


Abstract
Tumor receptors play an important role in carcinogenesis and tumor growth and have been some of the earliest targets for tumor-specific therapy, for example, the estrogen receptor in breast cancer. Knowledge of receptor expression is key for therapy directed at tumor receptors and traditionally has been obtained by assay of biopsy material. Tumor receptor imaging offers complementary information that includes evaluation of the entire tumor burden and characterization of the heterogeneity of tumor receptor expression. The nature of the ligand–receptor interaction poses a challenge for imaging—notably, the requirement for a low molecular concentration of the imaging probe to avoid saturating the receptor and increasing the background because of nonspecific uptake. For this reason, much of the work to date in tumor receptor imaging has been done with radionuclide probes. In this overview of tumor receptor imaging, aspects of receptor biochemistry and biology that underlie tumor receptor imaging are reviewed, with the estrogen–estrogen receptor system in breast cancer as an illustrative example. Examples of progress in radionuclide receptor imaging for 3 receptor systems—steroid receptors, somatostatin receptors, and growth factor receptors—are highlighted, and recent investigations of receptor imaging with other molecular imaging modalities are reviewed.
Cancer therapy is becoming increasingly directed and specific, taking advantage of biologic targets that are uniquely expressed or markedly overexpressed in tumors. Tumor receptors have been some of the earliest targets for cancer therapy, with notable successes in the treatment of endocrine-related cancers such as breast, prostate, and thyroid cancers (1–3). Advances in molecular cancer biology have revealed an ever-increasing number of tumor targets, many of which are receptors, such as the epidermal growth factor (EGF) receptor (EGFR) (4). The ability to measure the expression of tumor receptors is essential for selecting patients for receptor-targeted therapy (2). Although this information traditionally has been obtained by in vitro assay of biopsy material, recent studies have highlighted the complementary value of tumor receptor imaging for measuring regional tumor receptor expression, which can be quite heterogeneous.
Tumor receptor imaging emphasizes important emerging themes in molecular imaging: characterizing tumor biology, identifying therapeutic targets, and delineating the pharmacodynamics of targeted cancer therapy (5,6). The advantages of imaging include noninvasiveness, the ability to measure receptor expression for the entire disease burden and thereby to avoid the sampling error that can occur with heterogeneous receptor expression, and the potential for serial studies of the in vivo effects of a drug on the target. A very practical consideration is that imaging can assess receptor expression at sites that are challenging to sample and assay, such as bone metastases. This review discusses receptor pharmacology, the biology of tumor receptors, and special considerations for tumor receptor imaging, with the estrogen receptor (ER) as an illustrative example. This discussion is followed by highlights of recent work in the imaging of steroid receptors, somatostatin receptors (SSTRs), and growth factor receptors, as examples of 3 different types of tumor receptors. These topics were selected from a broad range of investigations in tumor receptor imaging (Table 1).
Selected Examples of Tumor Receptor Imaging Agents
RECEPTOR PHARMACOLOGY
Common to all receptors is the interaction of a ligand and the receptor, in which specific binding of the ligand to the receptor results in downstream biochemical or physiologic changes (7,8). Ligand–receptor binding can activate or inhibit downstream processes through a variety of mechanisms, such as G-protein activation (e.g., PAR1), tyrosine kinase activation (e.g., EGFR), or activation of transcription (e.g., ER) (9–11). Ligands that cause physiologic changes with receptor binding, typically the naturally occurring ligands, are called agonists. Ligands that bind to the receptor and block the binding of agonists but that do not activate changes are known as antagonists. Drugs directed toward tumor receptor systems most frequently use a receptor antagonist, for example, tamoxifen, which blocks estrogen binding to the ER. Alternatively, some drugs lower the level of an agonist to decrease ligand–receptor interactions, for example, levothyroxine, which suppresses thyroid-stimulating hormone levels in the treatment of thyroid cancer (12).
The ligand–receptor interaction is a bimolecular chemical reaction. The concentration of the receptor is typically quite low; for example, the level of ER expression in breast cancer is in the range of 3–100 fmol per milligram of protein (13). Furthermore, receptor-specific ligands bind to receptors with a high affinity and often with a very low ligand–receptor dissociation rate (7). The combination of a low receptor concentration and a high ligand–receptor affinity leads to a low overall capacity for ligand–receptor binding. This property is helpful for drug therapy, in which the goal is to saturate the receptor with an antagonist to prevent receptor activation by an agonist. However, the high-affinity, low-capacity ligand–receptor binding reaction presents a challenge for imaging in that the number of molecules that can contribute to the specific receptor image is small. Furthermore, nonspecific binding of ligands to plasma proteins and nontarget tissues can limit imaging agent delivery and contribute to nontarget image background. For these reasons, imaging of receptor binding, as opposed to imaging of an enzymatic reaction, such as glucose phosphorylation, in which it is difficult to saturate the uptake mechanism, is challenging. It is important for receptor imaging probes to have very low molecular concentrations. Even small molar quantities of imaging agents may saturate receptors, limiting the ability to visualize receptor expression and increasing the background of nonspecific binding (14). Therefore, molecular imaging of tumor receptors has been mainly confined to radionuclide imaging (PET and SPECT), with which it is possible to generate images with micromolar to picomolar concentrations of imaging probes.
It is important to note that the criteria for a suitable receptor imaging agent are different from those for a receptor-targeted drug. Although selectivity for the drug requires an effect on the tumor in the absence of appreciable toxicity from nontarget tissue drug action, the requirement for high target uptake and low image background in imaging places constraints on radiopharmaceutical selectivity and background clearance that can be even more stringent than those for therapeutic drugs.
The estrogen–ER system is an illustrative example of a receptor system with relevance to cancer (15). The ER is important in female reproductive physiology and is selectively expressed in a variety of normal tissues—most notably, breast, uterus, ovary, bone, and pituitary tissues (9). Estradiol is a naturally occurring agonist ligand for the ER. The molecular mechanism of estradiol action through the ER has been well studied (3,9). Estradiol is lipophilic, allowing access across cell membranes to the ER, a nuclear receptor. The ER has 2 receptor subtypes: ER-α and ER-β. ER-α serves mainly as an activator of downstream events related to breast and female sex organ function. The function of ER-β is less well understood; in some situations, ER-β may inhibit ER-α by forming a heterodimer with ER-α (16). Estradiol binding to ER-α in the nucleus results in dimerization of the receptor and allows interactions with specific DNA sequences known as the estrogen response elements (15), leading to the selective regulation of target gene transcription.
ER activation leads to different physiologic actions in different tissues. Much of the tissue specificity appears to be attributable to coregulators that interact with the ER homodimer and the estrogen response elements and that can affect the pattern of gene transcription (15). In the uterus, estrogens bound to the ER stimulate endometrial growth and are critical in maintaining a functioning uterine–placental unit during pregnancy. Estradiol promotes new bone formation and is important in maintaining bone mineral density, especially in women (9,17). Also, estrogens affect the cardiovascular system, mainly through their beneficial effect on serum lipids. In breast tissue, estradiol promotes ductal epithelial cell proliferation and is a key component stimulating lactation. Estrogens are established growth factors for endometrial and many breast cancers. Over 70% of breast cancers express the ER, and estradiol and other estrogens provide a key stimulus for tumor growth and a target for endocrine system–based therapy (endocrine therapy) (2,3,18–21). Tissue-specific coregulators interact with ligand–ER dimers and may affect the physiologic actions of different ligands when they bind to the ER. For example, drugs known as selective ER modulators (SERMs) exhibit various degrees of either ER agonist or antagonist behavior in different tissues, an effect thought to be based on the differential expression of ER coregulators in different tissues (9).
