


Abstract
Bombesin is a peptide exhibiting high affinity for the gastrin-releasing peptide receptor (GRPr), which is highly overexpressed on prostate cancer cells. In the present study, we developed an 18F-labeled bombesin analog, 18F-BAY 86-4367, which is currently being clinically tested for use in PET of prostate cancer. Methods: In vitro pharmacologic studies were performed to characterize the nonradioactive (19F) standard of the bombesin analog for binding affinity and selectivity for GRPr. The stability of 18F-BAY 86-4367 was determined in murine and human plasma. In vivo, the tumor-targeting potential and pharmacokinetic profile of the 18F tracer were analyzed with biodistribution experiments and PET studies of prostate tumor–bearing mice. Results: The nonradioactive (19F) standard of the bombesin analog showed subnanomolar and GRPr-selective binding affinity. The stability of the tracer in murine and human plasma was found to be high. In 2 prostate cancer xenograft models (PC-3 and LNCaP), 18F-BAY 86-4367 showed more specific and effective GRPr-based targeting in vivo than the benchmark radiotracers 18F-fluoroethylcholine and 18F-FDG. In addition, rapid tumor targeting and fast renal excretion (∼70%) and hepatobiliary excretion (∼10%) were identified in both xenograft models. Furthermore, PET studies provided clear and specific visualization of PC-3 tumors in mice. Conclusion: Favorable preclinical data showing specific and effective tumor targeting by 18F-BAY 86-4367 suggest that a clinical trial be undertaken to test its diagnostic utility in PET for prostate carcinoma patients.
Prostate cancer is the most common nonskin cancer among men in most Western populations, and it is the second leading cause of cancer death in men in the United States ( 1). Advances in the imaging of prostate cancer may help to achieve earlier and more accurate diagnosis and treatment ( 2).
The most widely used PET tracer, 18F-FDG, does not play a prominent role in the diagnosis and staging of prostate cancer, apparently because the use of glucose by prostate carcinoma cells is not enhanced significantly compared with that of normal cells ( 3). Compiled data on staging or restaging for 244 prostate cancer patients suggested an overall sensitivity of only 57% on a per-patient basis. Other PET tracers, such as 18F- or 11C-choline, 11C-acetate, and 18F-fluorothymidine, have shown promise for prostate cancer imaging ( 4). PET/CT with 11C- or 18F-choline is currently considered to be the standard diagnostic imaging tool for the clinical assessment of recurrent prostate cancer in Europe. However, like 18F-FDG, 11C- or 18F-choline is not specific for cancer cells; thus, using this tracer to distinguish benign from malign prostatic tissue is difficult.
The overexpression of the gastrin-releasing peptide receptor (GRPr), also called bombesin receptor subtype 2, on prostate cancer cells provides a potential target for the specific diagnosis and therapy of prostate cancer ( 5– 7). Several analogs of the natural GRPr ligand bombesin, a 14-amino-acid neuropeptide, have been labeled with various radionuclides for the diagnosis and treatment of GPRr-positive tumors in recent years ( 8– 12). Potent 99mTc-based (demobesin-1) and 111In-based (RM-1) bombesin analogs that were designed for GRPr-based targeting and tested in the PC-3 xenograft model showed high absolute tumor uptake in animals ( 13, 14). Few publications have reported on 18F labeling of bombesin peptides ( 15– 19). Our previous study indicated that the negatively charged bombesin derivative 3-cyano-4-18F-fluoro-benzoyl-Ala(SO3H)-Ava-Gln-Trp-Ala-Val-NMeGly-His-Sta-Leu-NH2 had much higher prostate tumor uptake than the corresponding positively charged analog (3-cyano-4-18F-fluoro-benzoyl-Arg-Ava-Gln-Trp-Ala-Val-NMeGly-His-Sta-Leu-NH2) ( 20). Increasing the hydrophilicity as well as the overall charge of the bombesin analog may lead to derivatives with improved sensitivity, specificity, and pharmacokinetics for the optimal targeting of GRPr-positive tumors. Therefore, 1 more L-cysteic acid [Ala(SO3H)] moiety was inserted in the linker of the previously mentioned negatively charged bombesin peptide. Here we report on the 18F radiolabeling of this bombesin analog, BAY 86-4367, and a detailed preclinical evaluation of 18F-BAY 86-4367 as a GRPr-selective and metabolically stable imaging agent for prostate cancer.
MATERIALS AND METHODS
General
9-Fluorenylmethoxy carbonyl (Fmoc)-amino acids were purchased from IRIS Biotech, except for Fmoc-statine and Fmoc-Ala(SO3Na)-OH, which were purchased from NeoMPS (now PolyPeptide Group). Peptide syntheses were performed with rink amide resin (0.68 mmol/g) in accordance with the standard Fmoc strategy. All amino acid residues were, if not further specified, L-amino acid residues. Statine (Sta) denotes (3S,4S)-4-amino-3-hydroxy-6-methylheptanoic acid, Ala(SO3H) denotes L-cysteic acid, and Ava denotes 5-aminopentanoic acid. High-performance liquid chromatography (HPLC) analyses were performed with an ACE C18 column (50 × 4.6 mm, 3 μm). Analytic HPLC chromatograms were obtained by use of an Agilent 1100 system equipped with multi-UV-wavelength and Raytest Gabi Star detectors and Gina Star software. Semipreparative HPLC purifications were performed with a semipreparative ACE C18 column (250 × 10 mm, 5 μm). A Merck-Hitachi L6200A system equipped with Knauer variable-wavelength and Eberline radiation detectors was used for semipreparative HPLC. A reversed-phase radio–thin-layer chromatography plate (aluminum sheets, silica gel, RP-18F254; Macherey-Nagel) was also used to analyze the 18F-labeled product with 10 mM K2HPO4 in water-acetonitrile (3:7) as the mobile phase.
