


Abstract
Radiolabeled peptides targeted against receptors on the cell surface have been shown to be remarkably specific and effective in the diagnosis and therapy of malignant disease. Much of the early work in this field took place outside the United States, but in recent years the research effort within the United States has accelerated. Most of the initial studies in the United States focused on somatostatin receptor ligands. 111In-pentetreotide was approved in 1994 and has been used extensively in the diagnosis and management of a wide variety of neuroendocrine tumors, particularly carcinoid. This work was extended to 99mTc-labeled analogs, and the most successful, 99mTc-depreotide, was approved in 1999. This agent was found to be accurate in the diagnosis of lung cancer, but it was not particularly successful because it was supplanted by 18F-FDG imaging with positron tomography. More recently, studies with 68Ga-labeled somatostatin analogs were initiated in the United States. This effort was delayed relative to that in other parts of the world because of difficulty in obtaining the necessary generators and regulatory uncertainty, both of which are less of a problem currently. Several ligands are being developed to image melanoma through targeting of the melanocyte-stimulating hormone receptor. Other ligands are being developed to use the arginine-glycine-aspartate oligopeptide to target angiogenesis and to use bombesin analogs to target the gastrin-releasing peptide receptor for the diagnosis and potential therapy of prostate cancer. Peptide dimers that target 2 receptors simultaneously are also being constructed, potentially increasing the selectivity of the approach significantly. Radiopeptide therapy has been explored with these ligands, initially with high-dose 111In-pentetreotide. This step has been followed by U.S. participation in several trials with 90Y-, 177Lu-, and 188Re-labeled analogs. Some of these agents are now available clinically outside the United States, and it is important to design and conduct the appropriate trials so that this therapy can be offered within the United States.
Radioactively labeled peptides have been recognized as an effective way to target cellular receptors in vivo for diagnostic identification and characterization and for targeted radiotherapy. Several peptides targeting a wide variety of receptors have been identified. Most of these have been relevant to tumors, but there are several potential applications of radiopeptides in nononcologic disease.
The 2 major advantages of peptides for receptor targeting are that they can be designed to essentially duplicate the critical region of the natural receptor ligand, thus achieving high specificity, and that they are relatively small, especially compared with antibodies, and therefore localize very rapidly. These advantages are significant for diagnostic imaging, leading to high sensitivity and specificity and to effective therapeutic agents that can achieve high tumor radiation doses.
The research and development of radiopeptides for diagnosis and therapy have been global efforts, with much of the early work taking place outside the United States. However, in recent years the research effort within the United States has accelerated. This article reviews that effort, both for diagnostic agents and for the use of radiopeptides in therapy. The agents discussed are summarized in Table 1.
Summary of Radiolabeled Peptides
IMAGING WITH SOMATOSTATIN RECEPTOR LIGANDS
The most widely used radiopeptide in the United States is the 111In-labeled somatostatin analog pentetreotide (OctreoScan; Covidien), which was approved by the U.S. Food and Drug Administration (FDA) in 1994 as an agent for the scintigraphic localization of primary and metastatic neuroendocrine tumors bearing somatostatin receptors. 111In-pentetreotide (also known as 111In-octreotide) has the highest affinity for receptor subtype 2, lower affinities for subtypes 3 and 5, and the lowest affinity for subtypes 1 and 4 (1).
Somatostatin in humans occurs as both 28-amino-acid and 14-amino-acid proteins. Octreotide is a diethylenetriaminepentaacetic acid conjugate of 8 of the amino acids from somatostatin that specifically target somatostatin receptors. The 8-peptide sequence of amino acids in octreotide is d-phenylalanine–l-cysteine–l-phenylalanine–d-tryptophan–l-lysine–l-threonine–l-cysteine–l-threonine.
The tumor types studied with octreotide include carcinoid, growth hormone–secreting pituitary tumors, endocrine pancreatic tumors, paraganglioma, medullary thyroid carcinoma, pheochromocytoma, small cell lung carcinoma, neuroblastoma, meningioma, and Merkel cell tumors. Octreotide has been used most successfully for carcinoid, endocrine pancreatic tumors, and neuroblastoma.
111IN-LABELED SOMATOSTATIN RECEPTOR IMAGING
The first published report on the use of 111In-octreotide imaging in humans in the United States was from a group of investigators at the Mayo Clinic in 1995 (2). They studied a single patient with cholangiocarcinoma and demonstrated uptake in the tumor.