The circulating levels of agonists for the ER are variable but are tightly regulated in normal human physiology (17). The agonist estradiol has 2 sources: synthesis in the ovary in premenopausal women and conversion from adrenal steroids, mainly through aromatization (and aromatase enzymes) present in a variety of tissues—most notably, fat, breast tissue, and breast cancers (22,23). Premenopausal levels of estradiol vary, depending on the phase of the menstrual cycle, reaching levels as high as 500 pg/mL (1.7 nM) at midcycle (17). In postmenopausal women and men, the levels are generally less than 30 pg/mL (0.1 nM). Estradiol is very lipophilic and is generally present at slightly higher concentrations in tissues with higher fat contents, providing an opportunity for nonspecific uptake. Circulating estradiol is mainly protein bound. This binding occurs with a high affinity but a low capacity to sex hormone–binding globulin (SHBG or SBP) and with a low affinity but a high capacity to albumin (24,25). Much of circulating estradiol is bound to SHBG, and the remainder is bound to albumin (24). Binding to both SHBG and the ER appears to be important for normal estrogen physiology and also appears to be important for ER imaging agents (25–27). One of the physiologic roles of SHBG appears to be the regulation of estrogen metabolism (28). Estradiol is highly metabolized in the liver to estrone and conjugates of both estradiol and estrone (29) and enters a cycle of enterohepatic circulation (30–32). Binding to SHBG, extraction by the liver, and reabsorption of conjugated estrogens in the small intestine all play important roles in regulating estradiol levels in normal physiology (24,28).
Considerations for imaging of the ER include the need for low injected molar doses to remain below physiologic levels (typically 30 pg/mL or higher), the need for imaging agents to bind to both the ER and the transport protein (SHBG), normal routes of estrogen metabolism and excretion, and nonspecific binding in the blood and lipophilic tissues (33). These considerations are discussed in more detail in the following sections as we examine other aspects of tumor receptor imaging. Overall, the complex nature of ER physiology emphasizes the need for a detailed understanding of receptor pharmacology and physiology in developing tumor receptor imaging approaches.
BIOLOGY AND PHYSIOLOGY OF TUMOR RECEPTORS
The biologic role of most receptor systems important in cancer is derived from their role in the tissue of cancer origin. In general, tumor receptors are expressed in the parent cell lineage and have an established physiologic function. For example, ER expression is essential to the function of normal mammary gland epithelial cells (9,15). When estradiol binds to ductal epithelial ER, it stimulates mammary gland growth, maintenance, and physiologic function (9,15). Many tumor receptors also play an important role in promoting carcinogenesis or tumor growth, as is the case for steroid receptors in breast and prostate cancer (34). The dependence on the receptor pathway for tumor growth makes the receptor an ideal target for therapy, because interruption of the receptor-initiated signal will result in a cessation of tumor growth and often tumor cell death (3,10).
For many tumor receptors expressed in the normal parent cell lineage, the receptors are expressed at levels comparable to those in normal cells, and tumor growth stimulation occurs in concert with other factors contributing to the dysregulation of tumor cell growth. This mechanism appears to be the case for steroid receptors, although gene amplification may occur in some cases (35). For other receptors, overexpression of the receptors leads to aberrant stimulation of the signaling pathway. Examples of this mechanism include EGFR in lung cancer and HER in breast cancer (3,10,36). In this situation, aberrant expression (overexpression) becomes an important marker for the activation of the pathway and predicts the likely efficacy of therapy directed against the target. For example, the overexpression of HER2 is highly predictive for a response to trastuzumab, a monoclonal antibody directed against HER2 (37). In all cases, knowledge of the levels of receptor expression, which may vary considerably in different tumors and even in different sites in the same tumor, is required to infer the likelihood that receptor-directed therapy will be effective.
For tumor receptor systems, considerable variability exists in both ligands and receptors. Ligands may be naturally occurring small molecules, such as estradiol and testosterone, synthesized and secreted in endocrine organs, such as the adrenal gland, ovary, and testis, or peptides assembled and secreted by different types of endocrine cells (38). Receptors can be located on the cell membrane, for example, EGFR and SSTRs (36), or localized within the cell, for example, steroid receptors in the nucleus (9). In some cases, such as steroid receptors, the binding of the agonist ligand to the receptor is well understood, and there is a clear causal relationship between binding and pathway activation (15). In other cases, such as HER2, the pharmacologic significance of ligand–receptor binding in activating the pathway is less well understood; however, receptor-targeted therapy can still be effective at interrupting the signaling pathway through antagonism (10,39).
The approaches to tumor receptor–targeted therapy vary. In many situations, receptor-targeted antagonists bind to the receptor and block the normal agonist ligand from binding and activating the pathway; for example, the drugs tamoxifen and flutamide block the ER and the androgen receptor (AR), respectively (9,40,41). Many receptor-blocking agents have structures relatively similar to that of the natural ligand and are designed to bind to the same site as the agonist; examples are antiestrogens and antiandrogens. Blocking agents can also bind through immune recognition; for example, the agents trastuzumab and cetuximab bind to HER2 and EGFR, respectively (36).
An alternative therapeutic strategy is to deplete the ligand. This approach has been extraordinarily effective for estrogens and the ER in breast cancer, for which the use of aromatase inhibitors has met with considerable success (22,23). With aromatase inhibitors, the drug is not a receptor antagonist ligand but rather is an enzyme inhibitor that blocks the synthesis of the naturally occurring agonist ligands estradiol and estrone (22). Aromatase inhibitors block the conversion of adrenal steroids to estrogens, the major source of estrogen in postmenopausal women, lowering estrogen concentrations both in the serum and in the local tumor environment (42). Aromatase inhibitors are now first-line adjuvant and primary metastatic breast cancer treatments in postmenopausal patients with ER-expressing tumors (43).
Despite the importance of tumor receptors in carcinogenesis and tumor growth, tumor receptors are not always effective targets for cancer treatment, because some cancers can sustain growth independently of receptor activation. In some situations, growth independence is accompanied by a loss of or a reduction in receptor expression, such as in ER-negative breast cancers (3). In such situations, the absence of receptor expression indicates a negligible chance of success of receptor-targeted therapy. In other situations, even though a receptor is still present, receptor pathway activation is not required for growth. For example, although 70% of breast cancers express the ER, only 50%–75% of ER-expressing primary breast cancers respond to endocrine therapy, and even fewer recurrent tumors respond (3). Redundant growth pathways may make the receptor system no longer necessary to sustain tumor growth. The latter situation appears to occur with breast cancers that both express ER and overexpress HER2 and that are often resistant to endocrine therapy, even when ER expression is preserved (44). These examples illustrate that although tumor receptor expression is necessary for a functional pathway, receptor expression does not necessarily guarantee the success of receptor-targeted therapy. The approach to patient selection is therefore sequential. The first step is the assay of receptor expression, because the absence of receptor expression invariably indicates that the receptor pathway is not a suitable target for therapy. After the selection of patients with receptor expression, the next step is to show that the pathway is functional and required, typically by assessing the response to receptor-targeted therapy. This clinical paradigm sets the stage for the goals of tumor receptor imaging.
