


Abstract
Ischemic insult to the myocardium is associated with cardiomyocyte apoptosis. Because apoptotic cell death is characterized by phosphatidylserine externalization on cell membrane and annexin-A5 (AA5) avidly binds to phosphatidylserine, we hypothesized that radiolabeled AA5 should be able to identify the regions of myocardial ischemia. Methods: Models of brief myocardial ischemia by the occlusion of the coronary artery for 10 min (I-10) and reperfusion for 180 min (R-180) for the detection of phosphatidylserine exteriorization using 99mTc-labeled AA5 and γ-imaging were produced in rabbits. 99mTc-AA5 uptake after brief ischemia was compared with an I-40/R-180 infarct model. Histologic characterization of both myocardial necrosis and apoptosis was performed in ischemia and infarct models. Phosphatidylserine exteriorization was also studied in a mouse model, and the dynamics and kinetics of phosphatidylserine exposure were assessed using unlabeled recombinant AA5 and AA5 labeled with biotin, Oregon Green, or Alexa 568. Appropriate controls were established. Results: Phosphatidylserine exposure after ischemia in the rabbit heart could be detected by radionuclide imaging with 99mTc-AA5. Pathologic characterization of the explanted rabbit hearts did not show apoptosis or necrosis. Homogenization and ultracentrifugation of the ischemic myocardial tissue from rabbit hearts recovered two thirds of the radiolabeled AA5 from the cytoplasmic compartment. Murine experiments demonstrated that the cardiomyocytes expressed phosphatidylserine on their cell surface after an ischemic insult of 5 min. Phosphatidylserine exposure occurred continuously for at least 6 h after solitary ischemic insult. AA5 targeted the exposed phosphatidylserine on cardiomyocytes; AA5 was internalized into cytoplasmic vesicles within 10–30 min. Twenty-four hours after ischemia, cardiomyocytes with internalized AA5 had restored phosphatidylserine asymmetry of the sarcolemma, and no detectable phosphatidylserine remained on the cell surface. The preadministration of a pan-caspase inhibitor, zVAD-fmk, prevented phosphatidylserine exposure after ischemia. Conclusions: After a single episode of ischemia, cardiomyocytes express phosphatidylserine, which is amenable to targeting by AA5, for at least 6 h. Phosphatidylserine exposure is transient and internalized in cytoplasmic vesicles after AA5 binding, indicating the reversibility of the apoptotic process.
Although it has been traditionally believed that most heart muscle cells during the ischemic insult to the myocardium are lost by necrosis, it is now being increasingly recognized that apoptosis plays an important role in the cell death. Apoptosis may either occur concurrently or precede the process of necrotic cell death (1). Unlike necrosis, apoptosis is associated with a genetically programmed cascade of intracellular events and may be amenable to intervention. Spontaneous recovery from an active cell death program has been reported in the nematode Caenorhabditis elegans, and the recovery may occur even after the shrinkage of the cells and activation of CED-3, a caspase-3 homolog (2). Although no experimental evidence is available, a similar recovery has been hypothesized in mammalian cell systems (3), and the use of caspase inhibitors in myocardial ischemia has resulted in the substantial salvage of cardiomyocyte death. The use of caspase inhibitors is associated with the reduction in the extent of both apoptosis and necrosis and suggests that these 2 components of cell death are interrelated (4). Thus, the feasibility of the noninvasive detection of apoptosis should help maximize the potential of salvage.
Caspase-3 activation during mammalian cell apoptosis is associated with phosphatidylserine expression on apoptotic cells (5). Annexin-A5 (AA5), which has a nanomolar affinity for binding to phosphatidylserine, has been used for the in vitro (6,7) and in vivo (8,9) detection of apoptosis. Radiolabeled AA5 has been successfully used, both clinically and experimentally, for the localization of apoptosis in the myocardial infarcts. This study evaluated whether apoptosis was initiated in brief episodes of myocardial ischemia and whether AA5 would be useful for the detection of ischemia. For this purpose, we performed molecular imaging with 99mTc-labeled AA5 in an experimental rabbit model of brief coronary occlusion. Because myocardial ischemia is a brief event, it was important to define the time window after the initial ischemic insult for the possible detection of apoptosis by imaging. As such, for the characterization of the length and duration of phosphatidylserine exposure we used AA5 labeled with biotin or fluorophores in mice after brief occlusion of a coronary artery.
MATERIALS AND METHODS
Preparation of AA5 Probes for In Vivo Use
Human AA5 was produced by expression in Escherichia coli (10). The recombinant AA5 retained its phosphatidylserine binding activity equivalent to the native AA5. In addition to the AA5, 2 mutants were used for the control experiments, one that had lost its phosphatidylserine binding capacity and another that did not internalize after surface phosphatidylserine binding. The cDNA of AA5 mutant M1234 (E72D, D144N, E228A, and D303N) (a kind gift by Dr. Françoise Russo-Marie) has 4 defective calcium binding sites (11). The AA5 mutant M23 was generated by subcloning the mutations D144N and E228A into the AA5 cDNA. The recombinant AA5 and the mutants M23 and M1234 were labeled with either Oregon Green or Alexa 568–succinimidylester according to the manufacturer's protocol (Molecular Probes). The labeled proteins, with 1:1 stoichiometry, were purified from the mixtures by MonoQ chromatography with Acta Explorer (Amersham-Pharmacia). In addition, hydrazinonicotinamide-derivatized AA5 was prepared as previously described, without affecting phosphatidylserine binding activity, for radiolabeling with 99mTc (12). The radiolabeled material retained specific activity, ranging from 3,700 to 7,400 kBq (100 to 200 μCi) of protein per microgram. The AA5 tracers used in this study are shown in Table 1, and protocols are shown in Table 2.
Recombinant AA5 Tracers
Experimental Protocols and Conclusions
Surgical Induction of Brief Ischemia in Rabbits and Mice
Myocardial ischemia and reperfusion models were produced in rabbits and mice as reported previously (13,14). In the New Zealand White rabbits, the heart was exposed by mid sternotomy. Brief myocardial ischemia was produced by ligation of the lateral branch of the left coronary artery for 10 min (I-10), followed by reperfusion for 180 min (R-180). The myocardial infarction model was created for comparison to brief ischemia by a 40-min occlusion of the coronary artery, followed by reperfusion for 180 min (I-40/R-180) (positive control). In addition, sham-operated animals were used as negative controls for imaging experiments.
