


Abstract
The prognosis of medullary thyroid carcinoma (MTC) varies from long- to short-term survival based on such prognostic factors as serum calcitonin and carcinoembryonic antigen (CEA) doubling times (DTs). This prospective phase II multicenter trial evaluated the efficacy and safety of anti-CEA pretargeted radioimmunotherapy (pRAIT) in rapidly progressing metastatic MTC patients and also how serum biomarker DTs correlate with clinical outcome. Methods: From June 2004 to January 2008, 42 patients were treated with anti-CEA × anti–diethylenetriaminepentaacetic acid (DTPA) bispecific antibody (hMN-14 × m734) (40 mg/m2), followed by 131I-di-DTPA-indium bivalent hapten (1.8 GBq/m2) 4–6 d later. Results: The disease control rate (durable stabilization plus objective response) was 76.2%. Grade 3–4 hematologic toxicity was observed in 54.7% of patients and myelodysplastic syndrome in 2, including 1 heavily treated previously. After pRAIT, 21 of 37 assessed patients (56.7%) showed a significant impact on DT (≥100% increase of pre-pRAIT calcitonin or CEA DT or prolonged decrease of the biomarker concentration after pRAIT). Pre-pRAIT DT and post-pRAIT DT were significant independent predictors for overall survival (OS) from pRAIT (pre-pRAIT: hazard ratio [HR], 0.46; 95% confidence interval [CI], 0.24–0.86; P = 0.016; and post-pRAIT: HR, 5.32; 95% CI, 1.63–17.36; P = 0.006) and OS from diagnosis (pre-pRAIT: HR, 0.21; 95% CI, 0.08–0.51; P = 0.001; and post-pRAIT: HR, 6.16; 95% CI, 1.81–20.98; P = 0.004). Conclusion: pRAIT showed antitumor activity, with manageable hematologic toxicity in progressive MTC. Increased biomarker DT after treatment correlated with increased OS.
Medullary thyroid carcinoma (MTC) has a variable prognosis, ranging from long- to short-term survival, depending on age, TNM stage, mutations in the RET protooncogene, and pre- and postoperative calcitonin or carcinoembryonic antigen (CEA) serum levels (1,2). Calcitonin doubling time (DT) is an independent prognostic factor: patients with a calcitonin DT < 6 mo constitute a high-risk group, with only a 25% 5-y survival; patients with calcitonin DT ≥ 6 mo and < 2 y (intermediate risk) experienced a 92% 5-y survival (3). In metastatic disease, cytoreductive therapeutic options are limited. Recently, disease stabilization has been reported with targeted therapy of multikinase inhibitors (MKIs) in advanced or metastatic MTC, and vandetanib has been approved in the United States (4–8).
Radioimmunotherapy has shown promising results in MTC using anti-CEA monoclonal antibodies (mAbs) (9,10). Pretargeted radioimmunotherapy (pRAIT) was developed to improve the therapeutic index (tumor–to–normal-tissue ratios) and to deliver increased tumor-absorbed doses to solid tumors, which are more radioresistant than indolent lymphomas (11,12). pRAIT consists of first administering a bispecific monoclonal antibody (BsmAb), followed a few days later by a radiolabeled bivalent hapten (12). With this technology, the radioactive bivalent hapten binds avidly to the BsmAb attached to the surface of targeted CEA-expressing cancer cells, whereas the nontargeted radioactive hapten in the circulation clears rapidly through the kidneys. Pretargeting has demonstrated a more favorable therapeutic index and antitumor efficacy than directly labeled anti-CEA mAbs in preclinical MTC models (12,13). Moreover, 2 phase I clinical trials assessing anti-CEA × anti-diethylenetriaminepentaacetic acid (DTPA)-indium BsmAb (murine F6 × 734 and chimeric hMN14 x 734) with 131I-di-DTPA-indium showed encouraging therapeutic results in patients with progressive, metastatic MTC, with an improved overall survival (OS) in intermediate- and high-risk (pre-pRAIT calcitonin DT < 2 y) patients, as compared with contemporaneous untreated patients, in a retrospective analysis (median value, 110 vs. 61 mo; hazard ratio [HR], 0.42; 95% confidence interval [CI], 0.19–0.94; P = 0.030) (14–16).
On the basis of our previous experience, we designed a prospective phase II single-arm trial to determine the efficacy and safety of pRAIT using a new construct comprising a humanized anti-CEA × murine anti-DTPA (hMN-14 × 734) BsmAb and 131I-di-DTPA-indium bivalent hapten in patients with rapidly progressing, metastatic MTC. We also assessed the use of a biomarker DT as a surrogate marker to predict clinical outcome.