SPECIAL CONSIDERATIONS FOR TUMOR RECEPTOR IMAGING
Tumor receptor imaging poses unique challenges for the design of radiopharmaceuticals and imaging approaches. Most receptors have high affinities for their ligands and are active at nanomolar concentrations of the ligands. For this reason, radiopharmaceuticals with high specific activity are essential. Even small molar quantities of an imaging agent may saturate a receptor and limit the ability to visualize receptor expression (14,33). For this reason, molecular imaging of tumor receptors has been most successful with radionuclide imaging (PET and SPECT), with which it is possible to generate images with nanomolar amounts of imaging probes. For larger molecules, such as peptides and monoclonal antibodies, other labels suitable for optical imaging, MRI, and ultrasound imaging are possible (Table 1); however, for small-molecule receptor imaging agents, such as labeled steroids for steroid receptors, radionuclide imaging appears to be the only feasible approach. The need for high specific activity (ratio of radioactive to nonradioactive molecules) also poses a challenge for radiopharmaceutical quality control (14,33). Specific activity often needs to be measured before each radiopharmaceutical administration to ensure that a failure to visualize the receptor is not the result of poor tracer-specific activity.
The choice of a label depends on the nature of the receptor imaging probe. For example, somatostatin imaging of neuroendocrine tumors entails the use of a labeled peptide closely related to the naturally occurring peptide hormone somatostatin (45). In this situation, the imaging molecule is large enough to enable the use of chelating groups and radiometal labels without a loss of receptor binding. A variety of somatostatin imaging agents have been successfully developed with both single-photon emitters (111In and 99mTc) and positron emitters (68Ga and 64Cu) (46–48).
For smaller molecules, the isotope label may significantly affect binding to the receptor, binding to transport proteins, and in vivo metabolism. In this situation, the choices of radionuclide and labeling position for imaging may be relatively limited, such as for ER imaging agents (14). Considerable work has been done with both single-photon–emitting and positron-emitting halides for ER imaging (49–51), but studies have suggested that 18F is the most attractive label for PET ER imaging (14). Fluorine is a small halogen in which substitutions can be made at several positions of the estrogen while preserving binding affinities for both ER and SHBG (52,53). Furthermore, 18F has a sufficiently long half-life to permit multistep synthesis of ligands (52,54) as well as uptake by target tissue and elimination by nontarget tissue during imaging (14,55); in addition, the use of PET permits quantitative imaging of regional receptor binding.
For small molecules, the location of the label can also be important. For ER imaging, 18F substitution in the 16-α position for the steroidal analog estradiol to yield 18F-16-α-17-β-fluoroestradiol (18F-FES) resulted in highly selective uptake by target tissue, with a uterus-to-blood ratio of 39 (52). Changing the molecule or labeling can have unexpected results. For example, 18F-labeled moxestrol (18F-FMOX), another ER imaging agent, was developed with the goal of decreased metabolism and increased ER binding. Preclinical studies in vitro and in rats demonstrated better ER binding in vitro and increased uterine uptake in immature rats for 18F-FMOX than for 18F-FES (26,56). However, 18F-FMOX performed poorly in human studies. The explanation for these findings was that poor binding of 18F-FMOX to the steroid transport protein, SHBG, likely limited its utility in humans. Rats lack SHBG (25); therefore, 18F-FMOX was an effective imaging agent in rats. In this example, a change in the imaging molecule that promoted increased ER binding unfavorably altered the binding to SHBG, resulting in a compound with poorer performance. This example illustrates the demanding nature of radiopharmaceutical design for tumor receptor imaging and the need for validation at each step of development, from the laboratory bench to the bedside.
For these reasons, considerable preclinical work and early testing in patients are necessary to develop and validate receptor imaging agents (33). In vitro studies must confirm the high-affinity receptor binding of a radiopharmaceutical as a starting point for development. Subsequently, in vitro and in vivo animal models must demonstrate that binding is specific to the receptor and that an excess of the nonlabeled natural ligand, or a suitable substitute specific for the receptor, can displace or block the binding of the radiopharmaceutical. In vivo clearance, metabolism, and biodistribution in preclinical models and early patient studies must confirm sufficient tracer clearance to visualize uptake in tumors but sufficiently slow blood clearance and metabolism to permit uptake in receptor-rich tissues. Defining the nature of labeled metabolites is also important, because some metabolites may bind to the receptor, whereas others may not. For example, in humans, 18F-FES-labeled metabolites are present mostly in the form of conjugates that do not bind to the receptor or have access to the nuclear receptor. These metabolites therefore contribute to nonspecific image background (27,55,57). In addition, assessment of nonspecific binding is important; nonspecific binding must be sufficiently low to avoid interference with the visualization and quantification of tumor uptake at target sites.
Receptor imaging poses some additional challenges for image acquisition and analysis. Because the absence of receptor expression may be even more important than the presence of a receptor, the imaging approach must be able to quantify low levels of radiopharmaceutical uptake and reliably identify situations in which a tumor is present but the uptake of a receptor imaging agent is low or absent. This approach requires multimodality imaging. Combined functional imaging and anatomic imaging, such as PET/CT or SPECT/CT, may be essential for localizing tumor sites at which the uptake of a receptor imaging probe can be quantitatively interrogated. Because it may be difficult to identify active tumor sites by anatomic imaging alone, it may be necessary to align tumor receptor images with images obtained with another tumor imaging probe, such as 18F-FDG, to identify viable tumor. We used this approach for the ER imaging of breast cancer; 18F-FDG PET identified sites of active breast cancer at which to evaluate ER expression by 18F-FES PET (Fig. 1). It may be beneficial to coregister different functional images; for example, for PET/CT and SPECT, the anatomic images may be used as the basis for coregistration. Finally, because radiopharmaceutical uptake may be at or close to background uptake in nontumor tissues, accurate correction for the imaging of physics-related background counts, such as scattered photons, is critical.