In the mouse model, the heart was exposed by left thoracotomy. The left coronary artery was ligated for 5 min (I-5) and reperfusion was established for 90 min (R-90), 3 h (R-180), 6 h (R-360), or 24 h (R-24 h) (Table 2). Ischemia and reperfusion were performed once only for all experiments, and no preconditioning or recurrent ischemia experiments were undertaken. For positive control, infarction was produced by coronary arterial occlusion for 30 min with reperfusion times of 90 min and 24 h (I-30/R-90 or I-30/R-24 h). In a subset of animals, pan-caspase inhibitor zVAD-fmk (3 mg/kg; BIOMOL) was injected through an intravenous line into the jugular vein 10 min before ischemia to assess whether the cascade of apoptosis could be aborted.
Imaging of Rabbit Myocardial Ischemia with 99mTc-AA5 and Pathologic Characterization
To investigate the potential of imaging myocardial ischemia, 99mTc-labeled AA5 was intravenously administered to the rabbits subjected to brief myocardial ischemia (I-10/R-180; n = 6). Concurrent studies were performed in the prolonged ischemia (I/R-40/180; n = 6) model for positive controls and sham-operated (I-0/R-0; n = 4) hearts for negative controls. 99mTc-AA5 (30–40 μg of AA5/kg; 444 ± 37 MBq of 99mTc [12 ± 1 mCi]) was administered at the time of reperfusion through the marginal vein of the ear. Animals were killed after 3 h with an overdose of pentobarbital, and explanted hearts were subjected to ex vivo planar imaging (X-SPECT; Gamma Medica, Inc.) for 15 min in a 128 × 128 matrix using a 1-mm pinhole collimator. Quantitative uptake of 99mTc-AA5 was determined by well-type γ-counter (PerkinElmerWallac Inc.) and reflected as percentage injected dose per gram (%ID/g). Tissue from the ischemic zone and remote myocardium were subjected to ultracentrifugal isolation of the cell membrane, cytoplasm, and other subcellular organelles to localize the 99mTc-AA5 uptake, based on the methodology described previously (14). In addition to subcellular analysis, myocardial tissue sections from the ischemic (or infarcted) and remote myocardial regions were stained by hematoxylin and eosin for standard histology. Tissue sections were also processed for the identification of apoptosis by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) (in situ cell death detection kit, POD; Roche) as described previously (15).
Targeting of Phosphatidylserine Exposure in Murine Models of Ischemia and Pathologic Characterization
For the characterization of surface phosphatidylserine exposure in response to ischemia, unlabeled recombinant human AA5 or fluorescent recombinant human AA5 was administered through a line inserted into the jugular vein of mice. The data were collected for 3 mice in each experimental group. To confirm the phosphatidylserine exteriorization, AA5 was used in I-5/R-90 and I-5/R-24 h mouse models. To assess the length of time for which the phosphatidylserine was expressed, I-5/R-90, I-5/R-180, I-5/R-360, I-5/R-24 h, I-30/R-90, and I-30/R-24 h models were studied with AA5–biotin injected at the onset of reperfusion. Conversely, to evaluate the fate of expressed phosphatidylserine, AA5–Oregon Green (2.5 mg/kg) and AA5–Alexa 568 (2.5 mg/kg) were used at 2 different time points after reperfusion, at the onset of reperfusion and 10 min before sacrifice. The hearts were removed and snap-frozen in liquid nitrogen for the histologic localization of exogenously administered AA5 and apoptosis. To study the internalization of AA5 occurring in vivo, a mutant of AA5 M23–Alexa 568, which binds to phosphatidylserine but does not induce its internalization, was injected before reperfusion. To test whether the internalization of AA5 was phosphatidylserine-dependent, a mutant of AA5 M1234–Oregon Green, which does not bind to phosphatidylserine, was injected before reperfusion. Both experiments with mutant AA5 were performed in the I-5/R-90 model (n = 3 each).