Solid-Phase Peptide Synthesis
Peptide synthesis was performed with Fmoc–rink amide–linker-functionalized polystyrene resin as previously described ( 16). The peptide chain was elongated in cycles of Fmoc deprotection followed by coupling of the subsequent Fmoc-amino acid. Eleven cycles were performed with Fmoc-protected and, if necessary, side chain–protected building blocks of the amino acids, in the following order: leucine (Fmoc-Leu-OH), statine (Fmoc-Sta-OH), histidine (Fmoc-His(Trt)-OH), N-methyl-glycine (Fmoc-NMeGly-OH), valine (Fmoc-Val-OH), tryptophan (Fmoc-Trp(Boc)-OH), glutamine (Fmoc-Gln(Trt)-OH), 5-aminopentanoic acid (Fmoc-Ava-OH), and cysteic acid monosodium salt (Fmoc-Ala(SO3Na)-OH). The Fmoc group of the terminal amino acid was removed to obtain the key intermediate, which was used in the final coupling steps to obtain the precursor (1a) as well as the reference standard, the 19F-bombesin analog (1b). (4-Carboxy-2-cyanophenyl)-trimethylammonium trifluoromethanesulfonate and 3-cyano-4-fluoro-benzoic acid were used as building blocks for this purpose.
The crude peptide precursor was dried under reduced pressure and purified by preparative HPLC with a Luna C18 column (5 μm; Phenomenex), a mobile phase of acetonitrile (plus 0.1% trifluoroacetic acid [TFA]) and water (plus 0.1% TFA), and a gradient of 5%–30% acetonitrile (20 min). UV peak data were collected and reanalyzed by ultra-performance liquid chromatography–mass spectrometry. Fractions containing the correct mass with a purity of greater than 95% were collected and lyophilized to yield the crystalline product. Starting with 350 μmol of resin, we obtained 112 mg of the product (19% overall yield).
The crude peptide reference standard was purified in a similar procedure by preparative HPLC with a Luna C18 column (5 μm), a mobile phase of acetonitrile (plus 0.1% TFA) and water (plus 0.1% TFA), and a gradient of 5%–40% acetonitrile (20 min). Starting with 100 μmol of resin, we obtained 60 mg of the product (39% overall yield) with a purity of greater than 95%.
Radiochemistry
No-carrier-added 18F-fluoride was produced via the 18O(p,n)18F nuclear reaction by irradiation of enriched 18O-water. 18F-fluoride was immobilized on an anion-exchange cartridge (QMA Light; Waters) and eluted with a solution of Kryptofix K222 (5 mg; Merck KGaA) and Cs2CO3 (2.3 mg) in acetonitrile (1.5 mL) and water (0.5 mL). The fluoride was dried by azeotropic distillation of acetonitrile at 110°C under vacuum with a stream of nitrogen. The azeotropic drying process was repeated 3 times with 1 mL of acetonitrile each time.
To the dried 18F-fluoride complex (typically 20 GBq), the precursor bombesin peptide (1a) was added. To find optimal radiolabeling conditions, we used various solvents (N,N-dimethylformamide and dimethyl sulfoxide [DMSO]), reaction temperatures (70°C–100°C), and reaction times (5–20 min), and aliquots of the reaction mixture under the various reaction conditions were analyzed by analytic HPLC. The identity of the tracer (18F-BAY 86-4367) was confirmed by coinjection of the authentic standard (1b) into the same analytic HPLC system ACE C18 column (50 × 4.6 mm, 3 μm). The elution conditions were as follows: 10 mM K2HPO4 in water (solvent A) and 10 mM K2HPO4 in water-acetonitrile (3:7) (solvent B); 0 min, 5% B; 0–7.0 min, 5%–95% B; 7.0–7.1 min, 95%–100% B; 7.1–8.8 min, 100% B; and 8.8–9.0 min, 100%–5% B; and a flow rate of 2 mL/min. The reaction mixture was diluted with water (4 mL) and injected into a semipreparative HPLC ACE column; the conditions were as follows: 0.1% TFA in water (solvent A) and 0.1% TFA in acetonitrile-water (9:1) (solvent B); isocratic elution with 23% solvent B; and a flow rate of 4 mL/min. The product (typically 2 GBq) was collected.
18F-FDG and 18F-fluoroethylcholine (18F-FCH) were purchased from University Clinic Charité and Eckert & Ziegler, respectively.