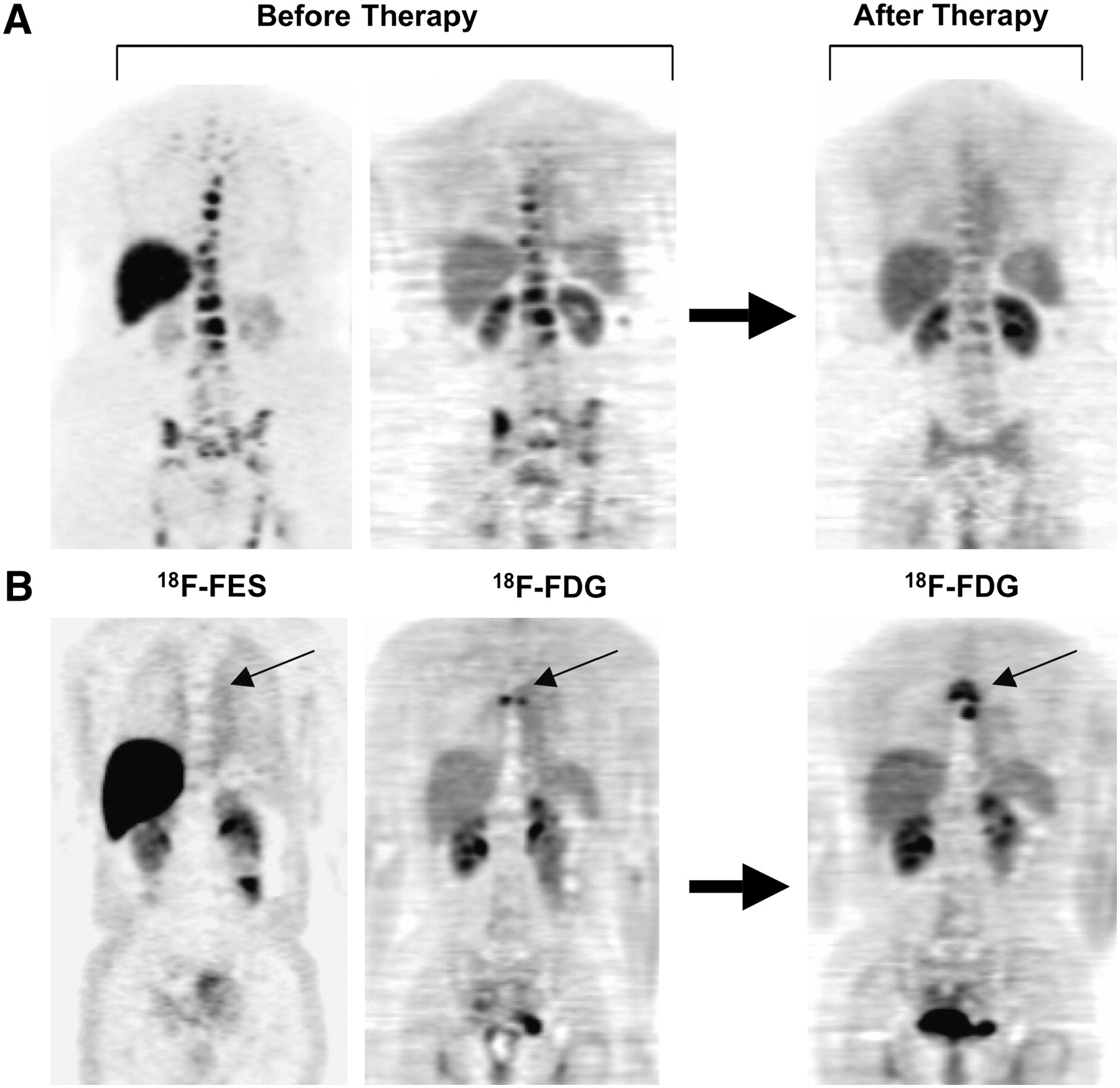
Examples of different patterns of ER expression measured by 18F-FES PET. Both patients had bone metastases arising from ER-expressing primary tumors, and both were treated with endocrine therapy. (A) For this patient, pretherapy 18F-FDG and 18F-FES PET scans showed 18F-FES uptake at all sites of active disease seen by 18F-FDG PET. Follow-up 18F-FDG PET scan showed response to treatment after initiation of aromatase inhibitor therapy. (B) For this patient, there was no uptake at site of disease seen on 18F-FDG PET scan (small arrow). Follow-up 18F-FDG PET scan showed subsequent disease progression (small arrow) with endocrine therapy. (Reprinted with permission of (84).)
Another potential issue is separating the effects of imaging probe transport and binding in determining the overall image. It is possible that transport barriers will limit the in vivo access of the imaging probe to the site of tumor receptor expression. Therefore, it may be difficult to determine from a single static image whether the absence of radiopharmaceutical uptake at a tumor site is attributable to a lack of receptor expression or to a lack of delivery of the imaging probe. This is less likely to be an issue for small, lipophilic molecules, such as steroid imaging agents, but may pose an important consideration for peptides and especially monoclonal antibodies. In such situations, more sophisticated, dynamic imaging acquisitions may be required, along with more detailed and kinetic analyses.
EXAMPLES OF TUMOR RECEPTOR IMAGING
Steroid Receptors
Steroid receptor targets in cancer include the ER and the progesterone receptor (PR) in breast cancer and the AR in prostate cancer. We first briefly review experience in AR and PR imaging and then discuss ER imaging, for which the most experience with steroid receptor imaging has been obtained.
Parallel to efforts for ER imaging, a variety of compounds have been developed for PET of the AR; these have been directed mainly toward prostate cancer imaging (58–64). AR imaging has proved to be somewhat more challenging than ER imaging, perhaps because of the relatively tighter binding of androgens than of estrogens to SHBG; the latter property may limit the delivery of the imaging agent over the time scale of PET, even though tight binding of the AR imaging agent to SHBG appears to be important in generating AR images (25,58). Preclinical studies in baboons, which have SHBG similar to that in humans, indicated that 16-β-18F-fluoro-5-α-dihydrotestosterone (18F-FDHT) was a promising compound for PET (58). Early studies with 18F-FDHT showed promise for the imaging of regional AR expression in prostate cancer (59,61). Larson et al. (61) reported significant 18F-FDHT uptake in 7 patients with prostate cancer. Like that of 18F-FES, 18F-FDHT metabolism was rapid, with 80% of radioactivity in the blood in the form of metabolites at 10 min after injection. Cancer treatment diminished 18F-FDHT uptake in the subset of patients re-imaged after therapy in that study. Dehdashti et al. (59) studied 20 patients with prostate cancer by 18F-FDHT PET and found evidence of uptake in 63% of the patients; 18F-FDHT PET revealed a substantial number of tumor sites that had not been identified by conventional imaging. Furthermore, in a subset of 12 patients who were re-imaged 1 d after the initiation of flutamide therapy, the imaging demonstrated a significant (>50%) average decline in 18F-FDHT uptake, indicating the ability to measure the pharmacodynamics of treatment with receptor antagonists. These encouraging results support the feasibility of PET AR imaging, and ongoing studies support the promise of imaging of AR expression in localizing prostate cancer and possibly predicting the response to antiandrogen therapy.
Efforts to image the PR have been less successful (56,65). In a study of 8 patients with primary breast cancer, Dehdashti et al. found that 21-18F-fluoro-16α-ethyl-19-norprogesterone was taken up in some tumors, but the level of uptake did not correlate with the level of PR expression (65). This result may have been attributable in large part to the relatively low affinity of progestins for the PR, with binding affinities that are orders of magnitude lower than those of androgens for the AR and of estrogens for the ER (56). Therefore, relatively high nonspecific binding compared with specific binding of imaging probes may limit their utility for PR imaging. Later studies also showed that the radiopharmaceutical tested had rapid metabolism in humans to a metabolite with poor receptor binding (66), a finding that was not predicted by preclinical models. Efforts to develop effective PR imaging agents continue (67).