For the localization of the fluorescent AA5, frozen 7-μm tissue sections were obtained, dried overnight, and mounted in 4,6-diamino-2-phenylindole–containing medium (Vector Laboratories) and examined by fluorescence microscopy. Intravenously administered human recombinant AA5 was tracked by immunohistochemical staining of frozen tissue sections. The tissue sections were dried overnight and fixed in dry acetone for 10 min at room temperature. After the blocking of endogenous peroxidase, sections were stained with an antibody against AA5 (rabbit polyclonal antihuman AA5; Hyphen Biomed). AA5 was detected by polyclonal horseradish peroxidase–labeled goat antirabbit antibody (Powervision peroxidase detection system; Immunovision Technologies). In contrast, biotin–AA5 was tracked by direct avidin/biotin conjugate peroxidase staining without the need for anti-AA5 antibody. Frozen tissue sections were also stained with hematoxylin and eosin for standard histology and stained by TUNEL (in situ cell death detection kit, POD; Roche) for the identification of apoptosis, as described previously (15).
RESULTS
Radionuclide Imaging of Brief Myocardial Ischemia Is Feasible
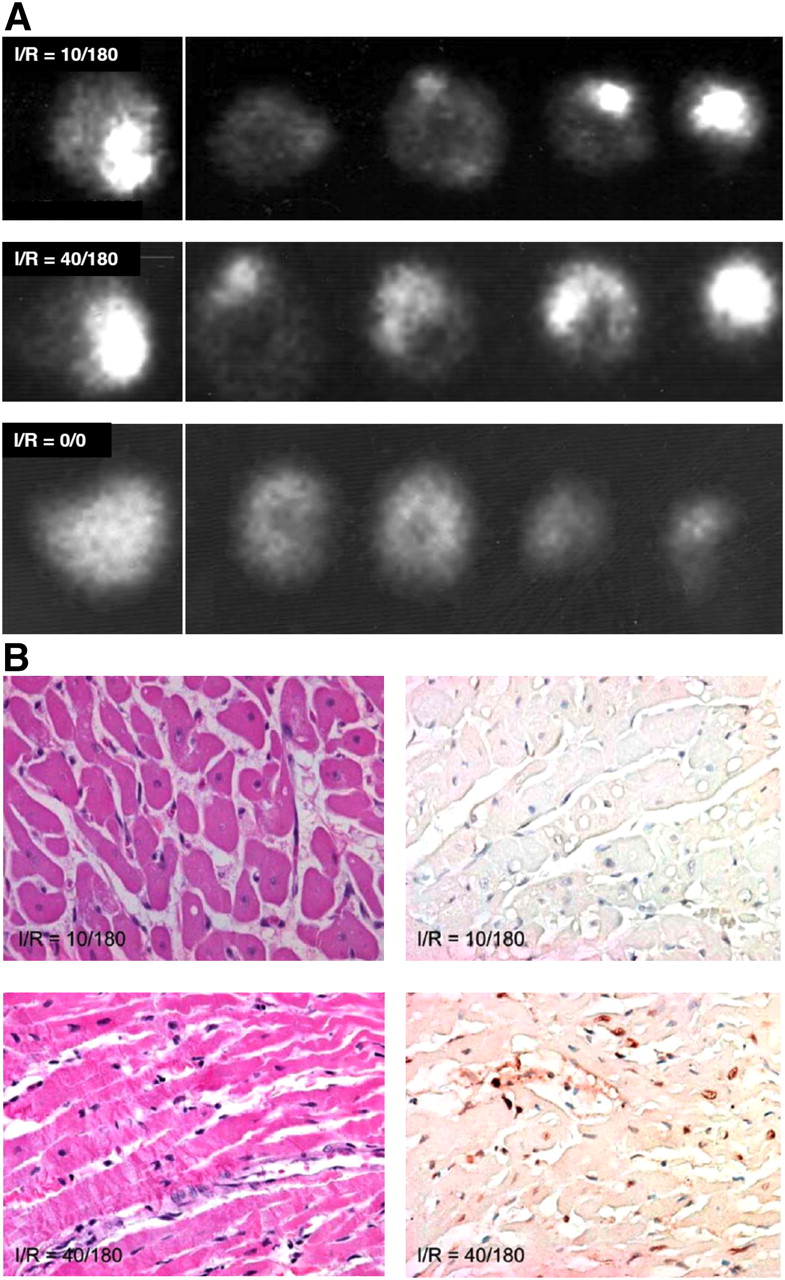
To explore whether phosphatidylserine exposure could be used potentially to detect recent ischemic events, 99mTc-AA5 was administered intravenously in the I-10/R-180 rabbit model at the time of reperfusion, and imaging of the explanted hearts was performed after 3 h (14). Significant radiolabeled 99mTc-AA5 uptake in the zone of initial ischemia was observed (Fig. 1A). 99mTc-AA5 uptake in the ischemic region was 9 ± 3-fold higher than in the normal myocardium (0.27 ± 0.16 %ID/g vs. 0.03 ± 0.01 %ID/g). Further, at least 85% of the radioactivity was recovered from the ultracentrifugally isolated cytoplasmic and organelle compartment of the cells, suggesting that 99mTc-AA5 bound to phosphatidylserine was internalized. Although enhanced uptake of 99mTc-AA5 had occurred in the ischemic regions, no histologic evidence of apoptosis or necrosis in the hearts was observed (Fig. 1B). Prolonged ischemia and reperfusion (I-40/R-180) was associated with prominent 99mTc-AA5 uptake (0.55 ± 0.3 %ID/g) (Fig. 1A), and apoptosis and necrosis (Fig. 1B). The 99mTc-AA5 uptake in the sham-operated animals (0.042 ± 0.012 %ID/g) was similar to the uptake in the remote nonischemic myocardium. The data from the rabbit model indicated that the phosphatidylserine exposure occurred after a brief episode of ischemia and may serve as a marker of ischemic memory in the myocardium. Data also suggested that 99mTc-AA5 accumulated intracellularly in ischemic cells. These data confirmed our previous in vitro observations (16) that AA5 mediated internalization of phosphatidylserine-positive membrane patches. To characterize the phosphatidylserine exposure observed in the rabbit imaging experiments, multiple sets of mouse experiments were conducted.
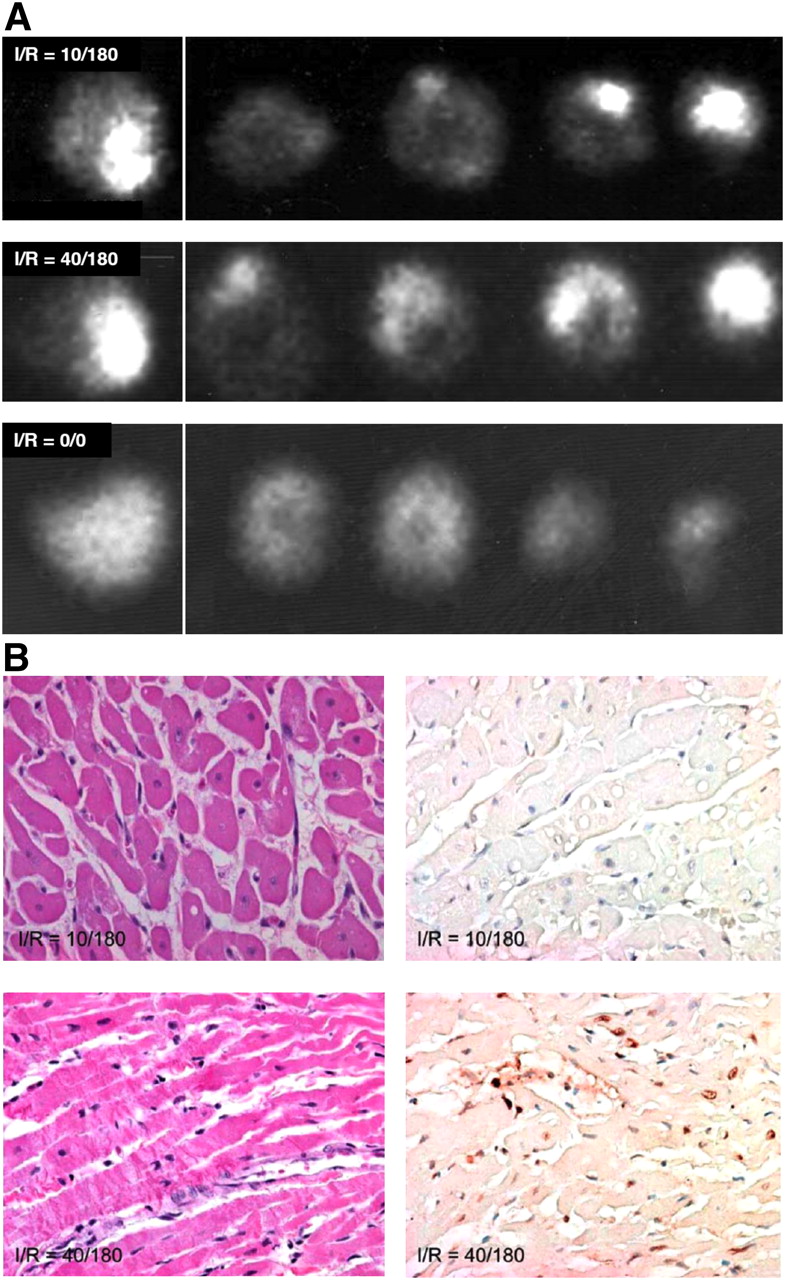
99mTc-AA5–based imaging of externalized phosphatidylserine in ischemic myocardium. (A, top) In I-10/R-180 model in rabbit, radiolabeled AA5 clearly delineates ischemic zone with 9-fold higher uptake than in remote myocardium. Uptake is seen in γ-image of whole heart and slices. (B, top) On tissue sections, no necrosis (left) or apoptosis (right) was observed in ischemic zone. In prolonged ischemic model (I-40/R-180) (A, middle), intense 99mTc-AA5 uptake is seen; pathologic characterization reveals (B, bottom) extensive evidence of necrosis (left) and apoptosis (right). Necrosis is represented by contraction bands and apoptosis by TUNEL staining. No 99mTc-AA5 uptake is seen in sham-operated animals (A, bottom). This experiment provides preliminary data generating hypothesis that hot-spot imaging of ischemia by radiolabeled AA5 is feasible.