Determination of Distribution Coefficient
The distribution coefficient was determined by the shake flask method ( 21). In brief, the peptide was dissolved in a mixture of phosphate buffer (600 μL, pH 7.4) and n-octanol (600 μL) at 20°C. The sample was equilibrated by 4 min of shaking followed by centrifugation. The concentration of the peptide in each phase was measured by HPLC-UV absorption. The distribution coefficient was calculated as the log ratio of the UV peak areas at 254 nm in the organic and aqueous phases.
In Vitro Characterization
Displacement Assays.
The binding affinities of the nonradioactive 19F-bombesin peptide for human GRPr and neuromedin B receptor were determined by use of a scintillation proximity assay (SPA) with transfected cellular membranes (RBHBS2 M and RBHBS1 M; PerkinElmer) and either 125I-Tyr4-bombesin or the universal ligand [D-125I-Tyr6,βAla11,Phe13,Nle14]bombesin (each at 81.4 TBq/mmol; PerkinElmer) as a radioligand. The membranes and agglutinin-coupled SPA beads (type A; polyvinyltoluene (PVT)- and polyethyleneimine-treated wheat germ; Amersham Bioscience) were mixed in incubation buffer (50 mM Tris-HCl [pH 7.2], 5 mM MgCl2, 1 mM ethylene glycol tetraacetic acid, protease inhibitor [1 tablet per 50 mL; Roche Diagnostics GmbH], and 0.3% polyethyleneimine) to produce final protein concentrations of 20 μg/mL (RBHBS2 M) and 28 μg/mL (RBHBS1 M) and a final concentration of PVT-SPA beads of 8 mg/mL. The radioligands were diluted to either 0.2 nM (125I-Tyr4-bombesin) or 0.5 nM ([D-125I-Tyr6,βAla11,Phe13,Nle14]bombesin) in incubation buffer. The inhibitor used for displacement was either nonradioactive 19F-bombesin (1b) or neuromedin B (Sigma). The test compounds were dissolved in DMSO to produce 1 mM stock solutions and further diluted in incubation buffer to 2 pM–1,000 nM. Nonspecific binding was determined with an excess (>1,000-fold) of either Tyr4-bombesin or [D-Tyr6,βAla11,Phe13,Nle14]bombesin (Sigma).
The SPA was performed in quadruplicate as follows. First, 10 μL of a test compound was placed in white 384-well plates (Lumitrac 200; Greiner). Next, 20 μL of the GRPr–PVT-SPA bead mixture and 20 μL of the ligand solution were added. After 120 min of incubation at room temperature, 50 μL of incubation buffer were added; the plate was sealed and centrifuged for 10 min at 520g and room temperature. Signals were quantified with a Top Count (PerkinElmer) for 60 s of integration per well. Nonspecific binding was subtracted from total binding to yield specific binding at each concentration. The 50% inhibitory concentration (IC50) and the inhibition constant (Ki) were calculated by nonlinear regression with GraFit 5 data analysis software (Erithacus Software Ltd.).
Scatchard Assays.
For saturation binding analysis, human PC-3 prostate carcinoma cells (American Type Culture Collection) at confluence were placed in 48-well plates (8 × 104 cells per well). Cells were incubated in triplicate with increasing concentrations of the 18F-labeled bombesin analog (0.05 nM–100 nM) in binding buffer (50 mM N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid), 1 mM ethylenediaminetetraacetic acid, 5 mM MgCl2, and 0.1% bovine serum albumin [pH 7.4] in Dulbecco modified Eagle medium with GlutaMAX I [Invitrogen]) to produce a total volume of 200 μL. The concentration range was selected on the basis of the IC50 of the nonradioactive peptide (see earlier description). Nonspecific binding was determined in the presence of 1 μM Tyr4-bombesin for each radioligand concentration. After incubation at room temperature for 1 h, cells were washed twice with cold phosphate-buffered saline (PBS) (plus 0.1% bovine serum albumin) and solubilized with 300 μL of 0.25% trypsin-ethylenediaminetetraacetic acid solution (incubation for 20 min at 37°C) and then washed with 1 mL of PBS. Radioactivity was measured with a γ-counter (Wizard; PerkinElmer). Saturation binding data were evaluated with KELL Radlig software (Biosoft). Three independent experiments were performed, and the dissociation constant was calculated by use of the Scatchard transformation.
In Vitro Stability Studies.
The stability of the radiolabeled bombesin analog was investigated in mouse and human plasma at various incubation times (0–120 min) and 37°C. PBS was used as a control. After incubation, plasma proteins were precipitated with acetonitrile and centrifuged for 10 min at 13,000g and 4°C. The PBS control was diluted with the same volume of acetonitrile. The supernatants and the PBS control were analyzed by analytic HPLC.
In Vivo Characterization
In Vivo Stability Studies.
All animal experiments were undertaken in compliance with the current version of the Swiss and German law concerning animal protection and welfare. Male NMRI nude mice were injected intravenously with approximately 25 MBq of the 18F-bombesin derivative. Animals were sacrificed at 1, 3, 5, 10, and 15 min after injection, and blood and urine were collected. Aliquots of plasma and urine were mixed with acetonitrile for protein precipitation. The samples were centrifuged at 5,000g (Megafuge R; Heraeus), and the supernatants were analyzed by radio-HPLC with a Luna C18 column (0.1% TFA in water [solvent A] and 0.1% TFA in acetonitrile-water [9:1] [solvent B]).
