


Abstract
Sex hormone–binding globulin (SHBG) is believed to play a key role in steroidal radiopharmaceutical delivery to target tissues in humans. To better understand the action of SHBG, we have synthesized and tested in vivo 2 novel 18F-labeled androgens: 7α-18F-fluoromethyl-dihydrotestosterone (7α-18F-FM-DHT) and 7α-18F-fluoromethyl-nortestosterone (7α-18F-FM-norT). Both 7α-18F-FM-DHT and 7α-18F-FM-norT have high affinity for the androgen receptor (AR); however, 7α-18F-FM-DHT has a high affinity for SHBG, whereas 7α-18F-FM-norT has a relatively low affinity. Methods: We developed an efficient radiochemical synthesis for both 7α-18F-FM-DHT and 7α-18F-FM-norT, producing them in good radiochemical yield and high specific activity. Biodistribution studies of both compounds were done on diethylstilbestrol-pretreated and DHT-blocked Sprague–Dawley male rats. Metabolism studies were done to determine the amount of intact ligand in the prostate. Results: We obtained 7α-18F-FM-DHT and 7α-18F-FM-norT in radiochemical yields of about 30% and radiochemical purities of greater than 99%. Rat biodistribution studies showed selective AR-mediated uptake in the prostate for both compounds. Both compounds showed relatively little defluorination, but the norT analog was more metabolically stable than the DHT analog. Conclusion: These studies show that 7α-18F-FM-DHT and 7α-18F-FM-norT have potential for use in human clinical imaging trials to evaluate more definitively the role of SHBG in radiotracer delivery of steroidal systems to target tissues.
Prostate cancer is the second leading cause of cancer deaths in men in the United States (1) and has been directly linked to the androgen receptor (AR), the target protein that mediates the stimulatory action of endogenous androgens. Sex hormone–binding globulin (SHBG) is an important human plasma protein, and in healthy men, between 40% and 65% of circulating testosterone is bound to SHBG (2). Only 1%–2% of circulating testosterone is free or unbound, the remainder being bound by serum albumin. According to the bioavailability hypothesis, it is thought that only unbound testosterone can enter the cell and reach the AR for activation (3). With this consideration in mind, AR radioligands for in vivo imaging have been designed to have high binding affinity for AR and low affinity for SHBG, to ensure that free steroid levels are adequate for good cellular uptake by AR-positive tumor cells.
In contrast to the bioavailability process by which SHBG might reduce the uptake of androgens are reports that SHBG facilitates target cell uptake of androgens directly, through its own SHBR receptor, also termed megalin (4), a plasma membrane protein present in androgen target cells. According to these reports, SHBG receptor is believed to mediate endocytosis of SHBG-bound androgens via clathrin-coated endocytic vesicles (4–8). Should an SHBG-mediated process be operative, radiopharmaceuticals with a high affinity for SHBG might, in fact, be more effectively delivered to the AR-rich target tissues and thus be better for PET than those with low SHBG binding.
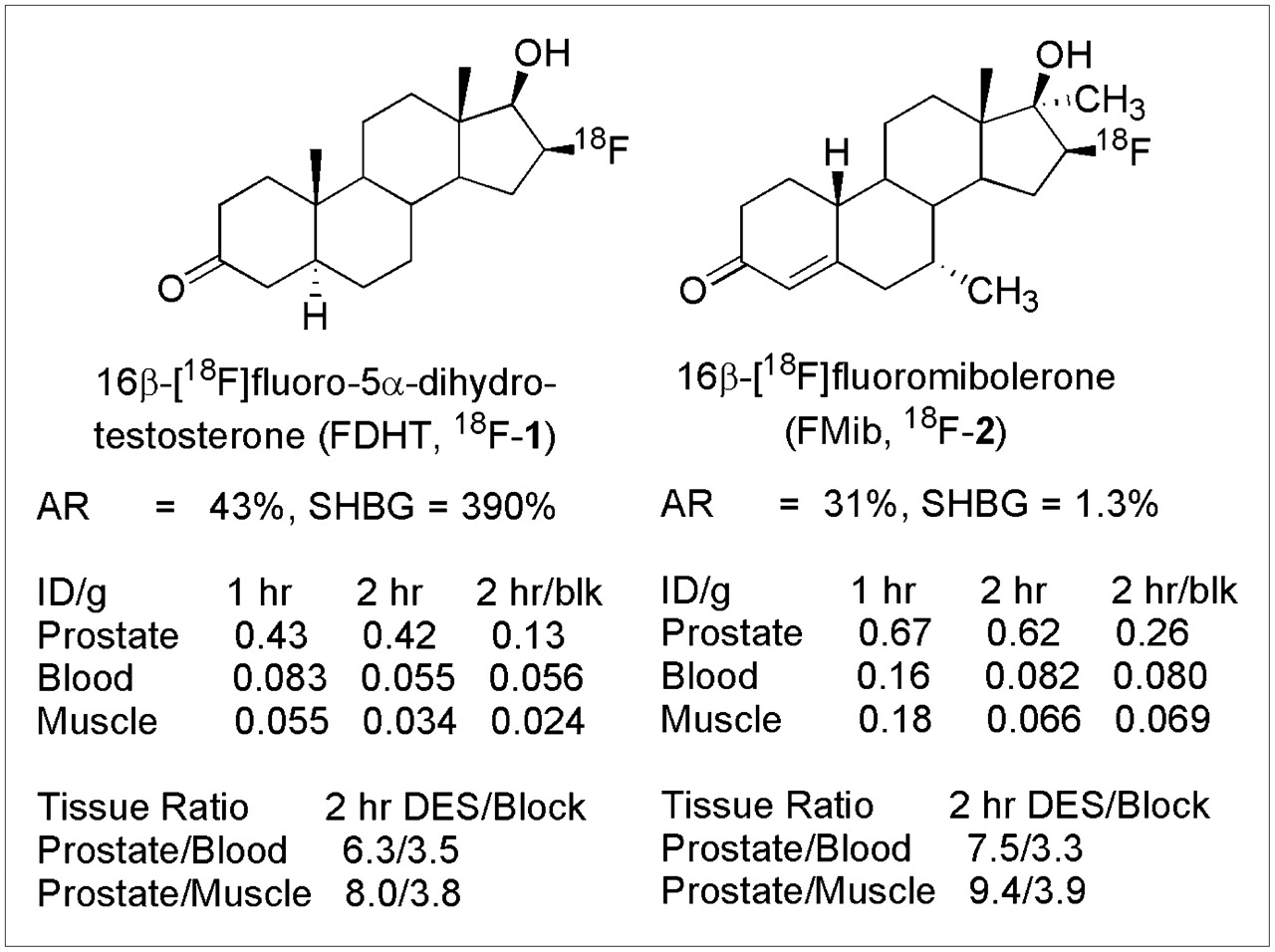