Phosphatidylserine Exposure Occurs After Brief Ischemia But Not Cell Death
To establish the phosphatidylserine exposure characteristics after brief episodes of ischemia observed in the rabbit model, a series of experiments was performed in mice. In the first set of experiments, I-5 hearts received human recombinant AA5 before coronary occlusion, and its localization was traced by immunoperoxidase staining using anti-human AA5 antibody in myocardial sections (13). AA5-positive cardiomyocytes were observed in the ischemic area of the hearts in I-5 animals that were subjected to reperfusion for 90 min and 24 h (Fig. 2). Cardiomyocytes of the nonischemic area and cardiomyocytes from sham-operated animals did not bind AA5. The AA5 was localized to the intracellular compartment. On TUNEL staining, only rare apoptotic cells were observed in the ischemic area of I-5/R-90 animals (Fig. 2). These findings suggested that the apoptosis program was initiated, phosphatidylserine was exposed that was available for targeting by AA5, and AA5 was internalized. However, because TUNEL-positive cells were not seen even after phosphatidylserine exposure had occurred, the apoptotic process was aborted on reperfusion after brief ischemia. As compared with the I-5/R-90 ischemia model, significantly more myocytes were AA5-positive in I-30/R-90 mice, and many AA5-positive cardiomyocytes demonstrated TUNEL-positive nuclei. This set of experiments confirmed the data obtained in the rabbit model that the phosphatidylserine externalization had occurred after brief episodes of ischemia and that AA5 ended up being internalized.
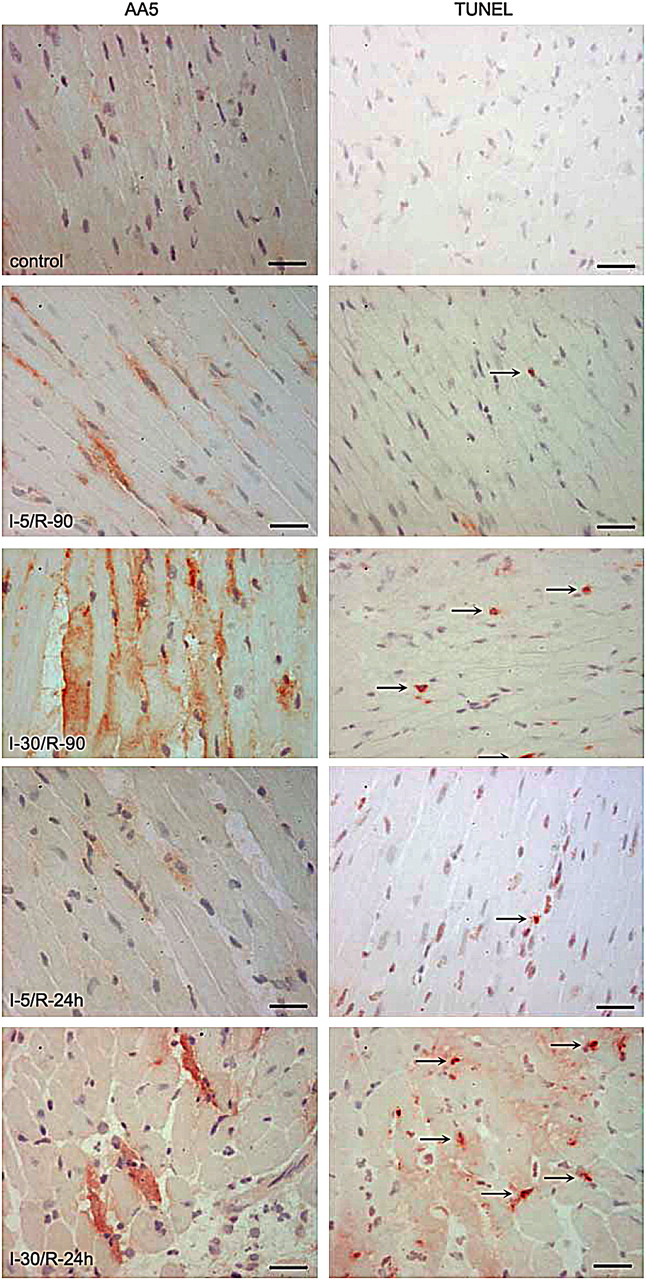
Cardiomyocytes expose phosphatidylserine after brief ischemia. Immunostaining for AA5 (×400) (left) and apoptosis detection with TUNEL staining (×400) (right) was performed on frozen tissue sections of mouse hearts subjected to different ischemia/reperfusion (I/R) time points. Scale bars represent 25 μm. AA5 was injected before coronary occlusion in I-5/R-90, I-5/R-24 h, I-30/R-90, and I-30/R-24 h mouse models. Note intracellular location of AA5, more so in prolonged ischemia. Rare apoptotic cells are seen in brief ischemia, unlike abundant apoptotic cells in infarcts. In I-5/R-90 model, preadministration of pan-caspase inhibitor zVAD-fmk prevents phosphatidylserine exteriorization. Arrows (right panels) refer to TUNEL-positive nuclei.
Apoptosis assays in vitro have shown that phosphatidylserine exposure is downstream of critical events in the cell death program such as the release of cytochrome c from mitochondria and the activation of caspase-3 (17,18). To evaluate the relationship between brief ischemia and caspase activation, immunoblotting of caspase-3 was undertaken in I-5/R-90 hearts. The presence of 12- and 17-kDa split active fragments of caspase-3 indicated the activation of caspase-3; densitometric analysis confirmed a 4-fold increase in activation of caspase-3 (data not shown). Increased densitometric intensity of activated caspase-3 was seen after longer ischemia.