Biodistribution Studies.
Biodistribution experiments were performed with male NMRI nude mice bearing subcutaneous PC-3 or LNCaP tumors next to the right shoulder. For induction of the xenografts, tumor cells (2 × 106 cells per mouse) were injected subcutaneously and allowed to grow for 4 wk. The sizes of the PC-3 tumors ranged from 40 to 450 mg, and the sizes of the LNCaP tumors ranged from 90 to 700 mg. Animals were injected intravenously in the tail vein with 45–238 kBq of the bombesin tracer (100 μL). Radioactive doses of the comparative tracers 18F-FDG and 18F-FCH were 180–225 kBq/100 μL per animal. Animals were sacrificed at various times (0.5–4 h; n = 3 for each time point). Organs and tissues of interest were collected and weighed. The amount of radioactivity was determined with the γ-counter to calculate uptake as the percentage injected dose per gram of tissue (%ID/g). In addition, 3 mice that received a coinjection of 100 μg of Tyr4-bombesin and the radiolabeled compound were sacrificed at 1 h after injection to determine nonspecific tissue uptake.
PET Studies.
PET of PC-3 tumor–bearing mice was performed with the 16-module variant of the dedicated small-animal PET quad-HIDAC tomograph (Oxford Positron Systems) ( 22). Animals were injected intravenously with 9–20 MBq (<1 nmol) of the radioligand and scanned in a whole-body configuration from 60 to 105 min after injection. Animals were anesthetized with isoflurane and monitored as reported previously ( 22). Two animals received a coinjection of 100 μg of bombesin (1 mg/mL), and 2 animals received a corresponding volume of vehicle. After termination of the PET scan, animals were sacrificed at 107 min after injection by decapitation for tissue sampling. PET data were reconstructed in a single 45-min time frame and normalized to the injected dose per gram of body weight.
RESULTS
Chemistry and Radiochemistry
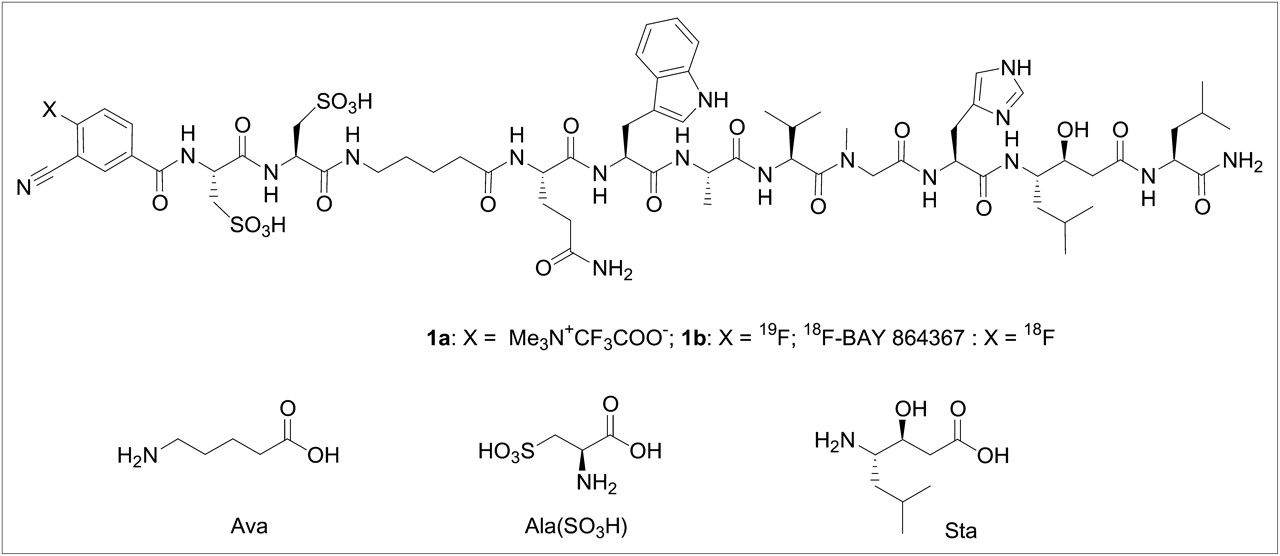
The bombesin peptide analog was synthesized by solid-phase peptide synthesis in accordance with standard Fmoc strategy. Coupling of the resin-bound peptide with 3-cyano-4-fluoro-benzoic acid and 3-cyano-4-trimethylamino-benzoic acid yielded the corresponding nonradioactive (19F) peptide standard (1b) and precursor (1a) for 18F labeling, respectively. The 19F-bombesin peptide served as a surrogate for the 18F-bombesin peptide in the determination of binding affinity and as a reference standard for radio-HPLC analysis. Figure 1 shows the sequences of 1a and 1b and the structures of the nonnatural amino acid derivatives. The synthesized bombesin precursor and reference standard were characterized by mass spectrometry:
-
3-cyano-4-trimethylammonium-benzoyl-Ala(SO3H)-Ala(SO3H)-Ava-Gln-Trp-Ala-Val-NMeGly-His-Sta-Leu-NH2-trifluoroacetate salt (1a, 112 mg, 19%)—m/z [M]+ calculated: 1,567.7, found: 1,567.5
-
3-cyano-4-fluoro-benzoyl-Ala(SO3H)-Ala(SO3H)-Ava-Gln-Trp-Ala-Val-NMeGly-His-Sta-Leu-NH2 (1b, 60 mg, 39%)—m/z [M+H]+ calculated: 1,528.6, found: 1,528.9.