At the University of Ohio, Martinez et al. conducted the first study of a series of patients with radiolabeled octreotide in the United States (3). They studied 6 children with neuroblastoma by using 125I-Tyr3-octreotide and intraoperative exploration with a γ-probe. Seventeen sites of radiotracer binding were identified, for a sensitivity of 100% and a specificity of 71%.
The first U.S. report on the use of 111In-octreotide in carcinoid imaging was from investigators at Vanderbilt University (4). They studied 69 patients with neuroendocrine tumors (among them 60 carcinoids) and showed that 111In-octreotide identified additional sites of metastasis in 12.
The first large clinical trial with 111In-octreotide in the United States was conducted at the National Institutes of Health and included 80 consecutive patients with Zollinger–Ellison syndrome (5). This trial showed that somatostatin receptor scintigraphy was markedly more sensitive than ultrasonography, CT, MRI, or angiography, detecting extrahepatic gastrinomas or liver metastases in 70% of the patients.
111In-octreotide imaging of paraganglioma in the neck has been a particularly successful application of the technique. In the United States, this application was recognized as being important because of a study done at the University of Miami and published in 2004 (6). Sixty patients with suspected neuroendocrine tumors of the neck were studied. 111In-octreotide scintigraphy had a sensitivity of 97% and a specificity of 82%.
Since the 1990s, numerous studies on the clinical utility of 111In-octreotide at sites in the United States have continued to be published. Notable recent articles described the added value of CT when combined with SPECT—now available in modern SPECT/CT systems (7)—and the observations that 18F-FDG uptake was higher in more poorly differentiated neuroendocrine tumors whereas 111In-octreotide uptake was higher in better-differentiated tumors (8). This point is important because tumors with high 18F-FDG uptake have a significantly worse prognosis that those with low uptake (9).
99MTC-LABELED SOMATOSTATIN RECEPTOR IMAGING
In the mid-1990s, several radiochemistry studies examining the feasibility of labeling octreotide and other peptides with 99mTc were reported by U.S. investigators (10–12). All of these reports, including both in vitro and in vivo studies, showed convincingly that 99mTc-labeled peptides were stable and that they bound to somatostatin receptors with a high affinity. The ratios of target uptake to nontarget uptake were generally better than those obtained with 111In-octreotide, and the tumor-to-blood ratios were lower.
A 99mTc-labeled octreotide analog developed as 99mTc-depreotide (NeoTect [Diatide Inc.]; NeoSpect [Nycomed]) was approved by the FDA late in 1999 for the characterization of pulmonary nodules. The first report of a clinical trial of 99mTc-depreotide in the United States was from Blum et al. in 1999 (13). The agent was designated P829 at that time. They studied 30 subjects who had indeterminate pulmonary nodules with an average size of 2.4 cm (SD, 0.9 cm). The sensitivity and specificity of 99mTc-depreotide for the diagnosis of malignancy in this pilot study were 93% and 88%, respectively. A larger trial was done at the University of Iowa with 166 subjects who had suspected lung cancer (14). The performance of 99mTc-depreotide for the diagnosis of malignancy in pulmonary nodules or masses and the detection of mediastinal nodal metastasis was compared with that of 18F-FDG. The median size of the lung lesions was 2.2 cm, with a range of 0.5–10.5 cm. The sensitivity and specificity of 99mTc-depreotide for the diagnosis of malignancy in pulmonary lesions were 94% and 51%, respectively; those of 18F-FDG PET were 96% and 71%, respectively. For the detection of mediastinal nodal metastasis, the respective sensitivity and specificity were 68% and 70% for 99mTc-depreotide and 81% and 77% for 18F-FDG PET (14).
In a small study of patients with lymphomas, good uptake of 99mTc-depreotide was observed in 7 of 9 patients. This finding led to the suggestion that β-emitting radiopeptides might be used for treatment in patients with lymphomas showing high uptake of the somatostatin receptor ligand (15).
99mTc-depreotide has not been widely used in the United States in recent years and is currently not available for clinical use, probably because it emerged at the same time as 18F-FDG PET, which grew rapidly and was apparently more accurate in characterizing lung cancer. Because of the limited approved indications, 99mTc-depreotide was never carefully studied for general neuroendocrine tumor imaging. There have been no recent reports of human imaging studies with 99mTc-labeled octreotide analogs from groups within the United States, although review of the literature reveals that these agents are being used in China for pheochromocytoma (16) and in Uruguay for neuroblastoma (17).
On October 28, 2010, the European Commission issued a decision to withdraw the marketing authorization for 99mTc-depreotide. This decision was apparently made in accordance with the manufacturer's request, which was based on commercial reasons.