MATERIALS AND METHODS
Patient Eligibility and Demography
Eligible patients were ≥ 18 y old, with histologically confirmed stage IVc MTC as defined by the American Joint Committee on Cancer—that is, occurrence of distant metastases—and disease progression before study entry as defined by increasing calcitonin or CEA serum levels and calcitonin or CEA DT ≤ 5 y. Patients were stratified in risk groups according to minimal DT (lowest value between calcitonin and CEA DT) as follows: high-risk patients had a minimal DT lower than 6 mo, intermediate-risk patients had a DT between 6 mo and 2 y, and low-risk patients had a DT longer than 2 y. The other inclusion criteria were no tumor lesion > 10 cm; calcitonin ≥ 100 pg/mL; Karnofsky performance status ≥ 70%; bilirubin and creatinine < 1.5 times normal; neutrophils ≥ 1,500/mL; platelets ≥ 150,000/mL; and normal serum human antimouse antibody (HAMA) and human antihumanized antibody (HAHA) titers at ≥4 wk beyond surgery, radiotherapy, or chemotherapy and ≥3 mo after targeted therapy. Previous treatment included total thyroidectomy and bilateral lymph node dissection in all patients.
Forty-five patients met the inclusion criteria and were subsequently enrolled in this trial, sponsored by the University Hospital of Nantes and involving 5 French collaborating institutions. Three patients did not complete pRAIT (2 had reactions during BsmAb infusions and one a hapten labeling failure) and were excluded. Thus, 42 patients completed pRAIT and were evaluated. Enrollment started in June 2004 and ended in January 2008.
Table 1 lists patient characteristics. All patients had distant metastases to lymph nodes (83%), liver (52%), lungs (43%), and bone or bone marrow (60%). Previous treatments included surgery in all patients, radiation therapy in 12 (28.5%), and radionuclide therapy using 131I-metaiodobenzylguanidine or pRAIT in 6 (14.3%). During the study, 19 patients died. The median follow-up was 3.7 y (range, 0.2–6.5 y) from pRAIT and 10.9 y (range, 1.7–31.5 y) from MTC diagnosis.
Patient Characteristics (n = 42)
All patients gave informed written consent in accordance with institutional guidelines, including the Declaration of Helsinki. The trial was approved by the responsible ethics committee and registered at ClinicalTrial.gov (http://www.clinicaltrial.gov, NCT00467506).
Baseline Studies
All patients enrolled into this trial were required to undergo a complete history, physical examination, complete blood cell count with leukocyte differential, platelet count, and liver and kidney function tests. Additionally, baseline serum CEA and calcitonin serum titers were measured with at least 4 time points to reliably determine DT, as described previously (3). Single exponentials were fitted to serum biomarker concentrations by nonlinear least-square regression. Data were weighted by the inverse of the measured concentrations, and SEs were calculated as asymptotic SEs. Imaging studies included contrast-enhanced CT of the neck, chest, abdomen, and pelvis; bone marrow MRI; 18F-FDG PET; and bone marrow biopsy within 28 d of pRAIT. Maximum standardized uptake values (SUVmax) were calculated on PET scans for the tumor lesions, as described previously (17).
Treatment Plan
Humanized anti-CEA × murine anti-DTPA (hMN-14 × m734) BsmAb and di-DTPA bivalent hapten were provided by IBC Pharmaceuticals, Inc. Radiolabeling of the hapten with 131I and quality controls were performed using the IODO-GEN (Pierce) method, as described previously (14). Patients were infused with BsmAb (40 mg/m2), followed by 131I-di-DTPA (1.8 GBq/m2) 4–6 d later. For hapten administration, patients were hospitalized for 4–7 d in accordance with the local French requirements for radiation protection. Whole-body immunoscintigraphy and SPECT were recorded 4–7 d after the 131I-di-DTPA infusion; tumor targeting was graded visually as poor (grade 1), heterogeneous with variable intensity (grade 2), and good/very good (grade 3).
Follow-up
Patients were restaged at 3 mo after pRAIT through a complete physical examination, blood testing including CEA and calcitonin serum concentration (twice at 3- to 4-d intervals), CT scan, bone marrow MRI, and PET scan. Disease status was evaluated again at 6 mo and then every 3 mo for CEA and calcitonin (twice at 3- to 4-d intervals for each time point) and every 6 mo for CT scan, bone marrow MRI, and PET until 36 mo. SUVmax was calculated for the lesions.
Morphologic tumor response was assessed using the Response Evaluation Criteria in Solid Tumors (RECIST). Metabolic response was evaluated using guidelines of the European Organization for Research and Treatment of Cancer (18). Biomarker DT impact after pRAIT was assessed as described previously; a significant impact was arbitrarily determined as a ≥100% increase of pre-pRAIT calcitonin or CEA DT or as a prolonged decrease of the biomarker serum concentration after pRAIT (3,16).
Safety was assessed during infusions and posttreatment over 12 wk by vital signs, physical examination, adverse events, serum chemistries, hematology, HAMA, and HAHA. Because of the transitory myelosuppression generally observed after RAIT, a complete blood cell count with leukocyte differential and platelet count was done in all patients on a weekly basis, from the second week after pRAIT until complete hematologic recovery. Beyond 12 wk, safety follow-up (physical examination, hematology, serum chemistry) continued semiannually until 36 mo. Toxicity grade was determined according to National Cancer Institute Common Terminology Criteria, version 2.0.