The most experience to date with steroid receptor imaging for tumors has been obtained with ER imaging for breast cancer. Considerable efforts have been directed toward the development of radiopharmaceuticals for ER imaging (51). Early efforts to develop a labeling approach for PET focused on steroids labeled with bromine; promising early results were obtained with 77Br-16α,17β-estradiol in animals and humans (68,69). Parallel efforts to develop 123I-labeled compounds for SPECT also yielded promising early results (70,71). Although a variety of ER imaging agents have been tested and continue to be developed and tested (14,53,72,73), the most successful ER imaging radiopharmaceutical to date has been 18F-FES (14,74). 18F-FES has binding characteristics similar to those of estradiol for both the ER and the transport protein SHBG (27,52). Typically, in humans, about 45% of 18F-FES in circulating plasma is bound to SHBG, and much of the remainder is weakly bound to albumin (27). The clearance and metabolism of 18F-FES have been studied in both animals and humans (55,57,75,76). Like other steroids, 18F-FES is rapidly taken up by the liver and metabolized shortly after injection. As a result, early blood clearance is rapid, reaching a plateau 20–30 min after injection (55). To date, no toxicity or significant adverse reactions have been reported for 18F-FES. Radiation dosimetry studies have shown that organ doses associated with 18F-FES PET are comparable to those associated with other commonly performed nuclear studies and that potential radiation risks are well within acceptable limits. The effective dose equivalent is 0.022 mSv/Bq (80 mrem/mCi), and the organ that receives the highest dose is the liver, at 0.13 mSv/Bq (470 mrad/mCi) (77). The recommended injection is 222 MBq (6 mCi) or less. 123I-labeled compounds have also produced images of regional ER expression with acceptable radiation dosimetry (70,78), albeit with somewhat lower image quality. The quantification of radiopharmaceutical uptake appears to be important for ER imaging, and this quantification is more challenging for SPECT than for PET.
18F-FES uptake has been validated as a measure of ER expression in breast tumors. In 1988, Mintun et al. (79) reported an excellent correlation between 18F-FES uptake measured in primary tumors on PET images and tumor ER concentrations measured in vitro by radioligand binding after excision for 13 patients with primary breast masses. A preliminary comparison of 18F-FES uptake with immunohistochemistry assays of biopsy material from patients with both primary and metastatic cancers also showed a good correlation (80,80a). Imaging results will not necessarily correlate perfectly with biopsy results. Differences between in vivo measures of ER expression by PET and in vitro assays of ER expression are to be expected, especially in comparisons of radioligand binding (e.g., 18F-FES PET) and immune recognition (e.g., immunohistochemistry assays of biopsy material).
In the earliest reported study of 18F-FES PET in patients, 18F-FES uptake was seen at sites of primary carcinomas, axillary nodes, and one distant metastatic site (79). The investigators then extended the use of this radiopharmaceutical for the imaging of metastatic breast cancer. Sixteen patients with metastatic disease underwent 18F-FES PET; increased uptake was seen in 53 of the 57 metastatic lesions, for a sensitivity of 93%, and there were only 2 apparent false-positive results (81). Imaging results were reported quantitatively as percentage injected dose per milliliter (uptake), ratio of lesion to soft tissue, and ratio of lesion to uninvolved bone. The same group of investigators obtained similar results in a later study of 18F-FES imaging in 21 patients with metastatic breast cancer; they reported 88% overall agreement between in vitro ER assays and 18F-FES PET (82). In addition to subjective analysis, 18F-FES uptake has been reported as a standardized uptake value (SUV). Using an SUV of greater than 1 to identify ER-expressing disease, Mortimer et al. (83) reported that the sensitivity of 18F-FES imaging was 76%, with no false-positive results, in 21 patients with metastatic breast cancer.
One of the chief advantages of ER imaging over tissue sampling for determining ER expression is the ability to evaluate the heterogeneity of ER expression. Mortimer et al. (83) found that for 4 of 17 patients (24%) with metastatic breast cancer, there was a discordance in 18F-FES uptake between sites in individuals. Mankoff et al. (80) reported the absence of 18F-FES uptake at one or more metastatic sites in 10% of patients who had ER-expressing primary tumors. In this same preliminary study, the quantitative site-to-site variability in 18F-FES uptake in individuals was high (coefficient of variation of approximately 30%). A total of 13% of patients (6/47) with ER-expressing primary tumors had one or more sites of 18F-FES–negative disease in a subsequent study by the same group of investigators (84). The rate of loss of ER expression at metastatic sites from ER-expressing tumors was comparable to (only slightly lower than) values obtained from tissue samples and reported in the literature (85,86), suggesting that sampling error may contribute to the apparent heterogeneity in tissue-based assay studies.
In clinical practice, in vitro ER assays are used primarily as predictive assays for endocrine therapy. Although 18F-FES PET has not been prospectively tested as a predictive assay in clinical trials, a comparison of 18F-FES uptake and the response to endocrine therapy in some groups of patients has indicated the likely performance of 18F-FES PET as a predictive assay. Mortimer et al. (87) showed that the level of 18F-FES uptake predicted the response to tamoxifen, demonstrating the potential utility of 18F-FES PET for predicting a response in the locally advanced and metastatic settings. Forty women with biopsy-proven ER-expressing breast cancer had 18F-FES PET before and 7–10 d after the initiation of tamoxifen therapy, and tumor 18F-FES PET was assessed with the SUV method. Both the percentage decrease in 18F-FES uptake (responders, 55% ± 14% [mean ± SD]; nonresponders, 19% ± 17%) and the absolute change in tumor SUV (responders, decrease of 2.5 ± 1.8 SUV units; nonresponders, decrease of 0.5 ± 0.6 SUV units) predicted the response to tamoxifen. The level of 18F-FES uptake before therapy also predicted the response to tamoxifen. The positive and negative predictive values for baseline 18F-FES uptake with an arbitrary SUV cutoff of 2.0 were 79% and 88%, respectively (87). No patient with an SUV of less than approximately 1.5 responded.
Linden et al. (84) showed that the initial 18F-FES uptake measurements in patients with ER-expressing tumors were correlated with subsequent tumor responses to 6 mo of hormonal therapy. Forty-seven patients with metastatic breast cancer from ER-expressing primary tumors, most of whom had been previously treated for breast cancer, many for some time and with several regimens, were given predominantly salvage aromatase inhibitor therapy. Objective responses were seen in 11 of 47 patients (23%). 18F-FES PET was assessed qualitatively and quantitatively with SUV and flux calculations. Although no patient without 18F-FES uptake at known tumor sites responded, qualitative 18F-FES PET results did not significantly predict responses to hormonal therapy. However, quantitative results were predictive of responses in that 0 of 15 patients with initial SUVs of less than 1.5 responded to hormonal therapy, compared with 11 of 32 patients (34%) with initial SUVs of greater than 1.5 (P < 0.01). Similar results were obtained when 18F-FES flux was used to measure uptake (P < 0.005). Interestingly, no patient whose tumor overexpressed HER2 showed an objective response, including patients with SUVs of greater than 1.5. In the subset of patients without HER2 overexpression, 11 of 24 patients (46%) with SUVs of greater than 1.5 responded to hormonal therapy. Hypothetically, the use of 18F-FES PET to select patients could have increased the response rates from 23% to 34% overall and from 29% to 46% in the subset of patients lacking HER2 overexpression. The timing of 18F-FES imaging may have been a confounder in that study, because patients underwent 18F-FES imaging while receiving aromatase inhibitor therapy, but preliminary data from the same group of investigators showed that serial 18F-FES measurements changed less than 20% in patients early after the initiation of aromatase inhibitor therapy (88).