68GA-LABELED SOMATOSTATIN RECEPTOR IMAGING
68Ga (half-life, 68 min) is a positron emitter and is obtained from a 68Ge (half-life, 271 d) generator. Several octreotide analogs have been labeled with 68Ga and used in research and clinical applications outside the United States. Although all of the early work and clinical trials with 68Ga-labeled somatostatin receptor agents took place outside the United States, effort in this area in the United States has not been entirely absent and is currently gathering momentum. Various reasons account for the delay of more than 10 y in implementing human studies in the United States. These include the availability of 68Ga generators and the regulatory requirements for first use in humans, uncertainty about the intellectual property ownership of various octreotide analogs and associated chelators, and general uncertainty about the regulations for manufacturing and using new radiopharmaceuticals, as well as recognition that the regulations are likely to change because the FDA was committed to updating the regulations under the Food and Drug Administration Modernization Act of 1997.
Radiopharmaceutical manufacturers have also delayed working toward approval of these agents for somewhat similar reasons, including uncertainty about intellectual property issues and the likelihood that regulations will change. In addition, economic analysis has suggested that, because of the relatively small numbers of patients with neuroendocrine tumors, the potential market may not be sufficient to justify all of the development expenses.
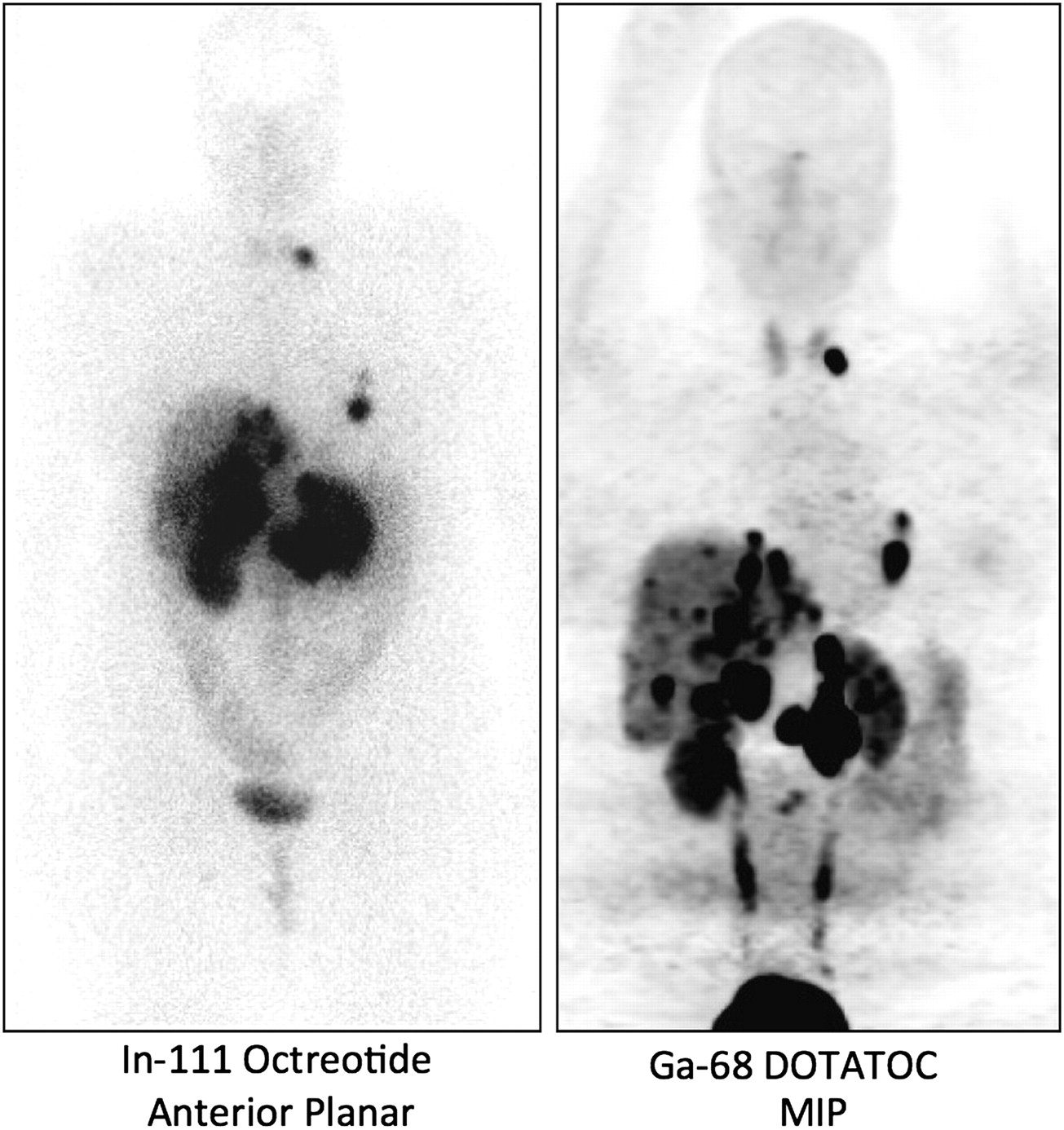
The current situation has definitely improved. The FDA has announced a new set of regulations for manufacturing PET radiopharmaceuticals, which will take effect in late 2011. 68Ga generators are now available from one company, and it is likely that other suppliers will enter the market in the United States in the near future. At least 2 U.S. academic sites are beginning to do human studies—Vanderbilt University under an Investigational New Drug authorization and the University of Iowa under Radioactive Drug Research Committee approval. Figure 1 shows an example from the group at the University of Iowa that illustrates the higher quality of 68Ga-DOTATOC images than of 111In-octreotide images. It is likely that the number of sites in the United States conducting human studies will grow significantly in the next few years and that these agents will become important tools in the diagnosis of and follow-up for neuroendocrine tumors.
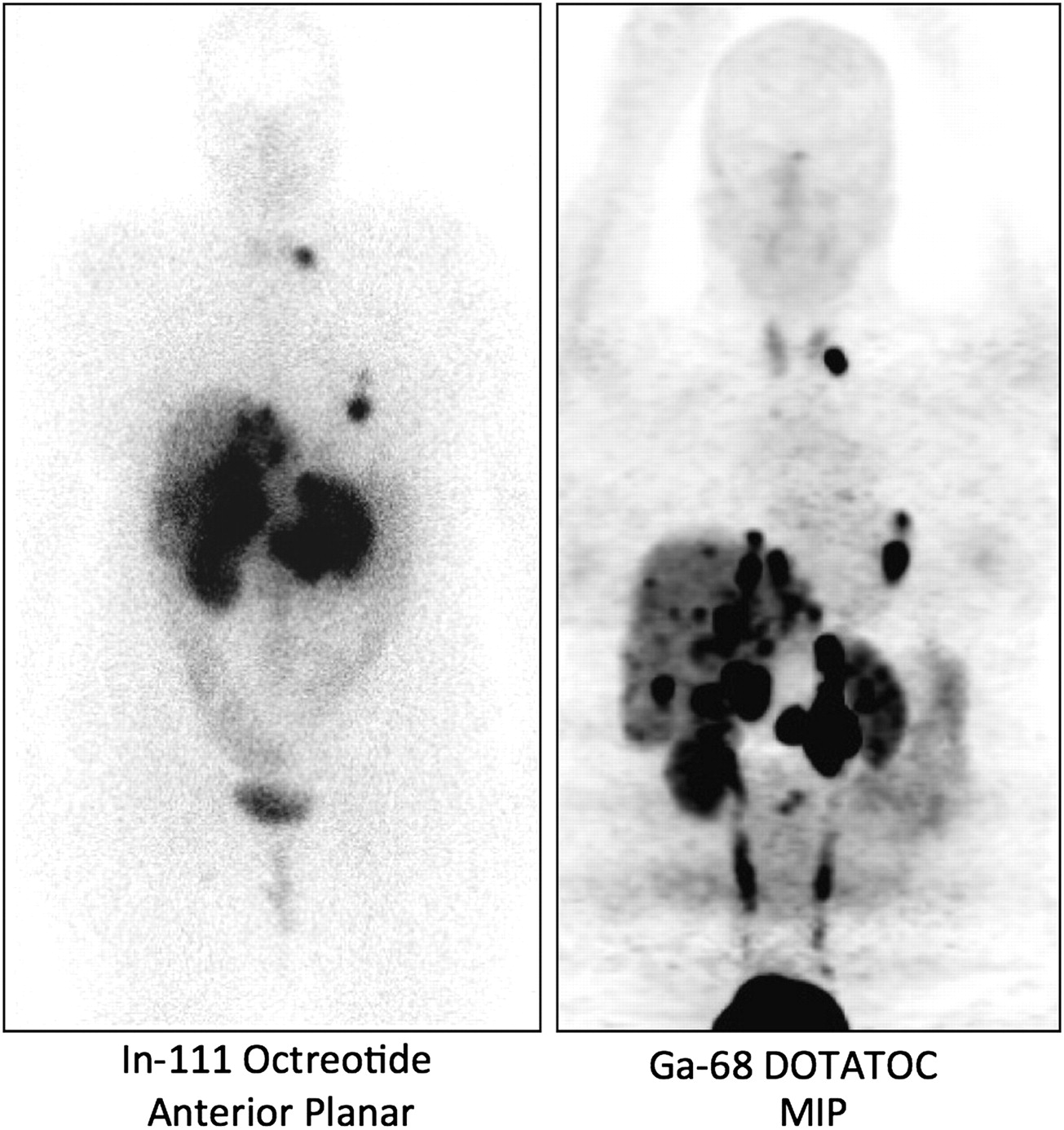
Comparison of 111In-octreotide image obtained at 24 h after injection with 68Ga-DOTATOC image obtained at 1 h after injection. Subject was 35-y-old man with metastatic gastrinoma. MIP = maximum-intensity projection.