Peptide sequences and structures of nonnatural amino acids. 1a = precursor; 1b = reference compound or 18F-BAY 86-4367.
Direct 1-step 18F labeling via aromatic nucleophilic substitution of 18F for +N(CH3)3—developed previously in our laboratories—was successfully applied to the bombesin peptide analog ( 19). Using DMSO as the solvent resulted in a higher level of 18F incorporation than using N,N-dimethylformamide at all of the tested reaction temperatures. The required reaction temperature and time for sufficient 18F labeling efficiency were either 80°C or 90°C and 5 min, respectively. For all of the in vitro and in vivo studies, 18F labeling of the peptide was performed with 2 mg of precursor in 150 μL of DMSO in the presence of a Cs18F-Kryptofix K222 complex. The reaction mixture was heated at 90°C for 10 min; after semipreparative HPLC purification, the product fraction was collected. The collected product was diluted with water (18 mL), immobilized on the cartridge, and washed with water (2 × 10 mL), and then the radioactive bombesin derivative was eluted with ethanol (1.5 mL). The solvent was evaporated at 80°C under vacuum with a stream of nitrogen and subsequently dissolved in saline-water. The decay-corrected radiochemical yield of the isolated product was approximately 15%, and the radiochemical purity was always greater than 95%. The specific activity after formulation was up to 170 GBq/μmol.
Distribution Coefficient
The distribution coefficient of the nonradioactive 19F-bombesin derivative was determined by the shake flask method and HPLC-UV absorption. This coefficient was found to be lower than −3.9 at pH 7.4, indicating a highly hydrophilic peptide.
In Vitro Characterization
Displacement and Scatchard Assays.
The affinity of the bombesin peptide analog for GRPr was in the subnanomolar range, with an IC50 (mean ± SD) of 0.94 ± 0.19 nM and a Ki of 0.13 ± 0.05 nM (n = 3) ( Fig. 2). Saturation binding assays revealed 40,000–60,000 binding sites per PC-3 cell and less than 10,000 binding sites per LNCaP cell. The selectivity of 18F-BAY 86-4367 for GRPr was demonstrated in a displacement assay with neuromedin B receptor–transfected cell membranes and [D-125I-Tyr6,βAla11,Phe13,Nle14]bombesin as a universal bombesin receptor ligand. The natural ligand neuromedin B had a high affinity for bombesin 3 receptors (IC50, 20.6 nM), but 18F-BAY 86-4367 exhibited no such affinity (IC50, >10,000 nM) (Supplemental Table 1) (supplemental materials are available online only at http://jnm.snmjournals.org).
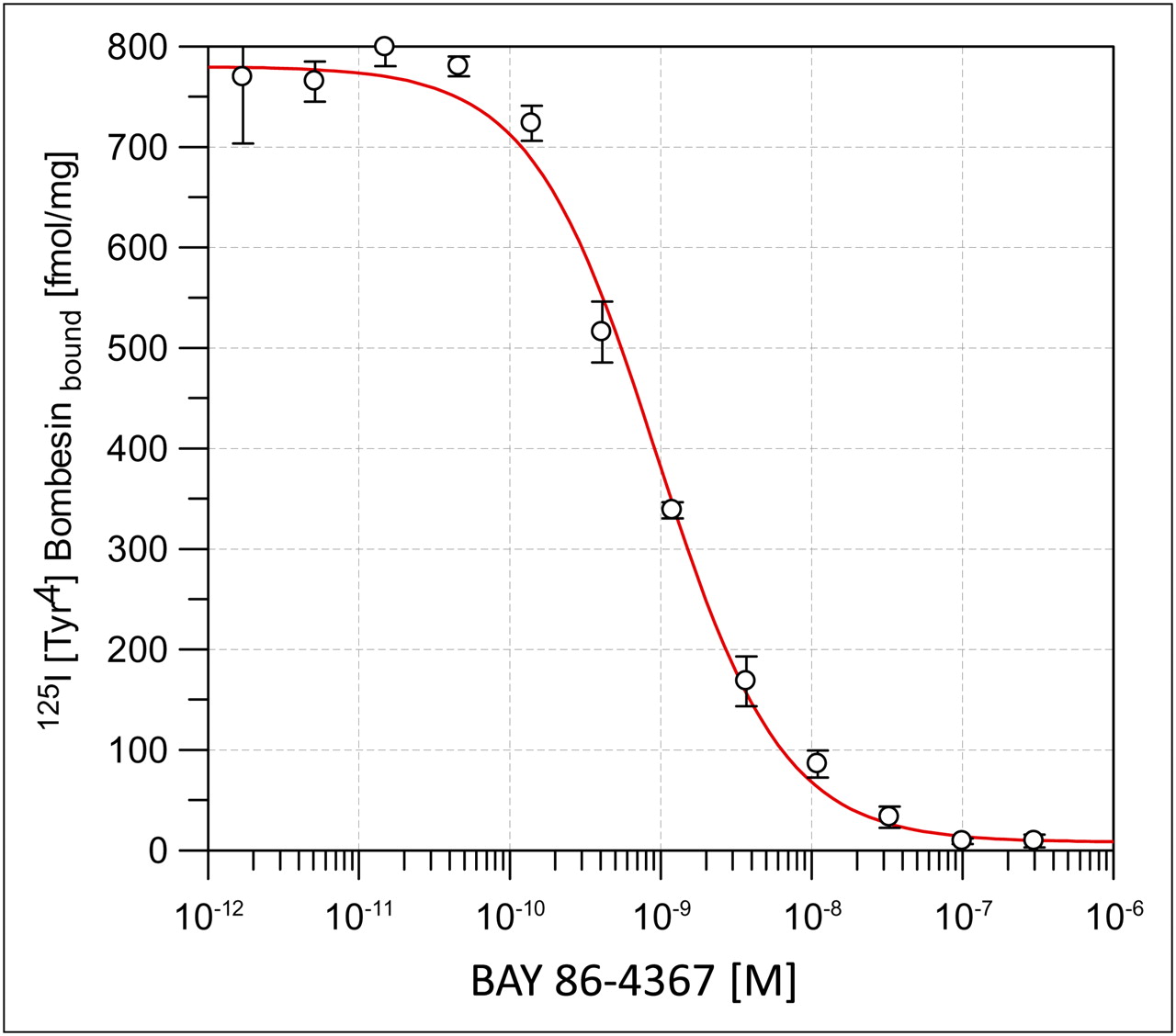
