of VND/fp obtained from a previous sample of control smokers may not be applicable to a study of nonsmokers. Further, the use of a fixed value for nondisplaceable binding across regions may not accurately reflect the observed regional differences in specific binding, especially for regions with lower levels of specific binding. Specifically, the change in thalamic VT/fp and BPf was 16% and 19%, respectively, whereas the change in a lower-binding region (i.e., cerebellum) was 17% and 36%, respectively. Thus, BPf values were reported with the purpose of showing how the estimate of VND/fp may be used in future studies. Finally, the small sample size allows drawing only preliminary conclusions and limits examination of sex or age differences.
CONCLUSION
We developed a paradigm to interrogate the acetylcholine system in vivo in human subjects and observed a significant decrease in total binding of 123I-5-IA after a physostigmine challenge, consistent with an increase in endogenous extracellular acetylcholine levels. This imaging tool may have enormous potential to facilitate the development of innovative medicines aimed at modulating the cholinergic system.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. Salary support was provided by a VA Career Award and K01MH092681 (Dr. Estelis), MH077681 (Dr. Picciotto), K12DA00167 (Dr. Hannestad), and K01DA20651 (Dr. Cosgrove). Studies were supported by the VA National Center for PTSD and Yale University. Dr. Seibyl has equity interest in Molecular Neuroimaging, LLC. Dr. Tyndale has participated in 1-day advisory meetings for Novartis and McNeil. Dr. Laruelle was a consultant for Amgen, Pfizer, and Roche and a GlaxoSmithKline shareholder at the time of completion of this study and is now a full-time employee of UCB Pharma. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank the technologists at the Institute for Neurodegenerative Disorders for conducting the scanning protocol and Louis Amici (Yale University) for metabolite and protein binding analyses of the radiotracer.
- © 2013 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication July 27, 2012.
- Accepted for publication September 27, 2012.





{kind=link}
{kind=link}