Human 68Ga studies were done in the United States as early as 1978 (18), but all of the early studies were done with 68Ga-ethylenediaminetetraacetic acid as a measure of vascular permeability in the brain. In the 1980s, 68Ga-labeled transferrin was used to study pulmonary blood flow and vascular permeability (19,20). At present there are apparently no reports from groups within the United States on human studies with 68Ga-labeled somatostatin receptor ligands. However, several preclinical studies have been reported.
The first published report on gallium-labeled somatostatin receptor ligands in the United States was from the Memorial Sloan–Kettering Cancer Center in 2002 (21). The investigators showed successful labeling of DOTATOC (DOTA-d-Phe1-Tyr3-octreotide) with 66Ga, 67Ga, and 68Ga and rapid uptake in a rat pancreas tumor implanted into nude mice.
Although several reports from groups in the United States described studies involving 68Ga-labeled peptides, the only other report on 68Ga-labeled somatostatin receptor ligands appears to be from researchers at the Memorial Sloan–Kettering Cancer Center in 2011 (22). They reported on the use of 68Ga-DOTATOC for the imaging of a reporter gene that results in the expression of somatostatin receptor subtype 2 in transfected cells.
PEPTIDE IMAGING OF MELANOMA
The first published study of peptide-based imaging of melanoma was from a group of investigators at Oregon Health Sciences University (23). Uptake of 111In-octreotide was shown in 12 of 16 patients with metastatic melanoma. Although their study clearly showed that such imaging is feasible and that melanoma often expresses somatostatin receptors, this line of research has not been significantly pursued. Rather, a more direct approach with ligands targeted to the melanocyte-stimulating hormone (MSH) receptor (also called melanocortin) is currently being pursued at several universities in the United States.
A group of investigators at Washington University in St. Louis, Missouri, reported tumor uptake in mice as well as the biodistribution of a 68Ga-labeled DOTA-rhenium–cyclized α-MSH analog (24). This agent showed promise for the early detection of malignant melanoma. A novel α-MSH peptide analog labeled with 111In, 86Y, and 68Ga also showed promise as a possible melanoma imaging agent in studies by the same group (25).
The radiochemistry group at the University of Missouri also investigated an analog for MSH (26). Their work involved a high-specific-activity 68Ga-labeled DOTA-rhenium–cyclized α-MSH peptide analog. The initial studies, with small-animal imaging, showed a high ratio of tumor uptake to nontumor uptake at 1 h after injection.
Researchers at Stanford University studied several approaches for imaging melanoma. Recently, they reported an animal study with 18F-labeled metallopeptides targeted to the MSH receptor (27). Good uptake was observed in tumor-bearing mice, suggesting that the agent may be useful clinically.
PEPTIDE IMAGING OF ANGIOGENESIS
Another oligopeptide that may become an important and useful clinical tool is the arginine-glycine-aspartate (RGD) sequence. Numerous studies have shown that this small peptide sequence binds avidly to the endothelium in regions of neoangiogenesis. The agent is likely to be useful in oncology—for visualizing where tumors are generating new blood vessels—but also in nonmalignant disease—when there is either too little (infarct repair) or too much (macular degeneration) angiogenesis.
The initial studies in the United States, investigating the feasibility of labeling the RGD sequence with 18F, 64Cu, and 125I, were done at the University of Southern California (28). Much of the more recently published work was done at Stanford University (29). In addition, investigations were also carried out at the University of Texas M.D. Anderson Cancer Center (30), Washington University (31), University of California—Davis (32), University of California—Los Angeles (33), and Purdue University (34). Most of these studies showed good tumor uptake with small-animal imaging.