Phosphatidylserine Is Expressed After Brief Ischemia and Internalized After AA5 Binding
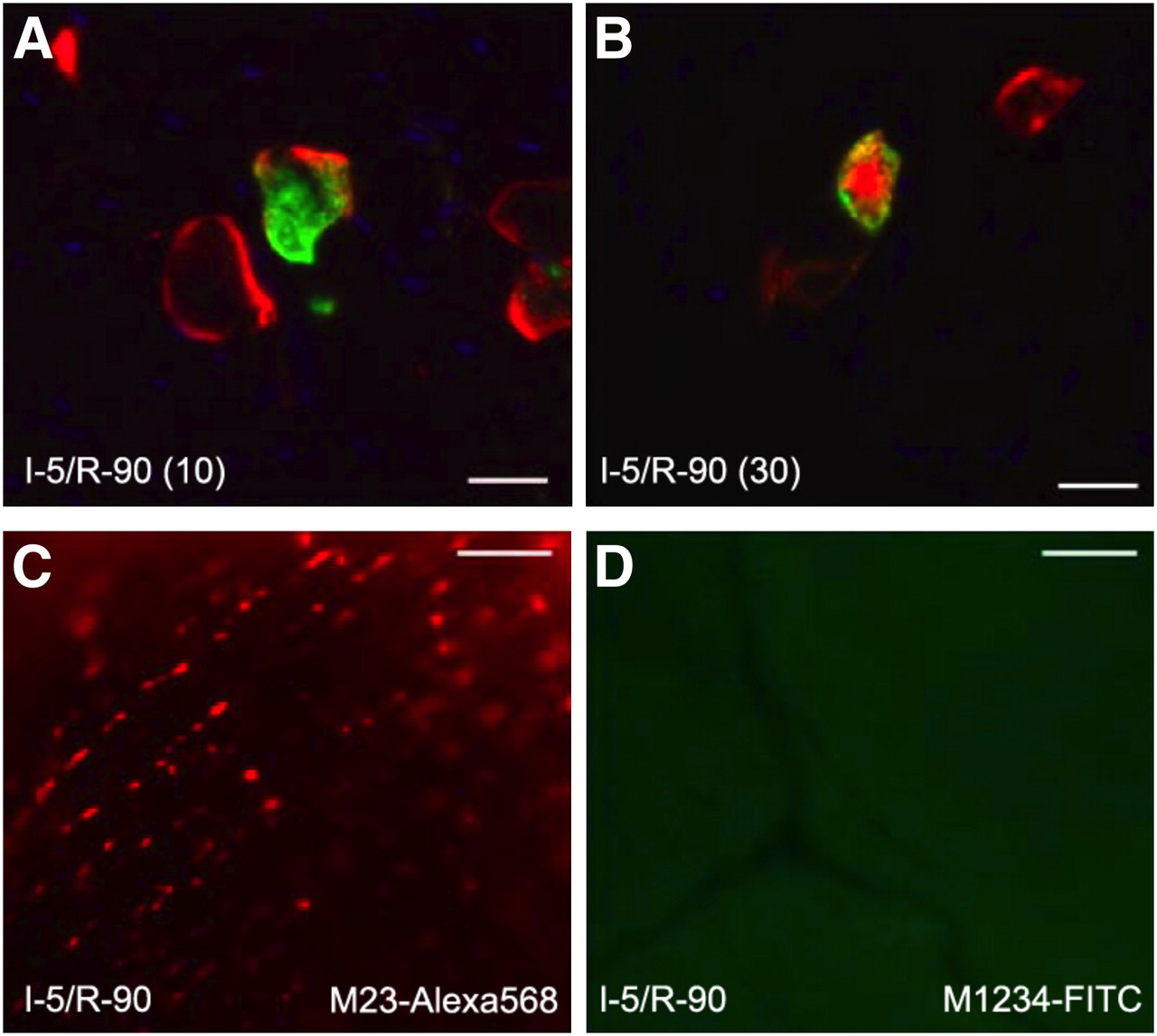
From the rabbit heart ultracentrifugation data and the first sets of mouse experiments, it was presumed that AA5 after targeting of the phosphatidylserine was internalized by the ischemic cells. To evaluate the kinetics of the internalization process in vivo after ischemia or reperfusion injury, we performed the second set of experiments. For this purpose, we injected AA5–Oregon Green at the beginning of reperfusion in the I-5/R-90 model. As expected, the green AA5 was localized intracellularly. In two separate groups of these I-5/R-90 animals that had already received AA5–Oregon Green at the start of reperfusion, AA5–Alexa 568 was injected either 10 or 30 min before the end of the reperfusion (or sacrificing the animal). AA5–Alexa 568 injected 10 min before sacrifice showed binding almost exclusively to the cardiomyocyte membrane; green AA5 was entirely intracellular (Fig. 3A). However, the injection of AA5–Alexa 568 30 min before sacrifice showed colocalization of AA5–Alexa 568 with AA5–Oregon Green within the cytoplasmic compartment of the cell (Fig. 3B), indicating that after AA5 targeting of the phosphatidylserine on the cell surface, the phosphatidylserine–AA5 complex was internalized between 10 and 30 min. These in vivo data confirmed our previously reported in vitro data that AA5 mediates the internalization of phosphatidylserine-positive membrane patches as endocytic vesicles, which detach from the plasma membrane and follow cytoskeleton-dependent trafficking into the cytosol (16).

Phosphatidylserine and AA5 are internalized in cardiomyocytes. (A) Tissue section (×400) of mouse hearts injected with AA5–Oregon Green at beginning of reperfusion and AA5–Alexa 568 10 min before end of reperfusion episode in I-5/R-90 model show surface binding and internalization of AA5–Oregon Green. In contrast, only surface binding but no internalization of AA5–Alexa 568 is seen. Parenthesis entry of 10 following I-5/R-90 denotes time of AA5–Alexa 568 administration before the end of R-90 reperfusion period. (B) Injection of AA5–Alexa 568 30 min before completion of reperfusion showed colocalization of AA5–Alexa 568 with AA5–Oregon Green within cytoplasmic compartment of cell. Scale bars represent 25 μm. (C) Ex vivo image (×160) of heart of mouse injected with M23–Alexa 568, before 5-min ischemia and 90-min reperfusion (I-5/R-90), demonstrates binding to cell membrane but no internalization. (D) Ex vivo image (×160) of heart of mouse injected with M1234–FITC before I-5/R-90 shows neither binding nor internalization. Scale bars represent 25 μm.
AA5 Binding to Phosphatidylserine Is Necessary for Phosphatidylserine Internalization
To demonstrate that the AA5 targets phosphatidylserine and is subsequently internalized, we conducted the third set of experiments with 2 fluorescently labeled mutant species of AA5. The AA5 mutant M23, by prediction from the available structural data (19), binds to phosphatidylserine but lacks the ability to crystallize on the phosphatidylserine-expressing membrane surface, the step necessary before internalization. M23–Alexa 568 was injected before reperfusion in the I-5/R-90 model. The mutant M23 was observed targeting the cell membrane of the cardiomyocytes in the ischemic zone but was not seen intracellularly (Fig. 3C). Furthermore, to test the phosphatidylserine dependence of the internalization process in vivo, we used yet another AA5 mutant M1234 that lacked the ability to bind to phosphatidylserine. M1234–fluorescein isothiocyanate (FITC) was injected before reperfusion in the I-5/R-90 model. The mutant M1234 did not bind to the cardiomyocyte surface, nor was fluorescence observed intracellularly (Fig. 3D).