Serial PET ER imaging can be used to measure the pharmacodynamic effect of ER-directed endocrine therapy. McGuire et al. (81) demonstrated tamoxifen blockade of the ER on serial 18F-FES PET scans in early studies in patients (81), similar to studies of AR blockade. Mortimer et al. (87) later showed a lower level of blockade occurring as early as 1 wk after the initiation of tamoxifen therapy. Linden et al. (88) analyzed serial 18F-FES PET scans of patients with metastatic disease undergoing treatment with different agents with different mechanisms of action: tamoxifen (n = 2), aromatase inhibitors (n = 14), and fulvestrant (n = 5). Patients were imaged a median of 29 d after the initiation of treatment. The decline in 18F-FES SUVs was greater with the antagonists (tamoxifen and fulvestrant) than with the aromatase inhibitors, which reduced the agonist concentration but did not block the receptor. Interestingly, qualitative 18F-FES scans obtained after treatment showed complete blockage with tamoxifen but incomplete blockage with fulvestrant in 4 of the 5 patients, despite complete blockage of uterine uptake, suggesting differential access of the drug to the uterus versus tumor sites.
Some preliminary studies have evaluated 18F-FES PET in settings other than breast cancer. Moresco et al. (75,76) studied 18F-FES uptake in normal brain tissue and meningiomas, using measures similar to the flux measure defined earlier. Although 18F-FES uptake in normal brain tissue was too low to quantify estradiol binding reliably by PET, significant 18F-FES uptake was seen in some meningiomas. Selective 18F-FES uptake by uterine endometrium has been shown in human imaging, with cyclic changes mirroring the menstrual cycle (89). 18F-FES uptake in endometrial cancer has been reported for a single patient studied by this method (90).
Overall, early studies of PET ER imaging have shown its promise as a tool for directing breast cancer treatment. The promising early results of the studies of Mortimer et al. (87) and Linden et al. (84) indicating the potential utility of 18F-FES PET as a predictive assay need to be confirmed in larger trials involving more institutions and objective determinations of the appropriate 18F-FES SUV cutoff for predicting a response to endocrine therapy. Ongoing studies, including prospective clinical trials, should define its potential use in clinical trials and clinical practice for breast cancer and possibly other ER-expressing tumors.
SSTRs
Somatostatin is a 28-amino-acid peptide agonist ligand secreted by endocrine D cells and neurons in the gastrointestinal (GI) tract and pancreas (91). SSTRs are membrane receptors for which 6 subtypes have been identified by molecular analysis (45,91). Somatic tissues most commonly express receptor subtypes 2 and 5 (91). Somatostatin binding to the receptor activates G proteins, leading to downstream physiologic actions that are tissue specific. Primary among these actions is the inhibition of the release of endocrine and exocrine factors in the GI system (91). Therefore, somatostatin plays an important role in the regulation of the GI tract. In addition to normal tissues, SSTRs are expressed in a variety of tumors, especially endocrine tumors, such as carcinoids, gastrointestinal and pancreatic neuroendocrine tumors, pheochromocytomas, paragangliomas, medullary thyroid cancer, and pituitary adenomas (45). Somatostatin analogs, such as the 8-amino-acid peptides octreotide and lanreotide, are used therapeutically to inhibit GI tract endocrine factor release in both nonmalignant disease (e.g., intractable diarrhea) and endocrine neoplasia, particularly pituitary and neuroendocrine tumors (91). Unlike somatostatin itself, which has a plasma half-life of approximately 2 min, octreotide has a half-life of 1.5–2 h, making it suitable for drug therapy (91,92). In the treatment of neuroendocrine tumors, somatostatin analogs reduce the symptoms associated with excess hormone secretion and may also have direct antitumor effects (91).
The main focus of SSTR imaging has been neuroendocrine tumors and related tumors, including carcinoids, gastrointestinal and pancreatic neuroendocrine tumors, pheochromocytomas, paragangliomas, medullary thyroid cancer, and pituitary adenomas (45). Other tumors expressing SSTRs have also been studied and include lymphomas, meningiomas, ethesioneuroblastomas, and lung cancers (93–95).
Most of the work carried out to date on SSTR imaging has used labeled somatostatin analogs, most commonly, octreotide or a closely related peptide (45). Octreotide is an 8-amino-acid peptide with stability in plasma, unlike somatostatin, which has a plasma half-life of less than 3 min (91). The labeling strategy is quite different from that used for a smaller molecule, such as estradiol. The first compound used in SSTR imaging, 123I-3Tyr-octreotide, was introduced in 1988 (96) and used direct halogenation of a tyrosine (45,96). However, the most widely used radiopharmaceuticals in current practice use a linked chelating group, such as diethylenetriaminepentaacetic acid (DTPA) or 1,4,7,10-tetraazacyclododecane-N,N′,N″,N″′-tetraacetic acid (DOTA), that binds to the peptide and chelates a radiometal, such as 99mTc, 111In, 68Ga, or 64Cu (45,97–100). Currently, most clinical SSTR imaging is carried out with the agent 111In-DTPA-pentetreotide (Octreoscan; Mallinckrodt), which is approved by the U.S. Food and Drug Administration for clinical SSTR imaging (101). Recent studies (48,98,100,102,103) examined octreotidelike compounds labeled with positron emitters (such as 64Cu- and 68Ga-labeled peptides), taking advantage of the higher spatial resolution and easier image quantification offered by PET than by SPECT (104–106). Of the 2 positron-emitting labels, 68Ga is conveniently available from a generator but has the disadvantage of a higher positron range than 64Cu.
Imaging of SSTR-expressing tumors has been applied to tumor detection and staging, prediction of the response to therapeutic somatostatin, and treatment planning for SSTR-directed radionuclide therapy (45,92). The most widely tested and clinically accepted indication is tumor staging, for which SSTR imaging has become part of the clinical routine for carcinoids and other neuroendocrine tumors (45,99,101). These tumors, frequently arising from the gut or other abdominal structures, can be challenging to identify by conventional anatomic imaging. Furthermore, well-differentiated neuroendocrine tumors have fairly limited 18F-FDG uptake, limiting the effectiveness of 18F-FDG PET in neuroendocrine tumor staging (104). Most comparisons have shown that SSTR imaging, mostly with 111In-labeled compounds, has higher sensitivity and accuracy for neuroendocrine tumor localization than other modalities, supporting its ongoing use in clinical practice (45,107). SSTR imaging has a significant impact on neuroendocrine tumor treatment, making it effective in clinical management (108). Combined-modality (SPECT/CT and PET/CT) imaging increases the impact of SSTR imaging, especially with regard to direct tissue sampling or surgical resection (109,110). Recent studies have suggested that the use of PET labels, such as 68Ga, along with PET/CT may be especially effective in this regard (47). Studies have suggested that binding of the labeled peptide to the SSTR and clearance from the plasma are sufficiently rapid to permit visualization and quantification of the regional SSTR concentration by 45 min after injection, necessary for the short half-life of 68Ga (111).