TUMOR IMAGING WITH PEPTIDE DIMERS
In many recent studies, the RGD peptide was combined with a second peptide with a different target to increase the specificity of tumor binding.
A group of researchers at Stanford University investigated cyclic RGD dimers in combination with bombesin, which targets the gastrin-releasing peptide receptor. They recently labeled the RGD–bombesin heterodimer with 18F, 64Cu, and 68Ga (35). The 3 agents were studied both in vitro and in vivo. The in vivo studies were done with breast cancer xenografts in nude mice. The 68Ga-labeled agent showed somewhat higher tumor uptake, but the ratio of tumor uptake to nontumor uptake was better with the 18F-labeled agent. The results are definitely encouraging, but clinical trials are required to determine the optimum approach.
A group at Purdue University is investigating RGD dimers labeled with 99mTc, 64Cu, and 111In (34). In both in vitro and in vivo animal studies, the dimers showed more specific uptake than the monomers. The distance between the 2 RGD elements in the dimers is a critical parameter that may be optimized. This aspect is a particular focus of all of the groups working with dimers.
BOMBESIN
Bombesin is a 14-amino-acid peptide that binds with a high affinity to the receptors for gastrin-releasing peptide and neuromedin B and is overexpressed on a variety of solid tumors. Various bombesin analogs have been studied, with much of the earliest work taking place within the United States. In 1997, a group at the University of Alabama described the process for labeling a 7-amino-acid analog of bombesin with 188Re and reported binding to prostate cancer cells (36).
In 1998, a group at Johns Hopkins University described the synthesis of a bombesin analog conjugated to 99mTc (37). That group continued to develop 99mTc-labeled bom-besin and, more recently, described in vitro binding to prostate cancer cells along with in vivo imaging in SCID mice with PC-3 prostate cancer xenografts (38). Other work with 99mTc-labeled bombesin analogs is under way at Missouri University. Researchers there developed a new method for conjugating 99mTc to bombesin analogs and reported good localization in breast cancer xenografts in mice (39).
Several other approaches for labeling bombesin have also been explored. Parry et al., at Washington University, evaluated a 64Cu-labeled DOTA linker for bombesin in both in vitro and in vivo animal studies of prostate cancer and reported excellent uptake and retention (40). The work with 64Cu-labeled bombesin analogs is reviewed in detail in an article from the group at the University of Missouri (41).
Radiolabeled bombesin analogs are also being studied as possible therapeutic agents. Initial studies with a 177Lu-labeled agent in prostate cancer cell lines and in in vivo studies of mice with prostate cancer xenografts showed good uptake and response to therapy (42).
OTHER RECEPTORS
Additional studies investigating several other receptor systems, generally those that are overexpressed in a variety of cancers, are under way. An example of such work was reported recently by Praharaj et al., from Columbia, Missouri (43). They studied peptides targeting guanylate cyclase C and the urokinase-type plasminogen activator receptor.
Other investigators studied radiolabeled analogs of vasoactive intestinal peptide and pituitary adenylate cyclase–activating peptide to target their respective receptors, which appear to be upregulated in breast cancer (44).
RADIOLABELED PEPTIDE THERAPY IN UNITED STATES
Because the only FDA-approved radiolabeled peptide for imaging in the Unites States is 111In-octreotide, the first clinical studies of peptide receptor radionuclide therapy (PRRT) were done with high-dose 111In-octreotide. In 2002, Anthony et al. reported on 27 patients with gastroenteropancreatic neuroendocrine tumors treated with 111In-octreotide in a phase II trial at Louisiana State University (45). For these patients, conventional therapies had failed and radiographic or clinical disease progression was evident. The initial dose of 111In-octreotide was 6,660 MBq, a second dose of 6,660 MBq was given 30 d later, and the therapy was repeated if patients responded to the initial dose, with a total dosage per patient ranging from 6,660 to 46,620 MBq (mean, 13,320 MBq). The investigators observed one case of grade 3 leukopenia, one case of grade 4 thrombocytopenia, and one grade 3 increase in the serum creatinine level, which were probably related to the therapy. Only 8% of patients showed a partial response radiographically; however, 62% of patients had clinical improvement (a decrease in the narcotic analgesic dose or an improvement in the Karnofsky score), and 81% had a biochemical response.