Several AR ligands labeled with 18F (9–11) have been prepared as PET agents, and a few of these androgens have been studied in vivo, both in rats, which have no SHBG (12,13), and in baboons, which do (14–16), with some curious observations. In rats, 16β-fluoromibolerone (2), a high-AR-binding derivative of 19-nortestosterone (19-norT) that has little affinity for SHBG, showed greater prostate uptake than 16β-fluoro-5α-dihydrotestosterone (DHT) (1), a high-AR-binding DHT derivative that has high SHBG binding (11,17). By contrast, in baboons, which have high SHBG levels, the high SHBG binder (16β-fluoro-DHT, 1) showed greater specific prostate uptake than did the low SHBG binder (16β-fluoromibolerone, 2). Also, 16β-fluoro-DHT (2), the only 18F-labeled androgen that has been studied in humans and the one with good SHBG binding, provided clear, AR-mediated images of prostate tumors (18,19). These findings suggested that when SHBG is present, it facilitates (or perhaps is even essential for) AR-mediated target tissue uptake of radiopharmaceuticals with good SHBG affinity, whereas when SHBG is absent, good uptake is observed regardless of SHBG affinity. The studies thus far, however, have been limited and have not involved the best possible comparisons between structurally related androgens, nor have the SHBG and experimental animal always been species-matched (e.g., human SHBG used in rats (20)).
The structures and the AR and SHBG relative binding affinities of compounds 1 and 2 and their rat prostate uptake values are shown in Figure 1 (17). Both have good AR binding but widely different SHBG affinities. The differences in SHBG binding probably reflect, in part, the different steroid skeletons on which these 2 radiopharmaceuticals are based: DHT (the steroidal core of 16β-fluoro-DHT, 1) is known to bind tightly to SHBG (2,130%; relative to estradiol = 100%), whereas 19-norT (the steroidal core of 16β-fluoromibolerone, 2) binds nearly 100-fold less well (10,14). Compounds 1 and 2, however, do not provide rational comparators for DHT and 19-norT, because in addition to the differences at C-19 and Δ4, they also differ in terms of substituents at C-7α and C-17α.

Structure, AR and SHBG binding, and rat prostate uptake of 2 previously reported radiolabeled androgens for PET (17). Binding is expressed relative to standards, R1881 and estradiol, respectively, with each set at 100% (20). Blk = receptor-mediated uptake blocked by coinjection of DHT; diethylstilbestrol indicates pretreatment to suppress endogenous androgen levels. DES = diethylstilbestrol.
Similarly, previous work with ER active ligands further supported the indication that SHBG, when present, is essential for good tracer delivery (21). Based on its excellent biodistribution behavior in rats (no SHBG), 16β-fluoromoxestrol (3) was selected for evaluation in humans as a potential replacement for 16α-fluoroestradiol (4), the well-established imaging agent known to be effective in visualizing ER-positive breast cancer (22–24). In immature female rats, 16β-fluoromoxestrol had a profile that appeared to be clearly superior for an imaging agent compared with that of 16α-fluoroestradiol: good ER binding affinity, lower nonspecific binding, high uterine uptake in rats, and slow in vivo metabolism, but it also had low SHBG affinity (structures, affinities, and rat uterine uptake values are shown in Fig. 2). In the rat (no SHBG), 16β-fluoromoxestrol appeared well protected from metabolism by its 11β and 17α substituents (compared with 16α-fluoroestradiol), and it demonstrated high uptake in the uterus. In humans (with SHBG), however, PET trials with 16β-fluoromoxestrol failed to detect a single estrogen receptor–positive tumor in the 3 patients with known estrogen receptor–positive status (26), suggesting again that when SHBG is present, SHBG binding is a prerequisite for good target tissue uptake.

Thus, contrary to the bioavailability hypothesis, these findings indicate that SHBG binding supports increased uptake of sex hormone–derived radiopharmaceuticals into ER- and AR-rich target tissues in primates, including humans. None of the previous studies, however, has been definitive.