Statistical Analysis
The primary endpoint was the assessment of tumor response rate according to RECIST. Secondary endpoints were toxicity, immunogenicity, PET metabolic response, biomarker response, progression-free survival (PFS) measured from time of pRAIT, OS from time of pRAIT, and OS from time of diagnosis.
The study was designed to provide a 97.5% power of detecting a disease control (objective response plus stabilization) rate of at least 25% after pRAIT at a significance level of α = 2.5%, when the response rate without treatment was less than 5%. We calculated that 44 eligible patients needed to be recruited, and a disease response or stabilization would need to be seen in at least 6 patients. The probability of 6 or more experiencing a response is 0.0216 if the response rate is 5%. The probability of 5 or fewer patients experiencing a response is 0.0215 if the response rate is 25%. Thus, if none of the first 11 patients enrolled had a response or stabilization, enrollment would be terminated because the probability that a response rate would be 25% was only 0.042.
Correlation studies between continuous parameters were conducted using a nonparametric Spearman test. Medians of continuous parameters were compared between groups using the nonparametric Mann–Whitney test (or nonparametric Kruskal–Wallis test if there were more than 2 groups). Relationships between qualitative parameters were determined using a Fisher exact test. PFS from pRAIT and OS from pRAIT and diagnosis were summarized using the Kaplan–Meier method. Survivals were compared using the log-rank test (or Breslow test if necessary). Finally, the Cox proportional hazards model determined independent prognostic effects on PFS and OS. Proportionality of hazards was verified. Statistical analyses were made using SAS (version 9.2; SAS Institute Inc.) and Stata 10.1 SE (StataCorp).
RESULTS
Treatment Response
Median pRAIT injected activity was 2.95 GBq (range, 2.32–4.04 GBq). Disease control according RECIST (objective response plus stabilization) was observed in 32 patients (76.2%), including a durable complete response of at least 40 mo in 1 patient (2.4%) and durable stable disease (≥6 mo) in 31 patients (73.8%). The prognostic values of all variables described in Table 1 for predicted RECIST response were determined; only tumor uptake assessed by immunoscintigraphy was a significant predictor, with 2 of 5 patients (40%) responding in grade 1, 12 of 17 (70%) in grade 2, and 17 of 17 (100%) in grade 3 targeting (P = 0.005, Fig. 1).

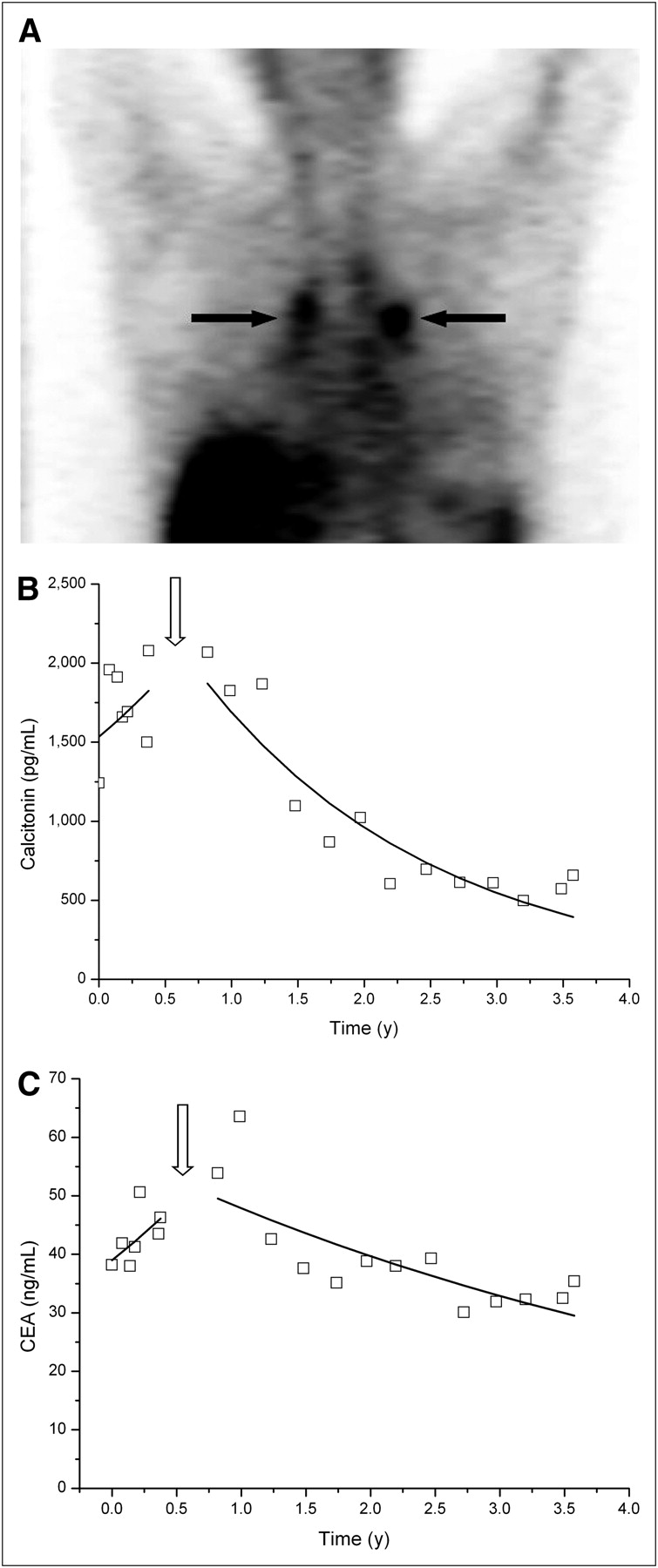
Patient with intermediate-risk MTC injected with hMN14 × 734 bispecific antibody (40 mg/m2) and 2.98 GBq of 131I-diDTPA-indium 5 d later. (A) Good tumor uptake (grade 3) on coronal SPECT slice recorded after 131I-diDTPA-indium injection (arrows). (B) Serum calcitonin kinetics. First part of curve (pre-pRAIT) shows biologic progression, with DT of 1.49 y. After pRAIT (arrow), impact was significant, with steady calcitonin serum level decrease corresponding to half-life of 1.22 y. (C) Serum CEA kinetics. First part of curve (pre-pRAIT) shows biologic progression, with DT of 1.56 y. After pRAIT (arrow), serum CEA decrease was observed, with half-life of 3.69 y.