Continuous Phosphatidylserine Externalization–Internalization Represents Ischemic Memory
The internalization experiments of the second and third sets raised another important possibility. If phosphatidylserine exposure occurred as early as 5 min after ischemia and internalized within 30 min after AA5 binding, then cell surface binding of AA5 injected at 80 min of reperfusion (10 min before the end of a 90-min reperfusion) would suggest an ongoing, continuous, and fresh expression of phosphatidylserine after a solitary initial episode of brief ischemia. To evaluate this possibility, a fourth and fifth sets of experiments were performed with AA5 administration at various time points after brief ischemia up to 24 h.
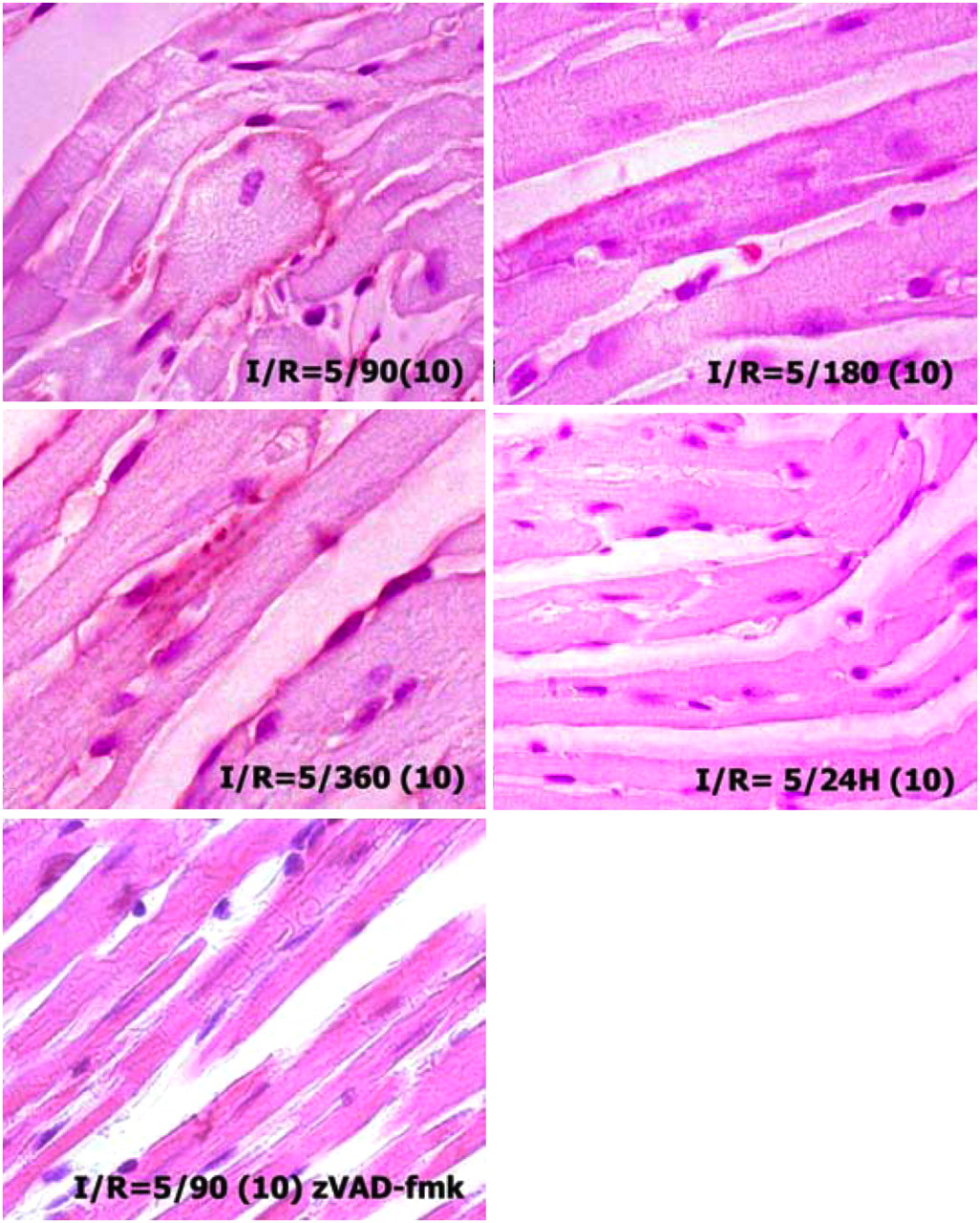
To demonstrate the length of phosphatidylserine exposure after brief ischemic insult, in the fourth set of experiments, I-5 animals were reperfused for 90, 180, and 360 min and 24 h. AA5–biotin was injected 10 min before sacrifice. Phosphatidylserine exposure was detectable up to 6 h after the ischemic insult (Fig. 4). No AA5 uptake was observed in the area at risk at 24 h of reperfusion. It seems that as long as apoptotic machinery remains active after the brief ischemic episode, phosphatidylserine continues being exposed and is amenable to removal from the cell surface, if AA5 is available. This concept was further substantiated by the pretreatment of animals with zVAD-fmk before the induction of brief ischemia (Fig. 4). In the pretreated I-5/R-90 mice, no AA5 binding was seen in these animals, suggesting that the inhibition of caspase successfully prevented phosphatidylserine externalization in response to brief ischemic insult. All experiments indicate that cardiomyocytes recovered and phosphatidylserine exposure resolved if reperfusion was restored in a timely manner or if caspase inhibition was applied.