Some studies have also examined the presence or absence of uptake of SSTR imaging probes as a predictor of the response to somatostatin therapy, akin to studies of 18F-FES PET as a predictor of the breast cancer response to endocrine therapy. Although there have been some promising studies (112), the results have been variable, and this indication is less common in clinical practice than simple tumor localization and staging. Several factors contribute to the lower success of SSTR imaging than of ER imaging in response prediction: SSTR imaging studies have been mainly nonquantitative, somatostatin therapy has limited efficacy in cytoreduction, and responses are difficult to measure in slowly growing tumors, such as neuroendocrine tumors (91). The effect of SSTR-directed therapy on SSTR expression may also be a confounding factor; some studies have suggested that somatostatin itself may affect receptor expression and the uptake of labeled analogs in a complex, rather than strictly competitive, fashion (113).
A rapidly emerging focus in radionuclide-labeled SSTR ligands is SSTR-directed radionuclide therapy (92). Neuroendocrine tumors can present with advanced, widespread disease that is often refractory to conventional chemotherapy (38). However, they are often slowly growing tumors, so that minor responses or disease stabilization can result in considerably prolonged survival (114). In this regard, radionuclide therapy with labeled SSTR ligands has been effective in treating advanced neuroendocrine tumors (92,115). The earliest studies used 111In-DTPA-pentetriotide and showed up to an 80% disease stabilization rate (92,116). More recent studies have used 90Y- and 177Lu-labeled compounds, which have some advantages over 111In-labeled compounds for radionuclide therapy. 90Y is a pure β-emitter with high-energy β-emissions; 177Lu has intermediate-energy β-emissions and has a longer half-life than 111In. Several studies have shown objective response rates higher than those obtained with 111In, and some recent studies have shown significant improvements in time to progression and survival (92,117,118). For guiding SSTR-directed radionuclide therapy, SSTR imaging has been key (92) in showing uptake of the labeled peptide at all known tumor sites and in estimating radiation dosimetry. For dosimetry, the use of PET radiopharmaceuticals has been especially helpful (97), given the ability of PET to quantify radiopharmaceutical biodistribution.
Recent studies have suggested that radionuclide SSTR imaging fused with anatomic imaging can be very effective in targeting meningiomas (119). This approach provides another novel application of SSTR imaging in directing therapy, namely, guiding conformal radiotherapy.
In summary, radionuclide SSTR imaging and SSTR-directed radionuclide therapy have been important in the diagnosis and therapy of SSTR-expressing tumors, especially neuroendocrine tumors. Although non–receptor-based imaging with compounds such as labeled metaiodobenzylguanidine and 3,4-dihydroxyphenylalanine has also played a role in the diagnosis of these tumor types (47,99), the specific imaging of SSTRs remains an important and evolving clinical tool with both diagnostic and therapeutic applications.
Growth Factor Receptors
A third and somewhat distinct receptor imaging example relates to the EGFR pathway, which has received considerable recent attention as a therapeutic and imaging target (10,36). EGFR is a membrane surface receptor that interacts with agonists such as EGF and transforming growth factor; however, receptor activation is complex, and the exact nature of the ligand–receptor interaction is not completely understood (4,39). It appears that activation of the EGFR pathway involves up to 4 related receptors, erbB1–erbB4 (also called HER1–HER4), which form homo- and heterodimers both in the presence and in the absence of a ligand (10,39). The result of these receptor interactions is the activation of a specific tyrosine kinase and a sequence of downstream reactions leading to diverse biologic consequences, which include cellular proliferation, angiogenesis, and resistance to apoptosis (10,39). The EGFR pathway has been implicated in the pathogenesis of many cancer types and has received considerable recent attention as a therapeutic target (4,10,39). The most notable and widely studied approaches are therapy directed to the EGFR (erbB1, HER1) for lung cancer and head and neck cancer and therapy directed to erbB2 (HER2 or HER2/neu) for breast cancer (4). Therapeutic strategies target either membrane receptors, typically with monoclonal antibodies (e.g., cetuximab for EGFR and trastuzumab for HER2), or tyrosine kinase, with small-molecule inhibitors (e.g., erlotinib for EGFR) (4).
Imaging approaches mainly parallel therapeutic approaches. Although there have been some studies of labeled receptor ligands (120–122), such as 111In-DTPA-EGF, most efforts to date have focused on receptor. Some progress has been made for specific tyrosine kinase probes (123–129); however, the multiplicity and ubiquity of tyrosine kinase expression sites as well as lipophilicity and associated nonspecific binding have made probe development challenging (123,127).
The most success to date has been achieved (and the largest number of studies have been carried out) for the targeting of EGFR or HER2 by immune recognition, in parallel with therapeutic agents. Specific imaging probes have been based on radiolabeled antibodies or fragments (130–134) or on novel constructs, such as Affibodies (135,136). Most studies have been preclinical (137–141); however, some studies have reported HER2 imaging in patients (133,142–144). Studies carried out by Smith-Jones et al. (145,146) with a 68Ga-labeled F(ab′)2 fragment of trastuzumab showed the feasibility of measuring regional HER2 expression in murine animal models. The imaging results clearly demonstrated alterations in HER2 expression accompanying experimental therapy with HSP90-directed agents (geldamycin analogs) to disrupt protein chaperone functions and reduce HER2 expression (Fig. 2) (145,146). Studies with 131I- or 111In-labeled trastuzumab demonstrated the ability of trastuzumab to image tumor expression of HER2 and tumor and normal tissue accumulation (133,142,147), although there has been some controversy about the significance of uptake in normal tissues prone to trastuzumab toxicity, such as the heart (133,142). A promising early study in patients was also reported for 89Zr-labeled trastuzumab (148). These early studies demonstrated the feasibility of HER2 and EGFR imaging, with the potential for directed therapy specific for HER2 and EGFR. In addition, recent studies suggested that altered glycolysis is an early event accompanying the interruption of the EGFR pathway (149,150), suggesting a role for combined HER2 or EGFR imaging and 18F-FDG PET in predicting responses to targeted agents.