Baseline PET SUVmax was 5.2 (range, 2.1–16.3). Metabolic PET response correlated well with RECIST response (P = 0.03), showing 1 patient with complete response (2.4%), 4 with partial responses (9.6%), and 24 with durable stable disease (57.1%). Thus, metabolic disease control was obtained in 29 patients (69.0%).
The variations of serum CEA and calcitonin concentration after pRAIT paralleled tumor size, but the correlation was not significant (P = 0.14): a decrease of greater than 50% of CEA or calcitonin was observed in 3 patients (7.1%; Fig. 1), and 29 patients (69.0%) showed durable stabilization (variation < 50%).
For the 39 patients included in the baseline biomarker DT analysis, the median baseline calcitonin and CEA DT were 1.25 y (range, 0.18–26.26 y) and 1.79 y (range, 0.42–23.76 y), respectively. calcitonin DT was positively correlated with CEA DT (Spearman R = 0.71; P < 0.0001). Minimal baseline DT was negatively correlated with age at diagnosis (Spearman R = −0.58, P < 0.001). Minimal DT also was lower in patients with an SUVmax ≥ 5 (0.61; range, 0.37–2.23) than in those with an SUVmax < 5 (1.69; range, 0.66–3.20, P = 0.0068). Baseline calcitonin and CEA DTs did not predict RECIST response (P = 0.509 and 0.836, respectively), and the efficacy did not differ between the 3 risk groups, with disease control in 6 of 8 (75%) high-risk patients, in 17 of 21 (81%) intermediate-risk patients, and in 8 of 10 (80%) low-risk patients (P = 1.00). After pRAIT, 21 of 37 assessed patients (56.7%) showed a significant impact on DT (≥100% increase of pre-pRAIT calcitonin or CEA DT), and DT impact tended to correlate with RECIST (P = 0.07).
Survival
Median PFS after pRAIT was 13.6 mo (range, 1.9–78.1 mo). In correlative studies of DT and PFS, a considerably longer PFS was shown in patients with a longer baseline DT (≥9 mo) than in those with a shorter DT (<9 mo; Table 2) and in patients with a significant impact on DT after pRAIT (≥100% increase) than in those with no significant impact (<100% increase, Table 3). The independent predictors for PFS were baseline DT with continuous values (HR, 0.61; 95% CI, 0.39–0.95; P = 0.031) and a ≥100% increase of DT after pRAIT (HR, 3.07; 95% CI, 1.32–7.14; P = 0.009), with the proportional hazards assumption not rejected (P = 0.54 and 0.20, respectively), using the multivariate Cox proportional hazards model.