In 2008, Delpassand et al. reported similar results, in terms of toxicity and efficacy, with 111In-octreotide in 32 patients who had progressive neuroendocrine tumors and who received 1 therapy (14 patients) or 2 therapies (18 patients) with 18,500 MBq of the radiopharmaceutical (46). There were no grade 4 toxicities, and only 1 patient developed grade 3 thrombocytopenia. A partial response was demonstrated radiographically in 11% of patients; the remaining patients achieved stable disease, with a radiologic response being observed for up to 17 mo after therapy. A clinical response was reported in 83% of patients, with 77% showing a significant decrease in the tumor marker or hormone level.
The main limitation of 111In-octreotide is the short range of the auger electrons emitted by 111In (<10 nm), which have very limited tissue penetration. This limitation has led to the development of somatostatin analogs radiolabeled with high-energy β-emitters (such as 90Y and 177Lu). Currently, no somatostatin analogs radiolabeled with β-emitters are approved by the FDA for clinical use; however, 90Y-DOTATOC and 177Lu-DOTATATE have been designated as orphan drugs by the FDA. The available clinical data from the United States on PRRT with these compounds are primarily from several multiinstitutional trials with participation by U.S. and European centers.
The University of Iowa was a major participant in a multicenter phase II trial evaluating the efficacy of 90Y-DOTA-Tyr3-octreotide (90Y-DOTATOC, also designated 90Y-edotreotide; Onalta [Molecular Insight Pharmaceuticals]) in 90 patients with metastatic carcinoid tumors treated in multiple centers in the United States and Europe. The results were published in 2010 (47). All patients had measurable disease and at least one symptom or sign refractory to optimal therapy with nonradioactive octreotide. Patients were treated with 3 cycles of 90Y-DOTATOC at 4,440 MBq per cycle, administered every 6–9 wk. Stable disease was observed in 70% of the patients after treatment; there were no complete responses, and there were 4.4% partial responses, according to Southwestern Oncology Group criteria (47). However, patients reported significant symptomatic improvement, with a mean duration of the clinical response of 8–12 wk. Furthermore, patients with a long-lasting improvement of diarrhea showed significantly better survival than patients without a long-lasting diarrhea response (18.2 vs. 7.9 mo). There were no grade 3 or 4 cases of leukopenia or thrombocytopenia. Side effects were mostly transient gastrointestinal tract symptoms (nausea, vomiting, and abdominal pain) related to the amino acid infusion (Aminosyn II; Abbott Laboratories; equivalent of 2 L over 4 h) used for renal protection.
The amino acid infusion is essential for reducing radiation injury to the kidneys in PRRT. Positively charged amino acids decrease the proximal tubular reabsorption of low-molecular-weight proteins and peptides, including radiolabeled somatostatin analogs (48). Bushnell et al. demonstrated a 32% reduction in the renal uptake of 90Y-DOTATOC with an amino acid infusion (Aminosyn II; 2 L over 4 h) that contained 29 g of both arginine and lysine and 140 g of additional amino acids (49). This solution was used in the United States because of its commercial availability; however, its higher osmolarity is likely responsible for the higher incidence of nausea or vomiting seen with this solution than with a solution that contains 25 g of both arginine and lysine and that is also more effective in reducing the renal uptake of radiolabeled peptides (50). Despite the infusion of an amino acid solution, renal toxicity remains the most common long-term side effect of 90Y-DOTATOC therapy, with approximately 10% of patients developing permanent grade 4 or 5 renal toxicity (51).
The clinical effectiveness of PRRT was also evaluated with a clinical response scoring system in 21 patients with neuroendocrine tumors treated at the University of Iowa and VAMC Iowa City as part of the multicenter phase II trial of 90Y-DOTATOC (52). The scoring system included evaluations of weight change, tumor-related symptoms (fatigue, abdominal pain, diarrhea, nausea or vomiting, and flushing), the Karnofsky score, and a patient-reported health state score. Of the 21 patients included in the analysis, 67% showed a favorable clinical response, with a significant decrease in the daily nonradioactive octreotide requirement.
Similar response rates were reported in a phase I study of 58 patients treated at Lee Moffitt Cancer Center at the University of Florida; Erasmus Medical Center, Rotterdam, The Netherlands; and Catholic University of Louvain, Brussels, Belgium (53). Of 36 symptomatic patients at baseline, 58% showed an improvement in clinical symptoms or the Karnofsky score; an objective response (Southwestern Oncology Group criteria) was noted in 57% of patients, who converted either from progressive disease to stable disease of from stable disease to a minimal response (53).