To elucidate further the role of the SHBG–ligand receptor complex in the uptake of radiopharmaceuticals directed at AR, we have designed and synthesized 2 new fluorine-substituted AR ligands as potential PET agents for AR in prostate cancer. Our design goals were to produce a pair of compounds that would have high AR binding affinity and markedly different SHBG binding affinity but otherwise be as close in structure as possible. In addition, we wanted to avoid fluorine substitution at C-16β, because, at least in rats, this site was highly prone to defluorination (17), which was presumed to occur by 16α-hydroxylation, a known mode of androgen metabolism (27). This last point led us to explore a new site of steroid radiolabeling, through a 7α-fluoromethyl group. Thus, our 2 target molecules for this study were 7α-fluoromethyl-dihydrotestosterone (5), for high binding affinity to both AR and SHBG (the latter due to its DHT core), and 7α-fluoromethyl-nortestosterone (6), to have high AR affinity but low affinity for SHBG (the latter due to its 19-norT core) (Fig. 3) (28). Both these compounds have a common (and novel) site of fluorine incorporation (the 7α-CH3 group) that is, in fact, known to be relatively metabolically stable in rats (29,30). As reported here, we have labeled these 2 compounds with 18F and studied their biodistribution and metabolism in adult male rats in preparation for human clinical imaging trials.

Binding affinities of various steroidal ligands for the AR and SHBG proteins (27).
MATERIALS AND METHODS
General
Unless otherwise stated, all chemicals were obtained from Sigma-Aldrich Chemical Co. and used without further purification. H218O was purchased from Rotem Industries. Screw-cap test tubes used for fluoride incorporation were purchased from Fisher Scientific (Pyrex no. 9825). Oasis HLB-6cc cartridges, 500 mg, were purchased from Waters Corp. (part no. 186000115). Vacutainer tubes (5 mL) were obtained from Becton-Dickinson (part no. 366434). 18F-fluoride was produced at Washington University by the 18O(p,n)18F reaction through proton irradiation of 95% enriched 18O-water, using either the JSW BC16/8 cyclotron (The Japan Steel Works Ltd.) or the CS15 cyclotron (The Cyclotron Corp.). Radiochemical purification used a semipreparative reversed-phase high-performance liquid chromatography (HPLC) column (Alltech Alltima C18 column; 10 × 250 mm). The mobile phase was acetonitrile-water. Analytic purity was determined using a reversed-phase HPLC (Phenomenex Luna C18 column; 4.6 × 150 mm). The eluant was monitored with a variable-wavelength detector set at 200 nm for 7α-18F-fluoromethyl-dihydrotestosterone (7α-18F-FM-DHT) and 254 nm for 7α-18F-fluoromethyl-nortestosterone (7α-18F-FM-norT) and the use of a flow-through sodium iodide scintillation detector, where appropriate. The radiochemical purity of the product was also checked by radio–thin-layer chromatography (TLC). The TLC plates were analyzed using a Bioscan Inc. System 200 imaging scanner. Radioactivity was determined with a dose calibrator. Radiochemical yields are decay-corrected to the beginning of synthesis time.
AR binding affinity assays were performed according to literature methods (11,31). AR preparation used in these experiments was purified rat AR ligand binding domain (Panvera/Invitrogen). Rodents for the biodistribution studies were obtained from Charles River Laboratories and were housed in a barrier facility with a corncob bedding that was changed twice a week. Animal handling techniques have been described previously (32).
Binding Affinities to the AR and Sex Hormone Binding Globulin (SHBG)
The binding of these compounds to the AR and to SHBG was determined using competitive radiometric binding assays (Fig. 3) as previously described (28).
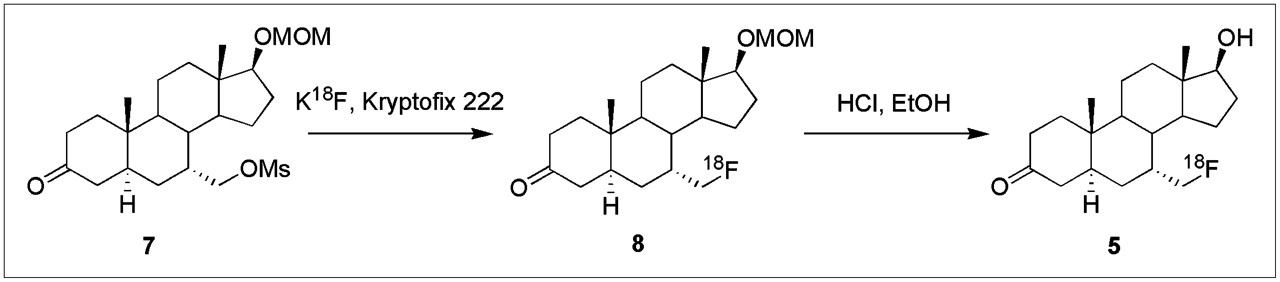
Radiochemical Synthesis of 7α-18F-FM-DHT
18F-fluoride ion (4.70 GBq, 127 mCi) was delivered in 200 μL of H218O and added to a clean test tube with a screw top lid containing K2CO3 (1.5 mg, 11 μmol) and Kryptofix 2.2.2. (6.1 mg, 16 μmol). Water was azeotropically evaporated from the mixture in an oil bath at 110°C using HPLC-grade acetonitrile (3 × 750 μL) under a stream of nitrogen. Meanwhile, in a separate small test tube, 7α-mesyloxymethyl-5α-androstan-3-one 17β-methoxymethyl ether (7) (2.5 mg, 5.7 μmol) was dissolved in dry dimethyl sulfoxide (350 μL). This solution was added to the test tube containing the K18F and Kryptofix and swirled vigorously. This mixture was then subjected to microwave irradiation at 65 W (3 × 30 s). Reaction progress was analyzed by radio-TLC (50% EtOAc/50% hexanes) and determined to have reached approximately 40% of tracer incorporation. Additional irradiation