Correlation of Serum Biomarker DT with Clinical Outcome
Grade 3 or 4 Hematologic Toxicity After Pretargeted Radioimmunotherapy
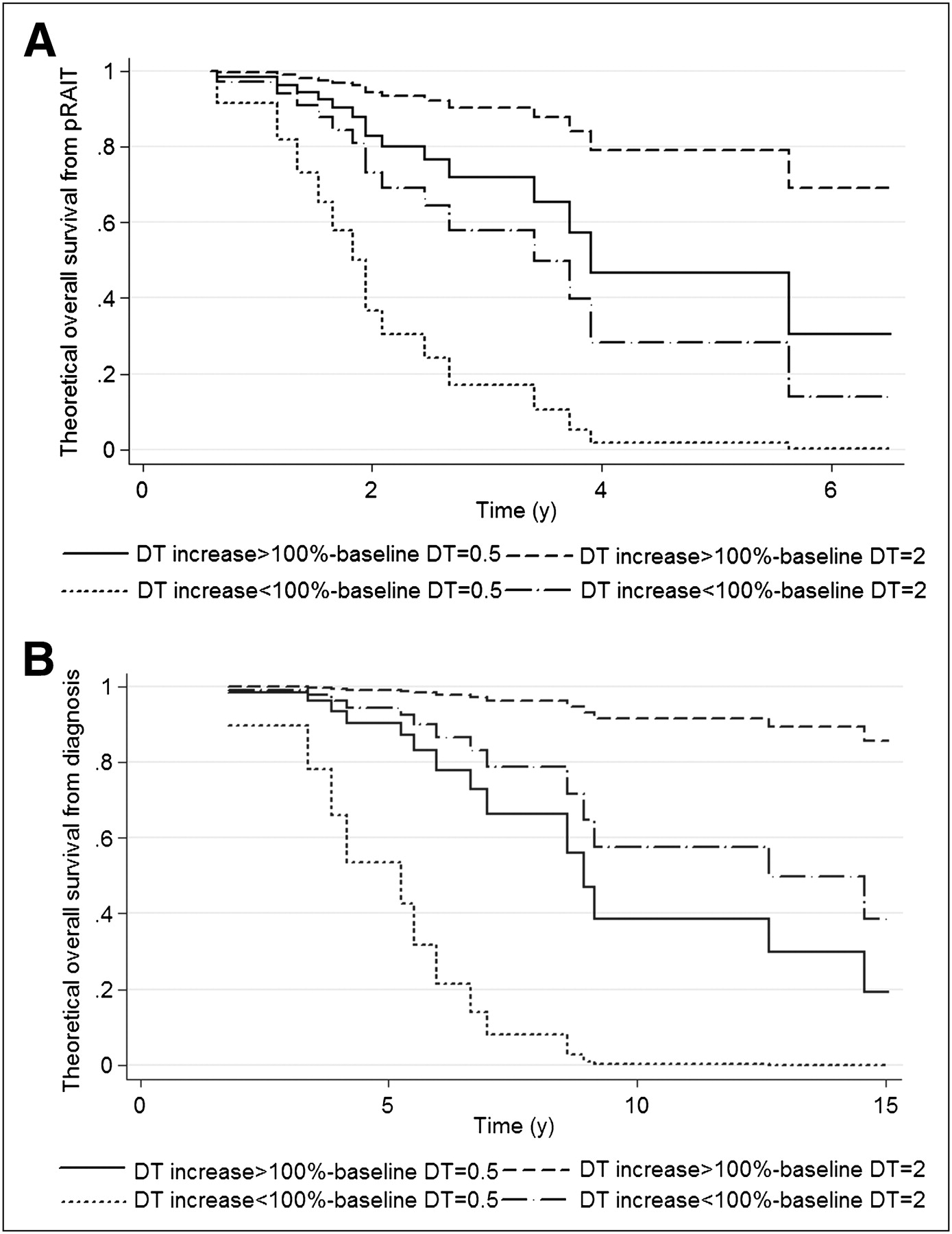
Median OS was 43.9 mo (range, 3.1–78.2 mo) after pRAIT and 136.4 mo (range, 21.2−234.4 mo) after diagnosis. In correlative studies of DT and OS from pRAIT, a considerably longer OS was shown in patients with a longer baseline DT (≥9 mo) than in those with a shorter DT (<9 mo, Table 3) and in patients with a significant impact on DT after pRAIT than in those with no significant impact (Table 3). Patients with a baseline SUVmax ≥ 5 had a significantly shorter OS than did patients with a lower SUVmax (5-y OS, 76.2% vs. 17.2%, P = 0.038; Fig. 2B). The independent predictors for OS from pRAIT were baseline DT using continuous values (HR, 0.46; 95% CI, 0.24–0.86; P = 0.016) and a ≥100% increase of DT after pRAIT (HR, 5.32; 95% CI, 1.63–17.36; P = 0.006), with the proportional hazards assumption not rejected (P = 0.06 and 0.50, respectively; Fig. 3), using the multivariate Cox proportional hazards model. SUVmax obtained in only 23 patients was not included in the multivariate analysis.

(A) OS after pRAIT according to minimal baseline biomarker DT. (B) OS after pRAIT according to pre-pRAIT SUVmax. (C) Theoretic survival from pRAIT according to minimal baseline biomarker DT. (D) OS after pRAIT according to post-pRAIT DT impact.