The results of a phase I clinical trial of PRRT in pediatric patients at the University of Iowa were recently published. Menda et al. reported on 17 children and young adults who had 7 neuroendocrine tumors, 4 brain tumors, 3 paragangliomas, and 2 neuroblastomas and who were treated with 90Y-DOTATOC in a dose escalation phase I trial (54). The dose of 90Y-DOTATOC started at 1,110 MBq/m2 per cycle and increased to 1,850 MBq/m2 per cycle, administered in 3 cycles with a 6-wk period between the cycles. There was no dose-limiting toxicity. There was significant variability in the renal radiation doses among the patients, suggesting that a dosimetry-based approach would be preferable to fixed dosing with 90Y-DOTATOC. The treatment responses—12% partial response, 29% minimal response, and 35% stable disease—warranted a phase II trial of 90Y-DOTATOC in children (54). More recently, Gains et al. reported promising results for 177Lu-DOTATATE in children, with 5 of 6 patients developing stable disease in cases of refractory or relapsing high-risk neuroblastoma (55).
Another radiolabeled peptide that has been reported in a therapeutic clinical trial in the United States is 188Re-P2045. This agent is an analog of 99mTc-depreotide (13,14). In a phase I trial, patients with metastatic or recurrent lung tumors that could not be treated surgically and that were demonstrated to have somatostatin receptors by positive 99mTc-P2045 imaging results were treated with escalating doses of 188Re-P2045 (56). Ten patients were treated with 188Re-P2045 at 1,110–3,330 MBq/m2; 5 patients did not receive the therapeutic dose because of a lack of uptake of 99mTc-P2045 in the tumor or a projected renal dose of greater than 20 Gy. Other than one case of grade 3 lymphopenia, there were no grade 3 or 4 toxicities definitely related to the study drug. Although there were no objective responses, 5 of 8 patients with progressive disease at baseline remained stable at 8 wk after therapy (56).
CONCLUSION
It is apparent that many basic and clinical research groups within the United States and elsewhere are studying a broad variety of radiopeptides that either have already been shown useful in the clinical setting or appear to have a significant potential to be shown useful in the future.
The breadth of effort and large number of agents are both encouraging and discouraging. It is encouraging that several promising ligands with a significant potential to make a real difference in the management of disease and the treatment of patients are being developed. However, the fact that the number of agents being studied is large is discouraging because it suggests that less effort is concentrated on bringing any given agent all the way through clinical trials to final approval.
Current research funding, particularly from the National Institutes of Health, overemphasizes the importance of novelty in research projects. This emphasis has the effect, in the radiopeptide area, of fostering the development of numerous new agents but does not provide the infrastructure for the translational effort to bring the agents to approval. The regulatory requirements in the United States also restrict access to several radionuclide peptides that are being used as clinical tools in a growing number of institutions across Europe. This issue is potentially critical for patients who may benefit from PRRT and need to travel to Europe for this treatment. Future attempts to balance the effort in the field with appropriate clinical trials are important so that at least some of these remarkable agents can be made available to patients in the United States.
Acknowledgments
No potential conflict of interest relevant to this article was reported.
- © 2011 by Society of Nuclear Medicine
REFERENCES
- Received for publication July 25, 2011.
- Accepted for publication October 25, 2011.





{kind=link}
Jump to section
- Article
- Abstract
- IMAGING WITH SOMATOSTATIN RECEPTOR LIGANDS
- 111IN-LABELED SOMATOSTATIN RECEPTOR IMAGING
- 99MTC-LABELED SOMATOSTATIN RECEPTOR IMAGING
- 68GA-LABELED SOMATOSTATIN RECEPTOR IMAGING
- PEPTIDE IMAGING OF MELANOMA
- PEPTIDE IMAGING OF ANGIOGENESIS
- TUMOR IMAGING WITH PEPTIDE DIMERS
- BOMBESIN
- OTHER RECEPTORS
- RADIOLABELED PEPTIDE THERAPY IN UNITED STATES
- CONCLUSION
- Acknowledgments
- REFERENCES
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- Bombesin Antagonist-Based Radiotherapy of Prostate Cancer Combined with WST-11 Vascular Targeted Photodynamic Therapy
- Improved Modeling of In Vivo Kinetics of Slowly Diffusing Radiotracers for Tumor Imaging
- Targeted Radionuclide Therapy: Proceedings of a Joint Workshop Hosted by the National Cancer Institute and the Society of Nuclear Medicine and Molecular Imaging
- A Comparative Study of Radiolabeled Bombesin Analogs for the PET Imaging of Prostate Cancer
- Peptide Receptor Radionuclide Therapy in the United States