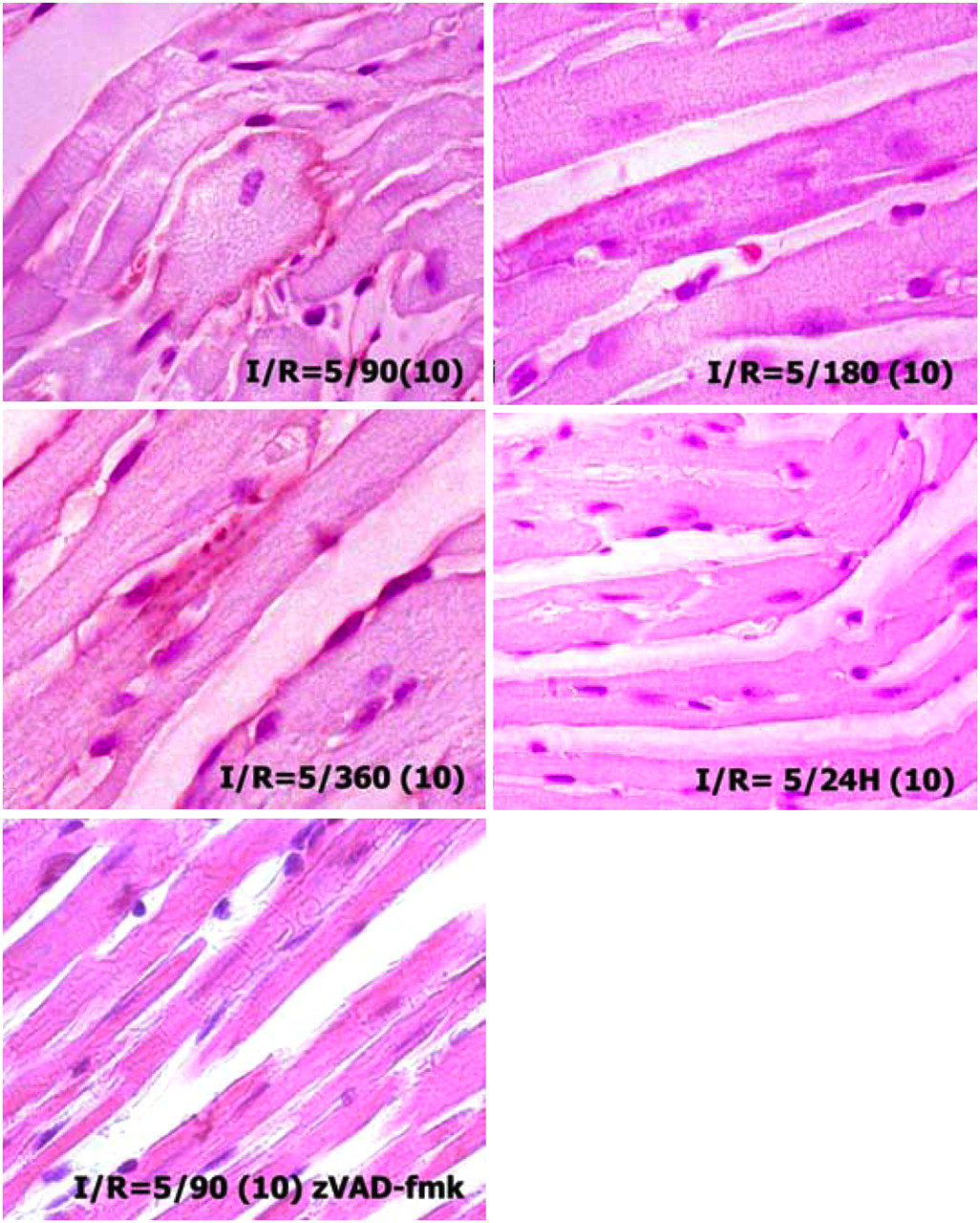
Cardiomyocytes expose phosphatidylserine up to 6 h after brief ischemia. Mouse hearts subjected to 5 min of ischemia and 90, 180, and 360 min and 24 h of reperfusion received AA5–biotin injected 10 min before sacrifice (parentheses entry). Cardiomyocytes in ischemic zone demonstrate AA5 binding up to 6 h; cardiomyocytes in I-5/R-24 h do not bind AA5–biotin (25 mg/kg) when injected 10 min before sacrifice. zVAD (3 mg/kg) was injected intravenously before application of ischemia in I-5/R-90 model.
To further demonstrate whether phosphatidylserine exposure was a continuous process and the fresh phosphatidylserine was available until 6 h after the initial brief ischemic episode, we again subjected I-5 mice to R-90, R-360, and R-24 h reperfusion (Fig. 5). AA5–Oregon Green was administered at the onset of reperfusion; green fluorescence was exclusively localized intracellularly. The administration of AA5–Alexa 568 10 min before sacrifice revealed that 90 min and 6 h after the brief ischemic insult cardiomyocytes with internalized AA5–Oregon Green still bound AA5–Alexa 568 on their cell surface. When AA5–Alexa 568 was injected 24 h after brief ischemia, the cardiomyocytes with internalized AA5–Oregon Green did not bind AA5–Alexa 568, indicating that these cardiomyocytes had already restored phosphatidylserine asymmetry. Although enhanced uptake of AA5 had occurred in the ischemic regions, no histologic evidence of apoptosis or necrosis in the hearts was observed. The data suggested that phosphatidylserine exposure occurred continuously at least up to 6 h after the ischemic episode and was amenable to AA5 targeting. These sets of experiments further highlighted the possibility of phosphatidylserine expression as a sign of ischemic memory.
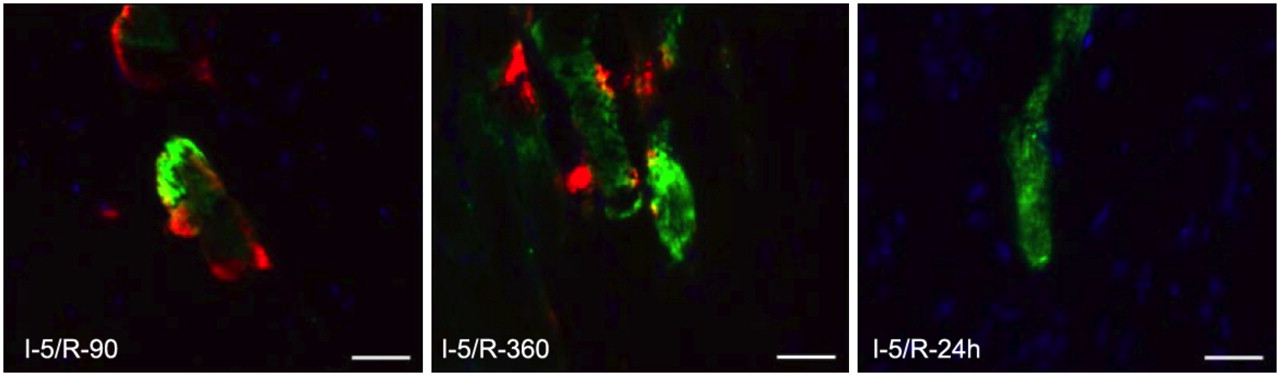
Phosphatidylserine exposure and internalization are transient processes. Tissue sections (×400) of I-5/R-90, R-360, and R-24 h mouse hearts, injected with AA5–Oregon Green at beginning of reperfusion, followed by injection of AA5–Alexa 568 10 min before completion of reperfusion. There is binding of AA5–Alexa 568 to plasma membrane of (internalization) AA5–Oregon Green–positive cells after 90 min and 6 h of reperfusion. After 24 h of reperfusion, AA5–Oregon Green–positive cells do not bind AA5–Alexa 568. This experiment suggests that after brief episode of ischemia, phosphatidylserine exposure, although transient, is continuously exposed for 6 h. Scale bars represent 25 μm.