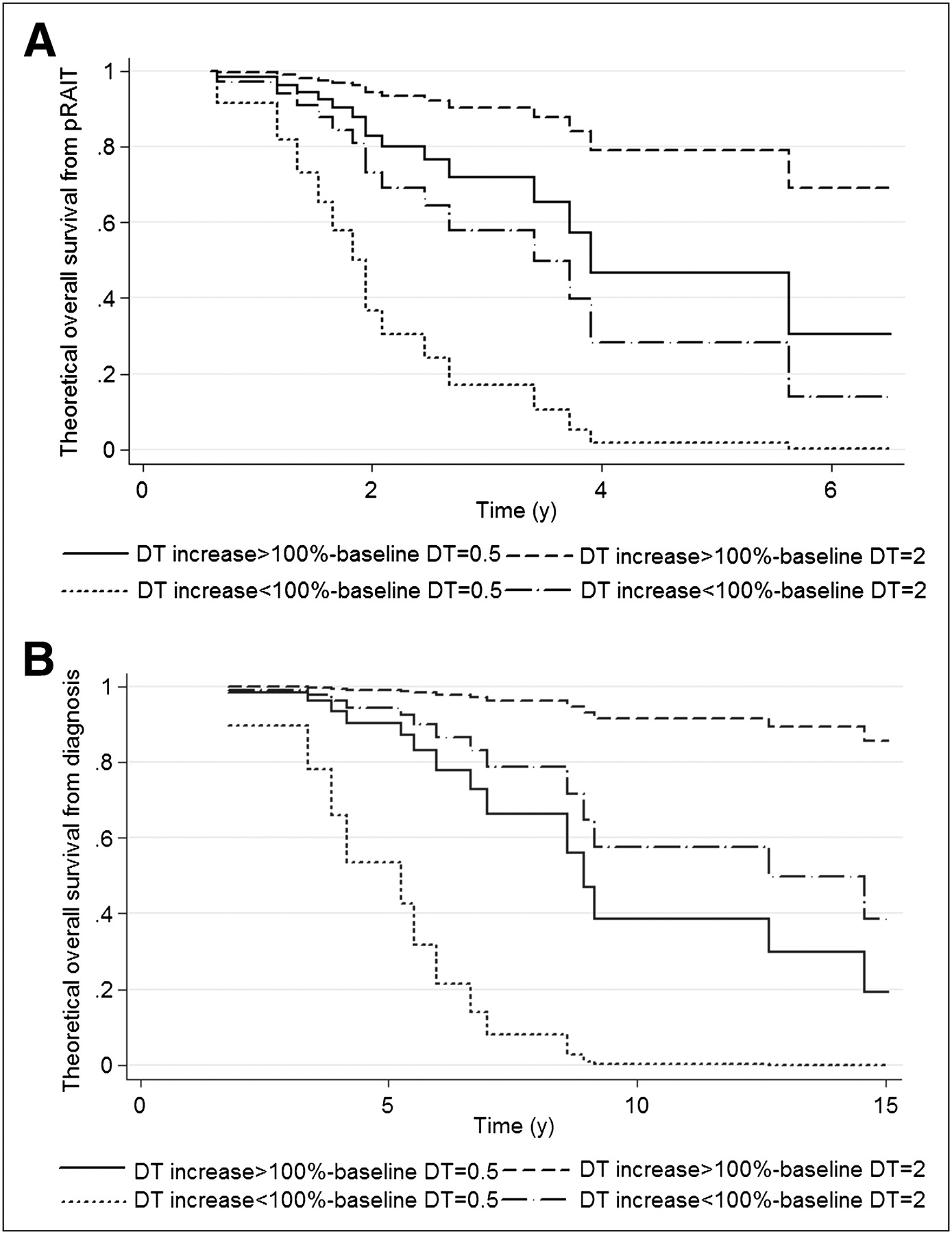
(A) Theoretic OS from pRAIT according to baseline biomarker DT and post-pRAIT DT impact. (B) Theoretic OS from diagnosis according to baseline biomarker DT and post-pRAIT DT impact.
An independent prognostic value for OS from diagnosis was also demonstrated for baseline DT using continuous values (HR, 0.21; 95% CI, 0.08–0.51; P = 0.001) and a ≥100% increase of DT after pRAIT (HR, 6.16; 95% CI, 1.81–20.98; P = 0.004), with the proportional hazards assumption not rejected (P = 0.54 and 0.41, respectively; Fig. 3).
Safety
The infusions of BsmAb and hapten were generally well tolerated, except in 2 patients, for whom the BsmAb infusions were stopped because of grade 3–4 immune response in one and grade 3 hypotension in the other; both recovered rapidly after adequate care. Six grade 1–2 (14.3%) and 1 grade 3 (2.4%) hepatic toxicities, as shown by aspartate or alanine aminotransferase increases, were reported 15–45 d after pRAIT (median, 28 d); the median duration was 76.5 d (range, 15–207 d). No other nonhematologic toxicity was observed. Grade 3–4 hematologic toxicity occurred in 23 patients (54.7%), 12 with known bone marrow involvement (Table 3). After adequate treatment (platelet transfusions, granulocyte colony-stimulating growth factor), all patients recovered from their grade 3–4 toxicities. Two patients with diffuse bone marrow involvement developed myelodysplastic syndrome 2 and 3.5 y after pRAIT. One patient previously received 2 courses of RAIT and high-dose mediastinal external-beam radiotherapy; the other did not receive any treatment before pRAIT. Antiantibody monitoring in 35 patients showed an increase of HAMA in 1 (2.3%) and HAHA in 11 (26.2%).