did not produce any significant increase in 18F incorporation. The crude radioligand 18F-8 was diluted with water (30 mL) and passed over a C18 Sep Pack (Waters) to separate the compound from dimethyl sulfoxide. The C18 column was washed with H2O (2 × 8 mL), and the product was then eluted with EtOH (2 mL). The EtOH solution containing the intermediate 18F-8 was treated with 2N HCl (500 μL), capped, and heated at 100°C for 10 min, reaction progress being confirmed by radio-TLC. The acidic solution was quenched with 1N NaOH (1 mL), and the solvent was removed under reduced pressure on a rotary evaporator and heat. The crude product was dissolved in a 1:1 mixture of acetonitrile and H2O (2 mL) and injected through a Teflon (Pall Life Sciences) filter onto a reversed-phase HPLC column (53% CH3CN/47% H2O, 4 mL/min) to obtain 799 MBq (21.6 mCi) of 7α-18F-FM-DHT (18F-5), which eluted at 8.27 min (34%, decay-corrected, 112 min) with a specific activity of greater than 48 GBq/μmol (1,300 Ci/mmol) by analytic HPLC. Product identity was verified by coelution of this activity with coinjected authentic product on analytic reversed-phase HPLC. An aliquot (111 MBq, 3.01 mCi) was taken and combined with CH3CN (1 mL), and the resulting solution was evaporated under reduced pressure on a rotatory evaporator with gentle heating. To the resulting dry flask was added EtOH (200 μL), followed by saline (1.8 mL). From this, an aliquot (14.8 MBq, 0.400 mCi) was diluted with EtOH (300 μL) and saline (3.0 mL), and the resulting solution was taken up into 20 fractions for rat biodistribution studies and quality control analysis.
Radiochemical Synthesis of 7α-18F-FM-norT
18F-fluoride (4.18 GBq, 113 mCi) was delivered in 200 μL of untreated H218O and added to a clean test tube with a screw-top lid containing K2CO3 (1.8 mg, 10 μmol) and Kryptofix 2.2.2. (6.4 mg, 17 μmol). Water was azeotropically evaporated from the mixture in an oil bath at 110°C using HPLC-grade acetonitrile (3 × 750 μL) under a stream of nitrogen. Meanwhile, in a separate small test tube, 7α-mesyloxymethyl-cyclic-3-(1,2-ethanediyl acetal)-estr-5,10-en-3-one 17β-methoxymethyl ether (9) (2.5 mg, 5.7 μmol) was dissolved in dry dimethyl sulfoxide (350 μL). This solution was added to the test tube containing the K18F and Kryptofix and swirled vigorously. This mixture was then subjected to microwave irradiation at 65 W (2 × 30 s). Analysis of the progress of reaction by radio-TLC (90% EtOAc/10% hexanes) showed that approximately 60% of the tracer was incorporated. Additional irradiation did not produce any significant increase in 18F incorporation. The crude radioligand 18F-10 was diluted with water (30 mL) and passed over a C18 Sep Pack to separate the compound from dimethyl sulfoxide. The C18 column was washed with H2O (2 × 8 mL), and the product was then eluted with EtOH (1.5 mL). The EtOH solution containing the intermediate 18F-11 was treated with 2N HCl (300 μL), capped, and heated at 105°C for 10 min, the reaction being confirmed by radio-TLC. The acidic solution was quenched with 1N NaOH (500 μL), and the solvent was removed under reduced pressure on a rotary evaporator and gentle heating. The crude product was dissolved in a 1:1 mixture of acetonitrile and H2O (2 mL) and injected through a Teflon filter onto a reversed-phase HPLC column (45% CH3CN/55% H2O, 4 mL/min) to obtain 654 MBq (17.7 mCi) of 7α-18F-FM-norT (18F-6), which eluted at 7.52 min (24%, decay-corrected, 73 min) with a specific activity of 26 GBq/μmol (700 Ci/mmol) by analytic HPLC. Product identity was verified by coelution of this activity with coinjected authentic product on analytic reversed-phase HPLC. The fractions were combined, diluted with water (60 mL), and passed over a C18 Sep Pack as before. The C18 column was washed with H2O (10 mL) and the product was then eluted with EtOH (600 μL). An aliquot (66 MBq, 1.80 mCi) was diluted with EtOH (200 μL), which was followed by saline (1.8 mL). From this, an aliquot (17.7 MBq, 0.481 mCi) was diluted with EtOH (300 μL) and saline (3.0 mL), and the resulting solution was taken up into 20 fractions for rat biodistribution studies and quality control analysis.
Biodistribution Study in Sprague–Dawley Male Rats
In the following experiments, animals were handled in accordance with the Guidelines for the Care and Use of Research Animals established by the Animal Studies Committee at Washington University, School of Medicine. A complete description of the animal-handling procedure, including animal care, anesthesia, and monitoring, has been published (32). After tracer administration, the animals were allowed to wake up, and normal husbandry was maintained until euthanasia by cervical dislocation.