Competitive binding of 18F-BAY 86-4367 and radioligand 125I-Tyr4-bombesin in SPA with membranes of GRPr-transfected cells.
The binding affinity of 18F-BAY 86-4367 for human PC-3 prostate carcinoma cells was analyzed by saturation binding assays and then Scatchard transformation. Radioligand binding was found to be saturable with a single high-affinity binding site; the dissociation constant was 1.43 ± 0.25 nM (n = 3) (Supplemental Fig. 2). These findings suggest that under the radiolabeling conditions (90°C for 10 min), the affinity of the peptide for GRPr was retained.
Additionally, the antagonistic properties of 18F-BAY 86-4367 were determined with 2 assays—receptor internalization visualized by immunofluorescence microscopy ( 23, 24) and cellular calcium mobilization ( 25)—performed in the laboratory of Jean C. Reubi (University of Bern). Bombesin-induced GRPr internalization in human GRPr–transfected HEK cells and calcium mobilization in PC-3 cells were efficiently antagonized by the nonradioactive bombesin peptide (Supplemental Fig. 3). These findings suggest that the candidate compound behaves as an antagonist for GRPr.
In Vitro Stability.
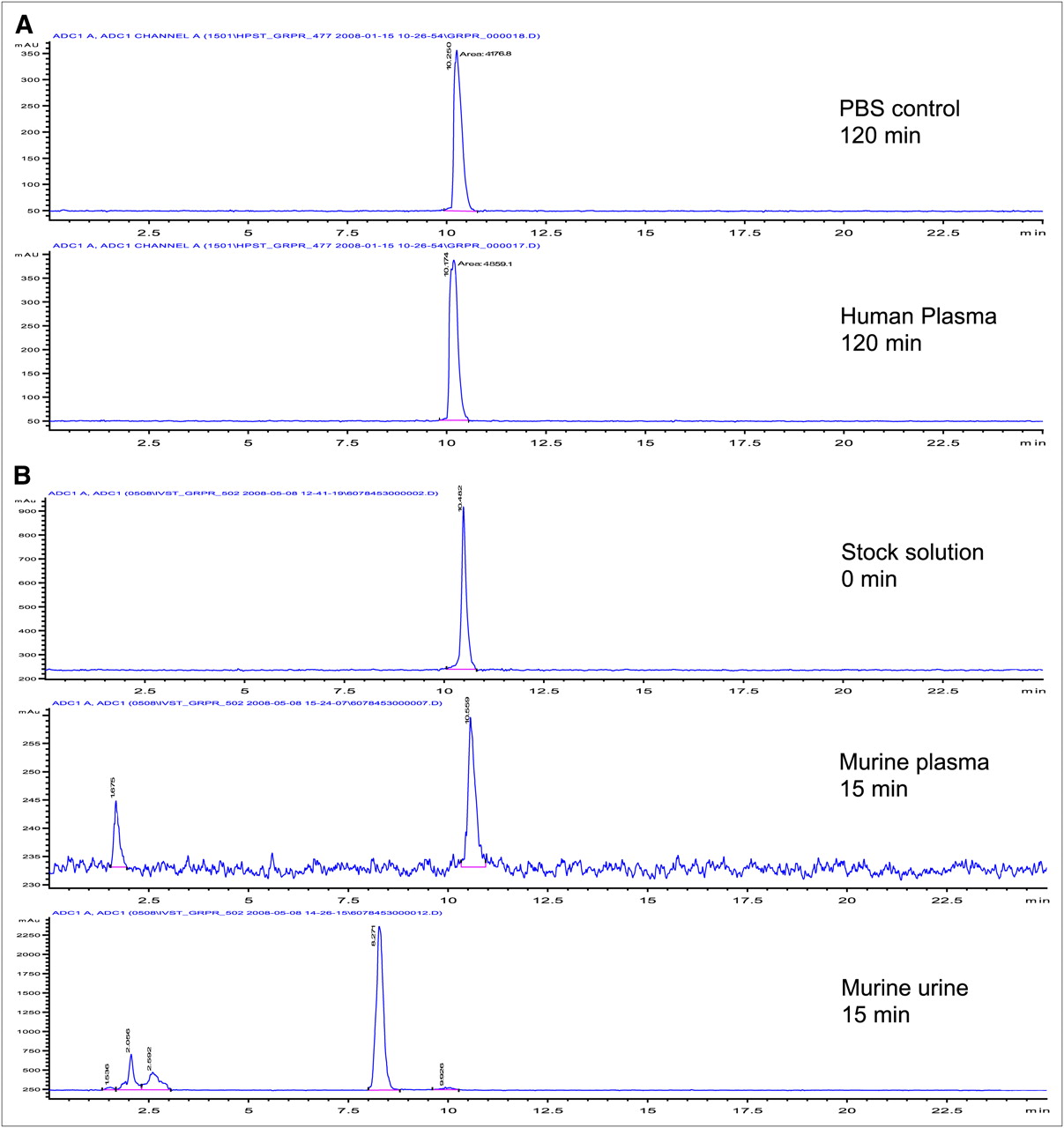
The radiolabeled compound did not show any degradation either in PBS or in mouse or human plasma up to 120 min ( Fig. 3A).

(A) In vitro stability of 18F-BAY 86-4367 in human plasma and in PBS. (B) In vivo metabolism of radiolabeled peptide in murine plasma and urine.
In Vivo Characterization
In Vivo Stability.
The in vivo stability of 18F-BAY 86-4367 was analyzed in murine plasma and urine by radio-HPLC. At various times after radiotracer injection into nude mice (1–15 min), blood and urine were collected for HPLC analysis. At 15 min after injection, only 1 minor polar radiometabolite could be detected in the plasma ( Fig. 3B). A prominent metabolite, more hydrophilic than the parent compound, was already found in the urine after 5 min.
Biodistribution Studies.
High PC-3 tumor uptake of the radiotracer was found at 1 h after injection (6.2 %ID/g), and this uptake could be blocked effectively (>80%) by coinjection of nonradioactive bombesin (100 μg per mouse) ( Table 1). The tumor-to-blood ratio at 1 h after injection was 19. The GRPr-expressing murine pancreas also showed high uptake of the radiotracer (>40 %ID/g). Other GRPr-positive organs showing natural specific uptake were the adrenal glands, stomach, and intestine. The radioactive peptide showed gallbladder uptake of 30–50 %ID/g even at early times, indicating rapid liver