DISCUSSION
This prospective phase II pRAIT trial confirmed the efficacy of anti-CEA pRAIT, with disease control in 76% of patients and a median PFS and OS of 13.6 and 43.9 mo, respectively. The disease control rate met the predefined endpoint of a disease control rate of at least 25%. MTC is a malignancy with a variable prognosis, and we showed, in a previous retrospective study, the independent prognostic value of biomarker DT, with a long life expectancy in patients with a calcitonin DT over 5 y (3,16). Therefore, in the present study, only patients with a DT less than 5 y were included and stratified into 3 risk groups according to minimal DT value (lowest value between calcitonin and CEA DTs). pRAIT efficacy did not depend on baseline DTs, and high disease control rates were obtained in the 3 risk groups, especially in patients with a DT shorter than 2 y and, even more, when DT was less than 6 mo, demonstrating that pRAIT should be considered as a new systemic therapy option in rapidly progressing metastatic MTC.
Subacute toxicity was mainly hematologic and related to bone marrow involvement, requiring careful post-RAIT blood monitoring. Myelodysplastic syndrome is a well-known serious adverse event after different forms of radionuclide therapy, such as 1% after ibritumomab tiuxetan RAIT or 177Lu-octreotate treatment (19,20). In the present series, the 2 myelodysplastic syndromes were observed in elderly men with diffuse bone marrow involvement, leading to a relatively high and protracted bone marrow irradiation after pRAIT. Moreover, 1 patient was heavily pretreated. Cytogenetic testing before RAIT may identify existing chromosomal abnormalities in previously treated patients who would be at higher risk for myelodysplastic syndrome.
Despite the use of chimeric BsmAb (human/mouse) in the present study, the immunization rate appeared high (26% HAHA/HAMA detection). However, new-generation pRAIT compounds are now available with humanized, recombinant, trivalent BsmAb (anti-CEA TF2) and histamine-succinyl-glutamine (HSG) peptides (21). The use of TF2, composed of a humanized anti-HSG Fab fragment derived from the 679 anti-HSG mAb and 2 humanized anti-CEA Fab fragments derived from the hMN-14 mAb (labetuzumab; Immunomedics, Inc.) by the dock-and-lock procedure, should reduce immunogenicity and facilitate repeated injections (22). Moreover, the HSG peptide allows facile and stable labeling with different radiometals, such as 177Lu or 90Y, having favorable physical features that could improve pRAIT efficacy (23).
Several targeted therapies have been assessed recently in MTC (4–10,24,25). Tumor responses have been reported with directly radiolabeled anti-CEA mAb, but the comparison with pRAIT remains difficult because the eligibility criteria were different (9,10,14). Moreover, this population with frequent bone marrow involvement does not allow demonstration of improved of safety with pRAIT, because the hematologic toxicity is due to the specific uptake of mAb by tumor cells in bone marrow. However, according to published clinical data, tumor doses and tumor–to–normal-tissue ratios seem to be higher with pretargeting than with directly radiolabeled anti-CEA mAb (9,10,14). 90Y-DOTATOC induced in 31 progressive metastatic MTC patients a biomarker response in 58% (24). However, only 60%–70% of MTC tumors express somatostatin receptors, whereas >90% of MTCs express CEA. As expected with a radiolabeled peptide, long-term renal toxicity occurred in a small number of patients. MKIs also have been evaluated, showing significant toxicity, including fatigue, nausea, vomiting, diarrhea, rash, hand–foot–skin reaction, or hypertension (4–8). Disease control after pRAIT compares favorably with that reported with MKIs, even if inclusion criteria before pRAIT were more severe, including confirmation of progression that was not required in trials with MKIs. In the phase II single-arm studies, motesanib, which targets all known vascular endothelial growth factor, platelet-derived growth factor, Kit, and RET receptors, induced 51% disease control (partial response plus durable stable disease) and vandetanib, which targets RET, vascular endothelial growth factor receptor, and epidermal growth factor receptor tyrosine kinases, showed a 73% disease control with decrease of calcitonin serum level of at least 50% in 80% of patients. However, no correlation was found between tumor shrinkage and calcitonin reduction. RET kinase can inhibit calcitonin gene transcription and protein secretion in a manner dissociated from inhibition of tumor growth (26). Vandetanib was just approved by the Food and Drug Administration for the treatment of adults with advanced MTC, with the phase III trial having shown an improved PFS (a median of 19.3 mo in the placebo arm and not reached in the vandetanib group but estimated at 30.5 mo) (7). Despite the impressive response rate and prolongation in PFS, this phase III study did not demonstrate that patients treated with vandetanib have a longer OS (25). The median PFS of the pRAIT series was lower (13.6 mo), reflecting probably the higher disease aggressiveness of included patients. By contrast to the phase III study assessing vandetanib, only patients with demonstrated disease progression with a DT lower than 5 y were enrolled in the pRAIT study.
The identification of predictive markers of response and survival appears to be important to selecting patients most likely to benefit from systemic therapy and to avoiding possible treatment-related adverse events and associated costs in patients who are unlikely to respond or who have a long life expectancy.