In both biodistribution studies, purified 18F-5 or 18F-6 was reconstituted in 10% ethanol-saline and injected (via the tail vein) into mature male Sprague–Dawley rats (250 g) that had been injected subcutaneously with 1 mg of diethylstilbestrol in 0.2 mL of sesame oil 24 and 3 h before the injection of the tracer to suppress endogenous androgen biosynthesis (33). Doses of radiotracer used were 0.48 MBq (13 μCi/animal) for 18F-5 and 0.55 MBq (15 μCi/animal) for 18F-6. Animals were sacrificed at 1 and 2 h after injection. To determine whether uptake was mediated by a high-affinity, limited-capacity system, one set of animals was coinjected with the radiotracer together with a blocking dose (36 μg) of DHT. At each time point, groups of 5 animals each were killed; tissues of interest were removed, weighed, washed with saline, and blotted dry; and the radioactivity was counted. The percentage injected dose (%ID) was calculated by comparison with dose standards prepared from the injected solution of appropriate counting rates, and the data were expressed as %ID/g of tissue.
Metabolism Study
To investigate the in vivo stability of 7α-18F-FM-DHT (18F-5) and 7α-18F-FM-norT (18F-6), prostate samples were taken from each time group of the diethylstilbestrol-treated rats used in the biodistribution studies and homogenized, and a portion of activity was extracted with acetonitrile (3 × 1 mL). The ratio of intact compound to metabolites was followed using radiometric normal-phase thin-layer chromatographic analysis of the acetonitrile extract.
RESULTS
Radiochemistry
The radiochemical synthesis of 7α-18F-FM-DHT (18F-5) was accomplished according to the following procedure (Scheme 1). 18F-fluoride ion was activated by addition of K2CO3 and Kryptofix 2.2.2. and incorporated into the methanesulfonyl (mesyl) precursor 7 to form the radiolabeled intermediate 18F-8, which was treated with acid to remove the methoxymethyl ether protecting group. The resulting radiolabeled product, 18F-5, was purified by reversed-phase HPLC, giving an overall radiochemical yield of 32% ± 5% (n = 4, decay corrected). Reanalysis by analytic HPLC indicated that this material was radiochemically homogeneous, and there was no evidence of chemical impurities with elution times different from that of the unlabeled analog (28). Specific activity was determined to be more than 48 GBq/μmol (1,300 Ci/mmol) by analytic HPLC comparison with an authentic sample.

Radiochemical synthesis of 7α-18F-FM-DHT.
The radiochemical synthesis of 7α-18F-FM-norT (18F-6) followed that of the DHT analog 18F-5 (Scheme 2). Addition of 18F-fluoride ion to the mesyl precursor 9 formed the labeled intermediate 18F-10, from which the methoxymethyl ether group was removed to afford the final product, 18F-6, in an overall radiochemical yield of 28% ± 7% (n = 3, decay corrected) after HPLC purification. Reanalysis by analytic HPLC indicated that this material was radiochemically homogeneous, with no evidence of chemical impurities with elution times different from that of the unlabeled analog (28). Specific activity was determined to be approximately 26 GBq/μmol (700 Ci/mmol) by analytic HPLC comparison with an authentic sample.
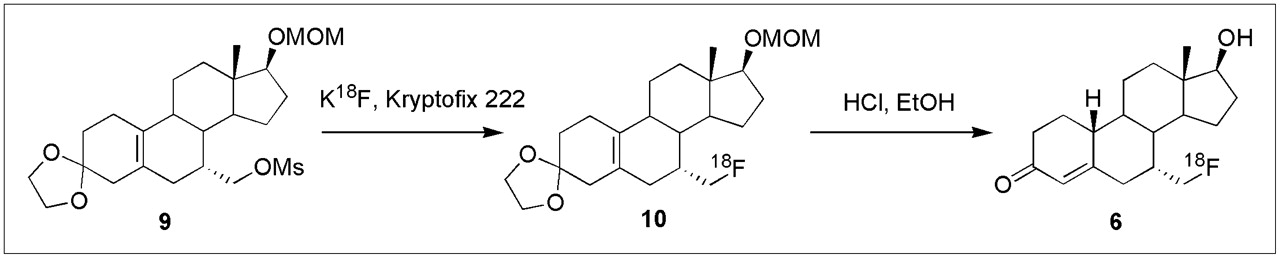
Radiochemical synthesis of 7α-18F-FM-norT.
Biodistribution and In Vivo Metabolism of 7α-18F-FM-DHT
The biodistribution of 7α-18F-FM-DHT (18F-5) at 1 and 2 h after injection into androgen-depleted mature male rats is shown in Table 1. 7α-18F-FM-DHT is found to be selectively retained by the prostate through the AR. Tissue samples at the 1-h point show high activity in the prostate relative to blood and muscle, a control tissue, with decreased activity in prostate and lowered prostate-to-blood and prostate-to-muscle ratios in the DHT-blocked animals. The extended time point of 2 h shows continued 18F activity selectivity in the prostate, with high prostate-to-blood and prostate-to-muscle ratios; all of these values drop in the 2-h DHT-blocked animals. Analysis of the tissue uptake data using Student's t indicates a greater than 80% confidence that the androgen-depleted prostate has a higher level of incorporation of 18F-5 than in the DHT-blocked rats at the 1 h-time point and a greater than 95% confidence interval at the 2 h-time point.
Biodistribution of 7α-18F-FM-DHT (18F-5)
Metabolism studies of 7α-18F-FM-DHT show that at the 1-h sample, 70% of activity present in the prostate tissue sample was extractable into the acetonitrile solution and at the 2-h time point, 85% of activity was extractable into acetonitrile. Of the activity in the prostate extract, approximately 30% at 1 h and 25% at 2 h is due to intact ligand (data not shown). The remainder of the activity is due to unidentified 18F-containing organic material with much greater polarity than that of the intact ligand. Little of the activity in the extract (<5%) is due to inorganic fluorine in either time group.