metabolism of parts of the circulating tracer. Subsequent intestinal excretion resulted in 10 %ID in the feces at 4 h after injection. In parallel, rapid urinary excretion of radioactivity was found, with 50 %ID already in the urine after 1 h. Tumor-to-tissue ratios at 1 h after injection were predominantly much greater than 1. The tumor uptake of 18F-BAY 86-4367 was also quantified for LNCaP tumors, which showed less than half of the activity concentration of PC-3 tumors, with 2.3 %ID/g at 1 h after injection because of a lower level of GRPr expression ( Table 2). All other organ uptake values were comparable to the biodistribution values obtained in PC-3 tumor–bearing animals.
Biodistribution of 18F-BAY 86-4367 in PC-3 Tumor–Bearing Mice at Various Times After Injection and After 60 Minutes of Blocking (n = 3)
Biodistribution of 18F-BAY 86-4367 (18F-BN), 18F-FCH, and 18F-FDG in PC-3 and LNCaP Tumor–Bearing Mice at 60 Minutes (n = 3)
In the next set of biodistribution experiments, the uptake of 18F-BAY 86-4367 was compared with that of 18F-FCH and 18F-FDG at 1 h after injection in prostate tumor–bearing mice. 18F-BAY 86-4367 showed the most favorable distribution in both prostate tumor models, with high tumor uptake, low nontarget uptake, and rapid excretion ( Table 2). Urinary excretion was lowest with 18F-FCH. Tumor-to-tissue ratios in PC-3 tumor–bearing mice at 1 h after injection were markedly higher for the radioactive peptide tracer than for 18F-FCH and 18F-FDG in the same tumor model.
PET of PC-3 Tumor–Bearing Mice.
Nude mice bearing subcutaneous PC-3 tumor xenografts next to the right shoulder were scanned from 60 to 105 min after injection in a dedicated small-animal PET system under both baseline and blocking conditions ( Fig. 4). The tumors were clearly imaged under baseline conditions, whereas no tumor was visualized under blocking conditions. As expected from the ex vivo biodistribution studies, the highest activity concentrations in the in vivo PET studies were identified in the urinary bladder, the gallbladder, and the intestine. Clear visualization of the GRPr-positive pancreas by PET was not possible because of the proximity of the pancreas to the bowel. Postmortem tissue sampling of the mice after PET (at 107 min after injection) confirmed the data obtained in the biodistribution studies. The absolute radioactivity uptake values in the GRPr-rich tissues were 4.7 %ID/g for the tumor and 15.6 %ID/g for the pancreas. The specificities of uptake for the tumor and the pancreas were 82% and 96%, respectively.