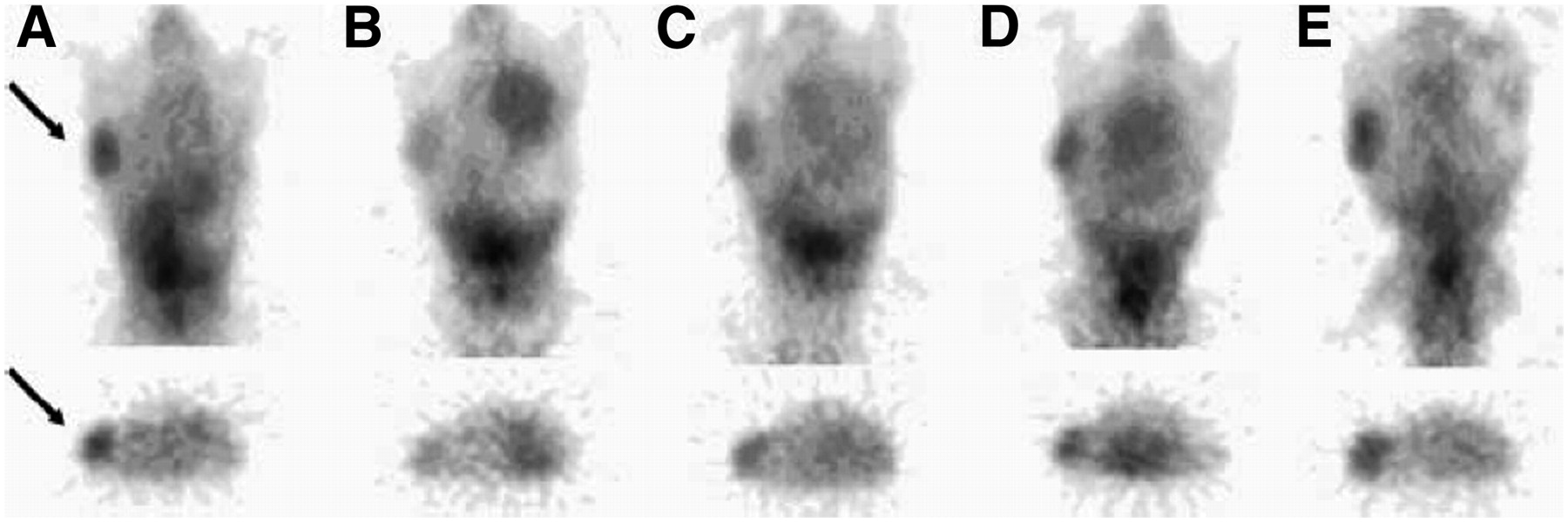
Images of mouse model of HER2-expressing breast cancer before treatment (A) and 1 d (B), 5 d (C), 8 d (D), and 12 d (E) after treatment with 17-allylamino-17-demethoxy-geldanamycin (17AAG) taken with 68Ga-DOTA-F(ab′)2-trastuzumab. Images show early decrease in HER2 expression shortly after 17AAG treatment, with reexpression of HER2 by day 12. Arrows indicate location of cancer. (Reprinted with permission of (145).)
OTHER RECEPTOR IMAGING MODALITIES AND APPLICATIONS
As emphasized so far, the requirement for low concentrations of receptor imaging probes limits the choice of imaging modalities for most applications. For small-molecule ligands with a high affinity and a low capacity for receptor binding, such as the ligands used for steroid receptor imaging, radionuclide detection methods are required; the most success has been associated with high-specific-activity PET compounds (Table 1) (33,56). Peptide ligand imaging, such as SSTR imaging, is somewhat more flexible, and some success has been achieved with optical imaging probes for SSTR expression (151–154) or MRI probes for targets close to the vasculature (155). Studies of optical imaging of SSTR-expressing tumors in animal models have supported the feasibility of optical imaging of peptide ligands. Adams et al. (156) demonstrated the optical imaging of another peptide, EGF, with a fluorochrome label (157).
For imaging with monoclonal antibodies and fragments, as in HER and EGFR imaging, an even wider range of probes, such as optical, ultrasound, and MRI probes, may be possible (158–163). Koyama et al. (164) demonstrated specific fluorescence imaging of HER2-expressing lung metastases in an animal model, near-infrared imaging of HER2 expression in vitro with gold nanoshell bioconjugates was demonstrated by Loo et al. (165), and fluorochrome labeling was demonstrated by Hilger et al. (166). Similar optical approaches have been tested for EGFR imaging (167–169). Multiparameter optical imaging combining HER2 and apoptosis probes revealed target expression and an early tumor response in a mouse model, and optical approaches were used to study the response to EGFR-directed therapy in glioma models (170). Preclinical studies with specific antibodies conjugated to gadolinium or magnetic nanoparticles demonstrated the feasibility of MRI antibody imaging in cells (171) and animal models (159). The feasibility of nanoparticle-based ultrasound probes conjugated to HER2-specific antibodies was demonstrated in early in vitro studies and simulated in vivo studies (160,172).
Besides detection and treatment guidance, a novel application of receptor imaging is gene therapy, in which receptors have been used as parts of PET reporter systems and receptor imaging has been used in some of the earliest studies designed to track gene transfection (173) and vector delivery (174–176). For this approach to be useful in tumor imaging, an appropriate choice of a receptor is required; typically, the receptor must be one that is not expressed by the nontransfected tumor or host tissue and for which uptake is not expected at sites at which the tumor will be imaged. Furthermore, it is important that the expression of the reporter does not elicit an immune response. Examples include the use of SSTR systems for vector delivery in non–SSTR-expressing tissues (174–176) and ER imaging for assessing gene transfection (177,178).
CONCLUSION
Tumor receptors are important in the biology of many malignancies. Receptor physiology is an important component of tumor pathogenesis, growth, and metastasis. The high-affinity, low-capacity nature of most ligand–receptor systems is ideal for therapeutic interventions. Receptor imaging can survey tumor receptor expression across the entire body and is therefore ideal for guiding receptor-targeted interventions. However, receptor imaging presents some challenges, most importantly, the requirement for a low molecular concentration of the imaging probe. This requirement has limited tumor receptor imaging mainly to radionuclide methods. A variety of SPECT and PET tumor receptor probes have been developed, with notable progress in peptide receptors and, more recently, in steroid receptors. Recent progress in receptor imaging for other modalities suggests that nonradionuclide receptor imaging is feasible for receptors that can be imaged with peptide ligands or antibodies. The importance of receptors in tumor biology and the abilities to predict responses to targeted therapy and to monitor drug interventions suggest that tumor receptor imaging will continue to be an important component of oncologic molecular imaging and will play a key role in cancer management.
Acknowledgments
This work was supported in part by NIH grants P01CA42045, R01CA72064, and RR17229.
Footnotes
-
COPYRIGHT © 2008 by the Society of Nuclear Medicine, Inc.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.
- 20.
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.
- 61.↵
- 62.
- 63.
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 80a.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.
- 125.
- 126.
- 127.↵
- 128.
- 129.↵
- 130.↵
- 131.↵
- 132.
- 133.↵
- 134.↵
- 135.↵
- 136.↵
- 137.↵
- 138.
- 139.
- 140.
- 141.↵
- 142.↵
- 143.
- 144.↵
- 145.↵
- 146.↵
- 147.↵
- 148.↵
- 149.↵
- 150.↵
- 151.↵
- 152.
- 153.
- 154.↵
- 155.↵
- 156.↵
- 157.↵
- 158.↵
- 159.↵
- 160.↵
- 161.↵
- 162.↵
- 163.↵
- 164.↵
- 165.↵
- 166.↵
- 167.↵
- 168.
- 169.↵
- 170.↵
- 171.↵
- 172.↵
- 173.↵
- 174.↵
- 175.
- 176.↵
- 177.↵
- 178.↵
- 179.↵
- 180.↵
- 181.↵
- 182.↵
- 183.↵
- 184.↵
- 185.↵
- 186.↵
- 187.↵
- Received for publication September 26, 2007.
- Accepted for publication January 2, 2008.





{kind=link}
{kind=link}