The prostate is a fairly fatty organ, facilitating diffusion of the hydrophobic radioligand into and out of the target organ and possibly resulting in high initial uptake of lipophilic compounds. Ligand binding to the AR, however, is required for retention of the radioisotope in the cellular nucleus. The relatively modest level of block of 7α-18F-FM-DHT by DHT most likely reflects the high level of non-AR binding metabolites that accumulate in the prostate, which would not be subject to DHT block.
Biodistribution and In Vivo Metabolism of 7α-18F-FM-norT
The biodistribution data of 7α-18F-FM-norT (18F-6) at 1 and 2 h after injection in androgen-depleted mature rats is shown in Table 2. 7α-18F-FM-norT is also taken up selectively by the prostate through the AR. Tissue samples at the 1-h point show high activity levels in prostate uptake versus blood and nontarget tissue (muscle), with a very marked DHT block in prostate, giving large reductions in prostate-to-blood and prostate-to-muscle ratios. At 2 h, prostate activity and prostate-to-blood and prostate-to-muscle ratios are all high, and these decline precipitously on DHT blockade. Analysis of the tissue uptake data using Student's t indicates greater than 99% confidence that the androgen-depleted prostate has a higher level of incorporation of 18F-6 than in the DHT blocked rats at both the 1- and 2-h time points.
Biodistribution of 7α-18F-FM-norT (18F-6)
Metabolism studies of 7α-18F-FM-norT show that at the 1-h sample, 78% of activity present in the prostate tissue sample was extractable into acetonitrile and at the 2-h time point, 84% of activity was extractable. At both the 1- and 2-h time points, approximately 100% of activity in the prostate extract is due to intact ligand (data not shown). Thus, little of the activity in the extract (<5%) is due to either inorganic fluorine or other 18F-containing organic materials in either time group. This finding corroborates the biodistribution finding that the reduced prostate activity on saturation of AR by the DHT coinjection actually reflects the displacement of AR-bound active ligand.
DISCUSSION
SHBG has the capacity to play a significant role in the transport and delivery of androgens to AR-rich tissues, although it is not clear whether one would expect its role to be beneficial or detrimental with respect to PET of AR in prostate tumors with androgen radiopharmaceuticals. SHBG binds androgens in plasma tightly, protecting them from metabolism and slowing their clearance, but this complex also interacts with the SHBG receptor, potentially facilitating steroid cellular entry. From our previous work, it appeared that in species where SHBG is found (primates), estrogen and androgen radiopharmaceuticals needed to have significant SHBG binding to show good target tissue or tumor uptake, whereas in species lacking this carrier (e.g., rats), good target tissue uptake occurred regardless of SHBG binding affinity (9,10,14,17,18,20,34–36). These prior studies, however, involved comparisons between pairs of estrogen or androgen radiopharmaceuticals that were not always as well matched structurally as they might have been, leaving open the possibility that the differences in target uptake behavior seen between species was due to structural effects other than ones engendering high or low SBHG binding.
Our aim in this study was to develop a pair of androgen radiopharmaceuticals—carefully designed to be different in SHBG affinity but otherwise as close in structure and AR binding as possible—that could be used to make a more definitive test of the importance of SHBG binding in AR-mediated uptake. We accomplished this aim by basing one of these compounds on the DHT skeleton, which engenders high AR and high SHBG binding, and one on the 19-norT skeleton, which maintains high AR binding but has low SBHG binding. To maintain structural consistency, both of these agents were labeled with 18F at the same position, on a 7α methyl group. Androgens labeled at the 7α position with halogens (fluorine and iodine) are known and have good AR binding affinity (37), but 7α-fluoromethyl androgens are novel. We chose this new site less for chemical novelty than to avoid fluorine labeling at the 16β position, a site known to be prone to defluorination (17), as well as to incorporate into both compounds a 7α-methyl group, which has been shown to slow metabolism of steroids (30). The 2 novel radiolabeled androgens, 7α-18F-FM-DHT (5) and 7α-18F-FM-norT (6), nicely fulfill these specified requirements: Their AR binding affinities are almost identical, and their structures are as well matched as is feasible, consistent with the need for their SHBG binding affinities to be different; this difference in SHBG binding is 22-fold.