In the present study, only tumor uptake on immunoscintigraphy was a predictor of response. Similarly, tumor uptake in somatostatin receptor scintigraphy predicted morphologic response after radiolabeled somatostatin analog therapy (19). The sensitivity of immunoscintigraphy to detect small MTC lesions, including bone marrow and liver metastases, has been shown previously (17,27). These results support the potential of pretargeting with short-half-life PET emitters, such as 18F or 68Ga, as a highly sensitive and specific immuno-PET method in MTC or other CEA-expressing tumors (28–31). No predictive value was determined for RET status, bone marrow involvement, and presence of nonparenchymal metastases, whereas RET status is involved in the cytotoxic effects of MKIs. Nonparenchymal target lesions could be less well perfused than parenchymal metastases, with a possible lower efficacy of systemic targeted therapy (32).
The present study again confirmed the value of serum biomarkers in selecting patients and monitoring therapy (3,16). Reliable DT determinations require repeated serum sampling. As reported in a retrospective study, baseline biomarker DT had a predictive value on OS (3). With 18F-FDG PET, SUVmax also had a prognostic value. A correlation between SUVmax and DT was established in a previous smaller patient series (17), and the current results can now reinforce the idea of using DT to select patients for systemic therapy: in low-risk patients with DTs longer than 2 y, watchful waiting may be the most appropriate strategy, whereas systemic therapies, especially pRAIT, should be proposed in high-risk patients with a calcitonin DT shorter than 2 y.
Additionally, there was a significant increase of DT (≥100%) in 56.7% of patients after pRAIT, related to clinical outcome, with an independent predictive value on PFS and OS. In advanced solid tumors, an objective response to treatment is the standard endpoint of trials aimed at assessing the anticancer activity of new drugs. However, its role as a surrogate marker of a beneficial effect of treatment has been questioned, both in clinical trials and in the individual patient (33). The most appropriate method to assess treatment response in MTC remains controversial. Morphologic evaluation using CT or MRI represents a standard in solid tumors, with obvious limits in the detection of disseminated microscopic disease, especially in bone or bone marrow. PET/CT is clearly useful for the early review and evaluation of cytostatic targeted therapy (therapy that is not correctly assessed on the basis of tumor size reduction). Our results suggest that biomarker DT impact after therapy could be considered as a relevant surrogate marker of survival in progressive MTC treated by pRAIT. Validation of response to a given treatment as a surrogate of survival in a specific cancer condition requires ideally the availability of large datasets of patients randomly assigned to the tested treatment or to a control (34). However, in rare diseases such as MTC, randomized trials are difficult to implement because of the need to include a sufficient number of patients over an acceptable time period.
Just like any other therapeutic agent, RAIT is not likely to cure metastatic cancer as a single shot of a single agent. In preclinical studies in an MTC animal model, a synergistic effect was demonstrated using the association of RAIT with paclitaxel and improvement of tumor response using a combination of RAIT with antiangiogenic agents such as thalidomide (35,36). Thus, multimodality therapy could be a step further in the development of pRAIT.
CONCLUSION
This prospective phase II study confirmed the efficacy of anti-CEA pRAIT in progressive, metastatic MTC. Disease control compared favorably with what is achieved by the limited number of alternative treatment modalities, which have not always been assessed in advanced, progressive disease. This study confirmed the possibility of serious hematologic toxicity of pRAIT, especially in patients with diffuse bone marrow involvement. Hence, pRAIT should be limited to the poor prognostic risk group of MTC patients who can be identified reliably by serum biomarker DT. A significant prolongation of DT after treatment correlated with clinical outcome, supporting the use of biomarkers to predict the response of advanced MTC to systemic therapy.
DISCLOSURE STATEMENT
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This study was supported by the French PHRC 2002 and was presented in part at the annual meeting of the Society of Nuclear Medicine, San Antonio, Texas, June 4–8, 2011. David M. Goldenberg has a leadership position and Chien-Hsing Chang is an employee of Immunomedics and IBC Pharmaceuticals, Inc. (Morris Plains, NJ). No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Jun. 28, 2012
- © 2012 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication December 15, 2011.
- Accepted for publication March 5, 2012.





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Current Landscape in Clinical Pretargeted Radioimmunoimaging and Therapy
- Anti-CEA Pretargeted Immuno-PET Shows Higher Sensitivity Than DOPA PET/CT in Detecting Relapsing Metastatic Medullary Thyroid Carcinoma: Post Hoc Analysis of the iPET-MTC Study
- Radioimmunotherapy: From Current Clinical Success to Future Industrial Breakthrough?
- Phase II Study of Lutetium-177-Labeled Anti-Prostate-Specific Membrane Antigen Monoclonal Antibody J591 for Metastatic Castration-Resistant Prostate Cancer
- Early changes in carcinoembryonic antigen but not in calcitonin levels are correlated with the progression-free survival in medullary thyroid carcinoma patients treated with cytotoxic chemotherapy