Series of horizontal whole-body slices (ventral to dorsal) from 2 nude mice bearing subcutaneous PC-3 tumor xenografts next to right shoulder (arrows). Mice were injected with 18F-BAY 86-4367 and scanned from 60 to 105 min after injection under baseline (A) and blocking (B) conditions. Image data were normalized to injected dose per gram of body weight.
DISCUSSION
Numerous bombesin analogs have been radiolabeled with a variety of radioactive isotopes for the detection and therapy of GRPr-expressing neoplasms ( 8– 12, 26– 29). Tetraamine-functionalized 99mTc-bombesin analogs with favorable in vivo profiles were reported by Nock et al. ( 28). Parry et al. also reported that 64Cu-DOTA-bombesin peptides exhibited tumor uptake and good imaging characteristics ( 30). Our laboratories have focused on labeling peptides for PET of both breast cancer and prostate cancer with 18F because this nuclide has ideal characteristics for peptide receptor imaging studies.
The precursor and reference standard were prepared by solid-phase peptide synthesis in optimal yields. The distribution coefficient of the nonradioactive 19F-bombesin derivative (1b) was lower than −3.9. The direct labeling of peptides with 18F in a single step was recently described by various groups ( 19, 31– 33). Schirrmacher et al. reported the direct radiolabeling of an organosilicon-modified peptide in an isotope exchange reaction ( 32). Several 1-step approaches for the facile 18F labeling of peptides have been developed in our laboratories ( 19, 31, 33). In the present study, 18F labeling of BAY 86-4367 was achieved in a single step via aromatic nucleophilic substitution. 18F-BAY 86-4367 was obtained in a good radiochemical yield and with excellent radiochemical purity after HPLC purification.
The affinity of 18F-BAY 86-4367 for GRPr was high—in the subnanomolar range—whereas the affinity of 18F-BAY 86-4367 for the other 2 bombesin receptor subtypes, neuromedin B and bombesin 3 receptors, was negligible. Tumor accumulation of 18F-BAY 86-4367 was investigated in PC-3 (androgen-independent human prostate cancer with a high level of GRPr expression) and LNCaP (androgen-dependent human prostate cancer with a lower level of GRPr expression than PC-3) tumor–bearing mice. High and persistent tracer uptake in PC-3 tumors and GRPr-expressing murine organs (pancreas, stomach, intestine, and adrenal glands) was identified. LNCaP tumors showed 50% less tracer uptake than PC-3 tumors because of a lower level of GRPr expression, as reported previously. Compared with 18F-FCH and 18F-FDG, the tracer candidate displayed the highest tumor-to-tissue ratios in PC-3 tumor–bearing mice. In line with the results of the biodistribution studies, PET images showed highly specific GRPr targeting of the radiotracer in PC-3 tumor–bearing mice, as demonstrated by effective blocking with nonradioactive Tyr4-bombesin.
Recent preclinical studies demonstrated that GRPr antagonists may be preferable to agonists for tumor targeting, yielding high tumor uptake and long tumor washout ( 23, 34). Our data indicated that 18F-BAY 86-4367 behaves as an antagonist for GRPr, making this peptide promising for further evaluation in a clinical trial.
On the basis of the aforementioned in vivo results, 18F-BAY 86-4367 has favorable pharmacokinetics, with rapid tumor targeting, rapid blood clearance, and fast and predominantly renal excretion (∼70%). This advantageous pattern of elimination, in combination with high affinity and specificity, suggest that 18F-BAY 86-4367 may have higher diagnostic accuracy than the standard tools 18F-FDG and 18F-FCH in PET for prostate cancer patients.
CONCLUSION
The favorable preclinical data obtained for 18F-BAY 86-4367 suggest that a clinical trial be undertaken to test its diagnostic utility for prostate carcinoma patients. A clinical proof-of-mechanism microdosing study in healthy volunteers and patients is currently under way to assess the tumor-targeting potential, radiation dosimetry, pharmacokinetics, and metabolism of the tracer candidate in a clinical setting.
Acknowledgments
We would like to thank Jean C. Reubi and his coworkers for performing the antagonist studies and Claudia Keller, Sabine Baumann, René Zernicke, Ingolf Weber, Volker Stickel, and Melanie Appel for their excellent technical assistance.
- © 2011 by Society of Nuclear Medicine
REFERENCES
- Received for publication July 23, 2010.
- Accepted for publication October 26, 2010.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Recent Advances in 18F Radiochemistry: A Focus on B-18F, Si-18F, Al-18F, and C-18F Radiofluorination via Spirocyclic Iodonium Ylides
- Dosimetry and First Clinical Evaluation of the New 18F-Radiolabeled Bombesin Analogue BAY 864367 in Patients with Prostate Cancer
- A High-Affinity, High-Stability Photoacoustic Agent for Imaging Gastrin-Releasing Peptide Receptor in Prostate Cancer