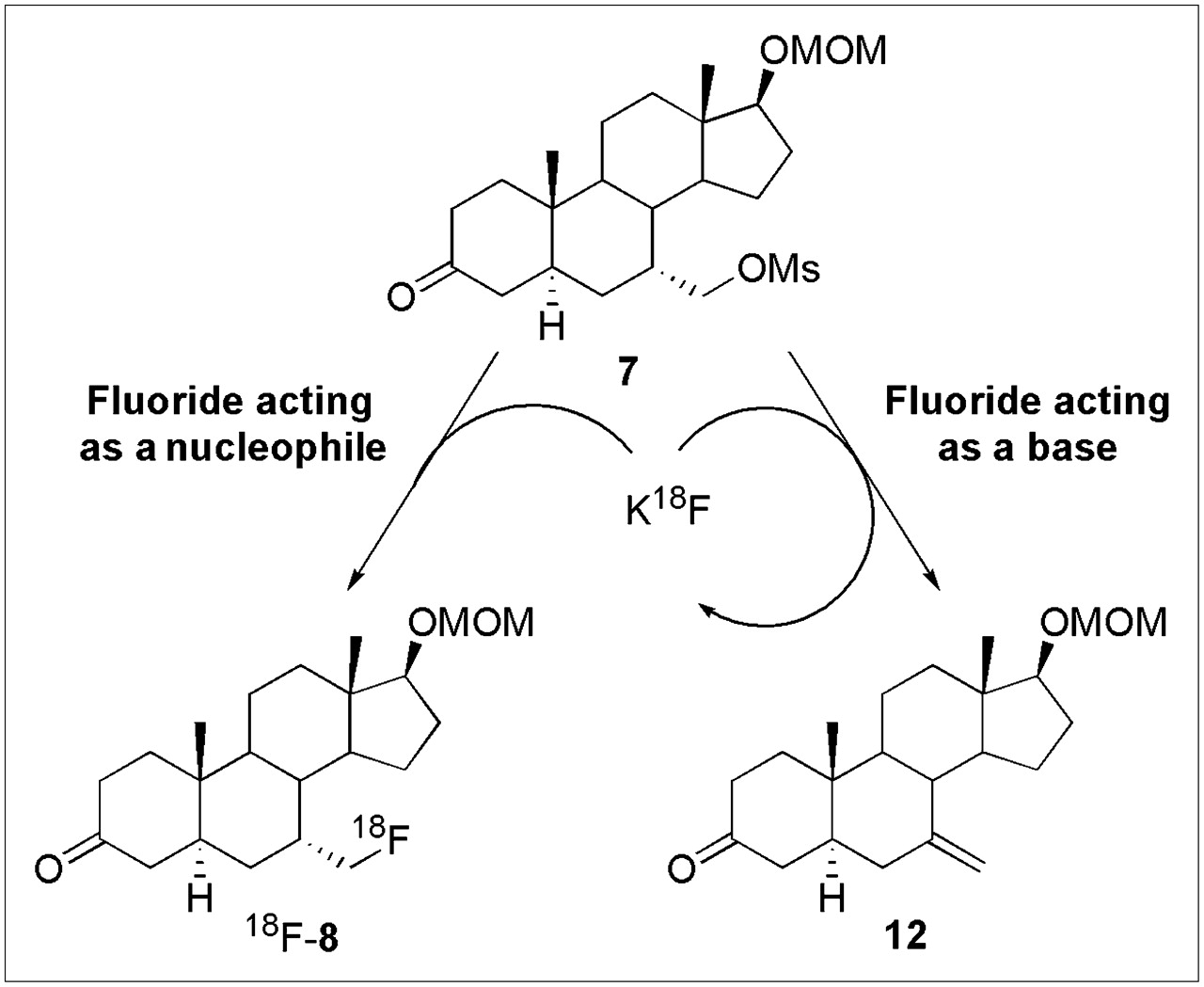
In our earlier work on the macroscopic scale synthesis of 7α-fluoromethyl-dihydrotestosterone (5) and 7α-fluoromethyl-nortestosterone (6), we found that the basic nature of fluorine required us to use an ionic liquid medium (38) to successfully effect fluoride ion displacement of the mesylate precursors 7 and 9; even so, the fluorine-substituted intermediates, 8 and 10, respectively, were obtained in only 7% and 11%, respectively, with base-induced elimination being the major course of reaction (28). In fact, use of other nucleophilic fluorine reagents, such as TBAF and Kryptofix 222–KF, in the case of 7α-fluoromethyl-dihydrotestosterone, led to complete elimination of the activated mesylate 7, giving alkene 12 as the sole product (Scheme 3).
Fortuitously, as is sometimes the case, we found that 18F radiolabeling at the tracer level worked in much higher yield, as can be understood by reference to Scheme 3. Although the bulk of the mesylate precursor 7 undergoes this elimination process to alkene 12 rather than substitution to form the desired intermediate 8, fluoride, acting as a base (right), is not consumed by the elimination route and is thus available to recycle back for further attempts in the nucleophilic substitution reaction (left). Eventually, and with sufficient excess of precursor 7 (which in practice is only 2.5 mg), the trace levels of 18F-fluoride ion eventually become incorporated by substitution. Thus, we were able to obtain decay-corrected radiochemical yields (based on fluoride ion as the limiting reagent) of 24%–34%, much greater than the yield of macroscopic products, which are based on the mesylate precursor. The efficiency of the radiolabeling process also allowed us to use other, more facile, labeling conditions, such as Kryptofix 222, instead of ionic liquids.