DISCUSSION
Our data show that brief episodes of ischemia result in phosphatidylserine exposure that normally lasts for at least 6 h after the ischemic insult and offers a target for 99mTc-AA5 for after-the-fact imaging of ischemia (ischemic memory). Phosphatidylserine exposure occurs continuously for the duration because the AA5–phosphatidylserine complex is continuously removed from the cell surface by internalization and new phosphatidylserine is exteriorized. Because the AA5–phosphatidylserine complex is internalized, the signal stays in situ after 99mTc-AA5 administration and may concentrate radiolabeled AA5 in the ischemic cells. This explains the possibility of imaging even if phosphatidylserine exposure may not be extensive or intense after brief ischemia. These data deliver another important message. Because phosphatidylserine exposure and caspase activation are the hallmarks of apoptosis (20), it appears that ischemia-induced apoptosis in cardiomyocytes is interrupted by reperfusion such that the cardiomyocytes survive. An escape from activated apoptosis has been reported previously in Caenorhabditis elegans (2,3). During developmental apoptosis, engulfment genes act in concert with CED-3 in cells destined to die in the anterior pharynx. In the absence of activation of the engulfment machinery, such cells show all the features of apoptosis, but cell death does not occur; these cells recover completely and remain viable thereafter. Our data confirm recovery from apoptosis in mammalian cells in vivo, despite activation of one of the key executioner caspases, caspase-3. Reversible phosphatidylserine exposure not associated with the execution of apoptosis has been observed in vitro with myocytic cell lines (21,22). Other examples include the activation of B-cell lymphoma cells, which in response to cross-linking of the membrane immunoglobulin receptor stained positive for AA5 but were able to restore their phospholipid asymmetry and resume growth on withdrawal of the stimulus (23). It seems that the phenomenon of recovery from apoptosis may be evolutionarily conserved. However, in contrast to C. elegans the absence of engulfment gene activity may not be critical in the recovery of cell death in the heart, because phagocytes enter the ischemic zone only after 24 h. An interesting aspect of the binding of AA5 to externalized phosphatidylserine is that this may offer the cells protection from being recognized by “professional” phagocytes, because externalized phosphatidylserine is necessary for the engulfment of apoptotic cells and serves as an “eat me” signal (20). Clinical studies have shown that plasma AA5 levels are elevated in patients with acute myocardial infarction (24). These data suggest that AA5 may serve as an acute-phase reactant, which may save cells that are susceptible to engulfment. It is also justified to hypothesize that exogenously administered AA5 may accelerate the internalization of externalized phosphatidylserine. The data on the internalization of phosphatidylserine in the presence of AA5 indicate that internalization is not the result of the inhibition of scrambling of phosphatidylserine molecules but results from invagination and vesicle formation of the patches of plasma cell membrane that expose phosphatidylserine (16). Together, our data suggest a novel model of phosphatidylserine exposure and internalization wherein phosphatidylserine exposure is continuously driven by caspase activation and phosphatidylserine internalization is mediated by AA5-aided formation of the endocytic vesicle (16). Cytoplasmic uptake of exogenous AA5 has been previously reported in animal studies of apoptotic cell death during embryogenesis (25), tooth development (26), and stroke (27). The investigators in these studies concluded that the intracellular uptake of exogenous AA5 was due to the ingestion of apoptotic vesicles coated with AA5 by neighboring cells. This mechanism in our model seems unlikely given the lack of apoptotic or necrotic cells in the hearts of animals subjected to brief ischemia. Our unpublished in vitro data further indicate that the perinuclear vesicles in an increased hypercalcemic intracellular milieu may prevent cytochrome c release from mitochondria, supporting the antiapoptotic role of AA5 at least in ischemic heart muscle cells. The reversible phosphatidylserine exposure constitutes an attractive target for the noninvasive detection of myocardial ischemia as a hot-spot imaging agent in addition to traditional stress myocardial perfusion imaging. Furthermore, persistence of phosphatidylserine externalization for at least 6 h after ischemic stress may allow retrospective diagnosis of cardiac chest pain and support the concept of ischemic memory. This concept would be somewhat similar to the metabolic stunning of myocytes after a myocardial ischemic episode, wherein fatty acid use is interrupted in favor of glucose use. These 2 metabolic alterations have been well characterized by the absence of 123I-BMIPP uptake (28) and enhanced 18F-FDG uptake (29) in the ischemic myocardial territories.
CONCLUSION
Our data demonstrate that caspase-3 is activated and phosphatidylserine is externalized in mammalian cells in response to brief ischemia of the heart. Cardiomyocytes destined to die recover from the initiation of the cell death program on restoration of blood flow. The transient exposure of phosphatidylserine by cardiomyocytes recovering from apoptosis may be exploited for diagnostic imaging. Further, AA5 binding to phosphatidylserine leads to the internalization of AA5 into vesicles. Although logically protective, the role of AA5–phosphatidylserine internalization remains to be understood.
Acknowledgments
The study was supported in part by Joseph DiPalma and Isadore Brodsky Research Funds to Dr. Narula; Dutch Heart Foundation grants NHS2000-D35 and 98.195; Dutch Science Foundation (NWO) grants 902-26-184, 014-80-103, and 912-03-013 to Dr. Reutelingsperger; and the Wynand Pon Foundation grant to Dr. Dumont.
Footnotes
-
↵* Contributed equally to this work.
-
COPYRIGHT © 2010 by the Society of Nuclear Medicine, Inc.
References
- Received for publication July 13, 2009.
- Accepted for publication October 22, 2009.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- A Molecular Signature for Anastasis, Recovery from the Brink of Apoptotic Cell Death
- Targeted Imaging for Cell Death in Cardiovascular Disorders
- Molecular Imaging in Drug Discovery and Development
- A molecular signature for anastasis, recovery from the brink of apoptotic cell death
- Cardioprotective Effects of HSP72 Administration on Ischemia-Reperfusion Injury
- Molecular Imaging: From Deep Pearl Diving to Enlightenment
- Fluorescence Tomography of Rapamycin-Induced Autophagy and Cardioprotection In Vivo
- Ischemic Episode and Hanging on to a Painful Memory...
- Dual Manganese-Enhanced and Delayed Gadolinium-Enhanced MRI Detects Myocardial Border Zone Injury in a Pig Ischemia-Reperfusion Model
- The Extent of Coronary Atherosclerosis Is Associated With Increasing Circulating Levels of High Sensitive Cardiac Troponin T