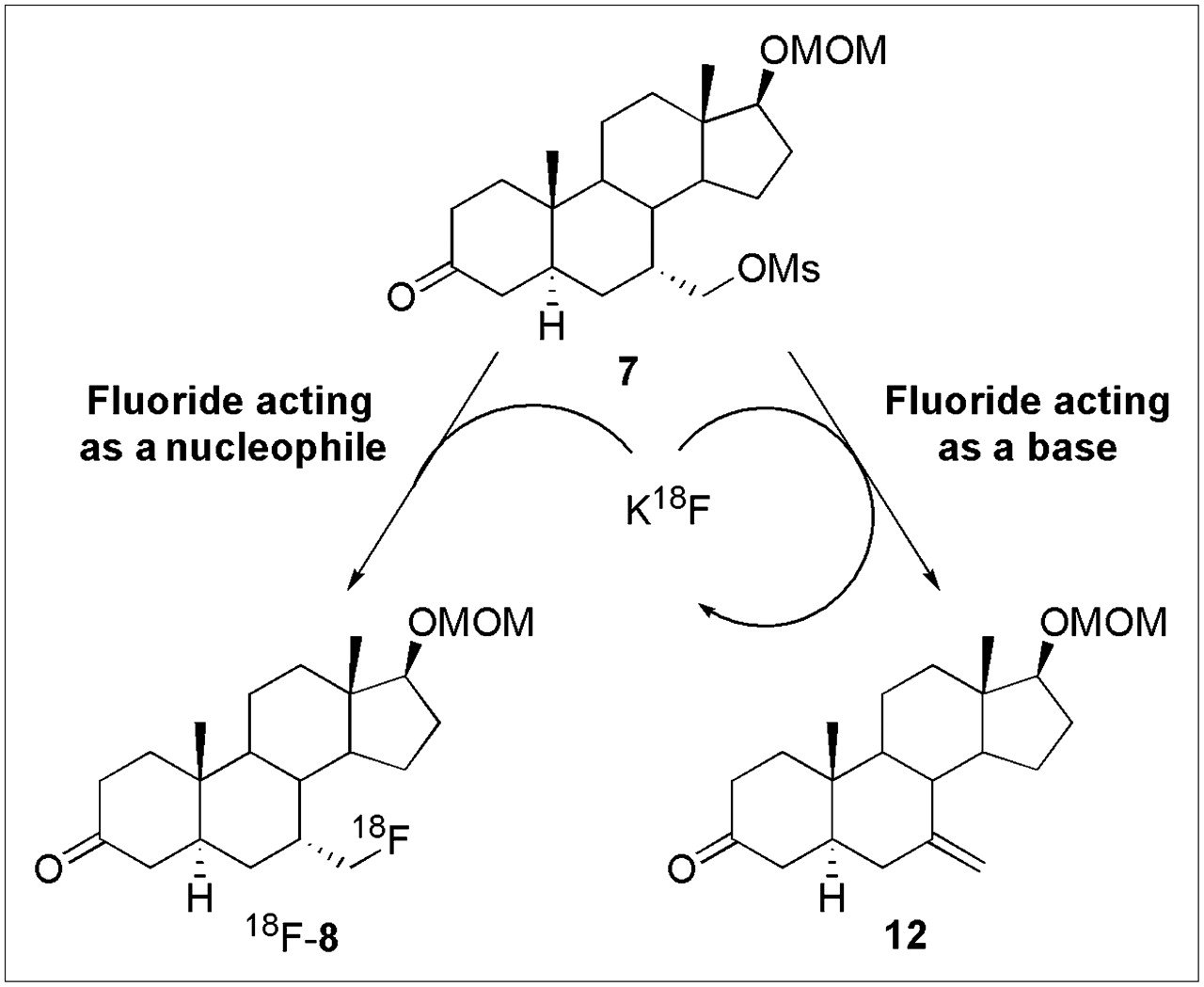
Cyclic use of 18F-fluoride ion in synthesis of 7α-18F-FM-DHT.
In biodistribution studies in androgen-depleted rats, both 7α-18F-FM-DHT (18F-5) and 7α-18F-FM-norT (18F-6) show the hallmarks of AR-mediated uptake: greater uptake in a target tissue (prostate) than in blood or a control tissue (muscle), but there are also significant quantitative differences between the 2 compounds. As %ID/g, 7α-18F-FM-norT (18F-6) shows nearly twice the prostate activity of 7α-18F-FM-DHT (18F-5), and this norT-based radioligand (6) is blocked much more completely by DHT than is the DHT-based ligand (5). On the other hand, ratios of prostate uptake to muscle or blood uptake at 2 h are somewhat greater for the DHT-based ligand (5) than for the 19-norT-based one (6).
The compounds also differ considerably in their metabolic stability in the rat, with the DHT-based one showing more rapid formation of polar metabolites, some of which were found in the prostate. The fact that prostate activity with the 19-norT-based ligand (6) is almost all unmetabolized compound, whereas only 25%–30% of prostate activity is authentic ligand 5, could account for the reduced level of prostate uptake and less complete DHT block with the DHT-based ligand. Notably, neither of these compounds underwent defluorination to a major degree, which was certainly the case with other 18F-fluoroandrogens with D-ring radiolabels (17).
Although the 2 compounds are based on somewhat different androgen steroid skeletons, DHT for compound 5 and 19-norT for compound 6, these differences were necessitated by the need for one compound to bind well to SHBG and the other poorly. Both compounds showed clear evidence of AR-mediated uptake in the prostate, although target uptake selectivity of the DHT-based ligand (5) was compromised to some degree by its relatively rapid metabolism. It is likely, as we and others have found for some other 18F-labeled androgens, that metabolism of compound 5 in nonhuman primates (14) and humans (18,19) is likely to be less rapid than it was in rats, likely making these 2 compounds better comparators for assessing the role of SHBG on AR-mediated uptake in prostate cancer in humans.
CONCLUSION
In conclusion, in this preliminary study we report the development of 2 structurally paired 18F-labeled androgens, 7α-18F-FM-DHT and 7α-18F-FM-norT, whose in vitro binding and in vivo biodistribution properties appear well suited for making a definitive study of the importance of SHBG binding in the AR-mediated uptake of 18F-fluoroandrogens in AR-positive prostate cancer. Human clinical imaging studies are being considered to further evaluate these compounds for their potential in PET of prostate cancer.
Acknowledgments
We thank Kathy Carlson for her assistance in AR binding affinity analysis and Nicole M. Fettig for her technical support with the biodistribution studies. This work was supported in part by grants from the DOE (FG02 86ER60401) and (FG02 84ER60218) and the NIH (PHS 1R24 CA86307).
Footnotes
-
COPYRIGHT © 2008 by the Society of Nuclear Medicine, Inc.
References
- Received for publication November 12, 2007.
- Accepted for publication February 26, 2008.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}