


Abstract
Reporter gene imaging has great potential for many clinical applications including the tracking of transplanted cells and monitoring of gene therapy. However, currently available reporter gene–reporter probe combinations have significant limitations with the biodistribution of the reporter probe and the specificity and immunogenicity of the reporter gene. The objective of the present study was to evaluate a new approach for reporter gene imaging based on cell surface expression of antibody fragments that can irreversibly bind to radiometal chelates. Methods: We developed a new reporter gene, designated 1,4,7,10-tetraazacyclodocecane-N,N′,N″,N‴-tetraacetic acid (DOTA) antibody reporter 1 (DAbR1), which consists of the single-chain Fv (scFv) fragment of the anti-Y-DOTA antibody 2D12.5/G54C fused to the human T cell CD4 transmembrane domain. The corresponding reporter probe is yttrium-(S)-2-(4-acrylamidobenzyl)-DOTA (*Y-AABD), a DOTA complex that binds irreversibly to a cysteine residue in the 2D12.5/G54C antibody. U-87 glioma cells were stably transfected with a DAbR1 expression vector. Binding of *Y-AABD to transfected and wild-type cells was studied in vitro and in vivo. Results: Flow cytometry revealed high expression of the DAbR1 protein on the cell surface of tumor cells. Uptake of 90Y-AABD in DAbR1-expressing human U-87 glioma xenografts was 6.2 (±1.3) percentage injected dose per gram (%ID/g) at 1 h and 4.9 (±0.62) %ID/g at 24 h after injection. The corresponding tumor-to-plasma ratios were 45:1 and 428:1, respectively. Uptake by U-87 tumors without the DAbR1 gene was 0.16 (±0.02) %ID/g at 1 h and 0.05 (±0.03) %ID/g at 24 h. PET images in mice with 86Y-AABD demonstrated intense uptake in DAbR1-positive tumors and low background activity in the liver. Conclusion: These findings indicate that cell surface expression of radiometal chelate binding antibodies such as 2D12.5/G54C is a promising strategy for reporter gene imaging.
Transplantation of genetically engineered cells is a field of intense research in almost all areas of medicine and may lead to the treatment of a large number of common diseases. Prominent examples include transplantation of stem cells after myocardial infarction (1,2), adoptive cell immunotherapy of melanoma and other malignant diseases (3), islet cell transplantation in insulin-dependent diabetes (4), and treatment of Parkinson disease and other neurodegenerative disorders with neuronal cells (5,6). Imaging techniques are valuable to these diverse applications, to study noninvasively and quantitatively the location, viability, and function of the transplanted cells in animal models and patients. Such techniques have to be both highly sensitive and highly specific, because small amounts of transplanted cells need to be identified and the signal from the much larger number of host cells should not interfere with the imaging of the transplanted cells.
Currently, reporter gene imaging with PET appears best suited to meet these stringent requirements. PET imaging of reporter genes involves the administration of a probe that is selectively bound or metabolized via interaction with the gene product in the reporter gene–transduced cells. Herpes simplex virus type 1 thymidine kinase (HSV1-tk) and its mutant HSV1-sr39tk are the most widely used reporter genes for PET and have been intensively studied for the monitoring of cancer gene therapy (7–9). The corresponding reporter probes are 124I-fluoro-deoxy-arabinofuranosyl-5-iodouracil (124I-FIAU), 18F-fluoro-hydroxymethybutyl-guanine (18F-FHBG), and 18F-2′-fluoro-2′-deoxy-1-β-d-β-arabinofuranosyl-5-ethyluracil (18F-FEAU). Because HSV1-tk is a viral protein, immune reactions are a serious concern for the use of this reporter gene for imaging of stem cells or other cells that are intended to survive for longer periods after transplantation. Indeed, immune responses specific to the HSV-tk protein were reported in 5 of 6 patients who received anti-HIV cytotoxic HSV1-tk–transduced T lymphocytes, and the transduced T cells were eliminated (10). Furthermore, currently available reporter probes for HSV1-tk or HSV1-sr39tk are, to a small extent, also phosphorylated by mammalian thymidine kinases. Thus, rapidly proliferating mammalian cells, such as cancer cells, show some uptake of these reporter probes. This nonspecific signal has been shown to limit the detection of lymphocytes expressing HSV1-sr39tk within the tumor tissue (11).
Several other reporter gene combinations have been evaluated. These include the sodium iodide symporter (NIS) (12,13), the norepinephrine transporter (NET) (14,15), and somatostatin (16) and dopamine D2 receptors (17). Although these mammalian proteins avoid the problem of immunogenicity, their physiologic expression pattern limits the organ systems that can be studied with these reporter genes. Furthermore, currently available reporter probes for these reporter genes have unfavorable biologic (high hepatobiliary clearance) or physical properties (low branching ratio and long physical half-life of the radiolabel) for PET.
To overcome these limitations, we explored the feasibility of using cell surface–bound antibody fragments as reporter genes that can bind covalently to small haptens such as radiometal chelates, which act as reporter probes (Fig. 1A). Two earlier studies (18,19) have used related strategies, but without irreversible capture of the probe by the antibody. In general, the antibody-probe approach has 3 advantages, compared with other reporter gene–reporter probe combinations. First, the pharmacokinetics of the reporter probe can easily be optimized. Haptens can be designed to exhibit rapid blood clearance and low nonspecific binding to normal tissues. Once a hapten demonstrates favorable properties for imaging, an antibody can be raised against this hapten and used as a reporter gene. With the current reporter gene–reporter probe combinations, optimization of the pharmacokinetic properties of the reporter probe is limited because any modifications can also affect target binding. As a consequence, it is difficult to optimize both target binding and pharmacokinetic properties, and the number of available reporter gene–reporter probe combinations has remained limited despite intensive research. Second, antibodies can provide an extremely high specificity for reporter probes. Furthermore, the reporter probes can be purely synthetic molecules with no natural analogs, making it straightforward to avoid problems such as modification by naturally occurring enzymes or competition for target binding by endogenous molecules. Also, the antibody can be engineered to bind the reporter probe irreversibly (with infinite affinity), so that the signal persists in the target while the probe clears from normal tissues, whereas antibody complexes with small molecules may dissociate too quickly for optimal results (20). In developing an infinite affinity system, it is essential that the reporter probe clears from the subject without nonspecific attachment to normal proteins. We accomplished this by incorporating a mild acryloyl group into the reporter probe, which reacts efficiently in the engineered antibody binding site but has low reactivity with blood components (21). Finally, the number of antibodies that can be used as reporter genes is virtually unlimited. In contrast, only a limited number of suitable viral or mammalian reporter genes exists. Cross-reactivity must be considered because, for example, the antibody used here can bind irreversibly to 1,4,7,10-tetraazacyclodocecane-N,N′,N″,N″′-tetraacetic acid (DOTA) complexes containing any of the lanthanide elements (22), but synthetic chemistry can easily meet this challenge by preparing nonbinding analogs. Therefore, antibody-based reporter genes are better suited for the multiplexed imaging of the expression of several genes.

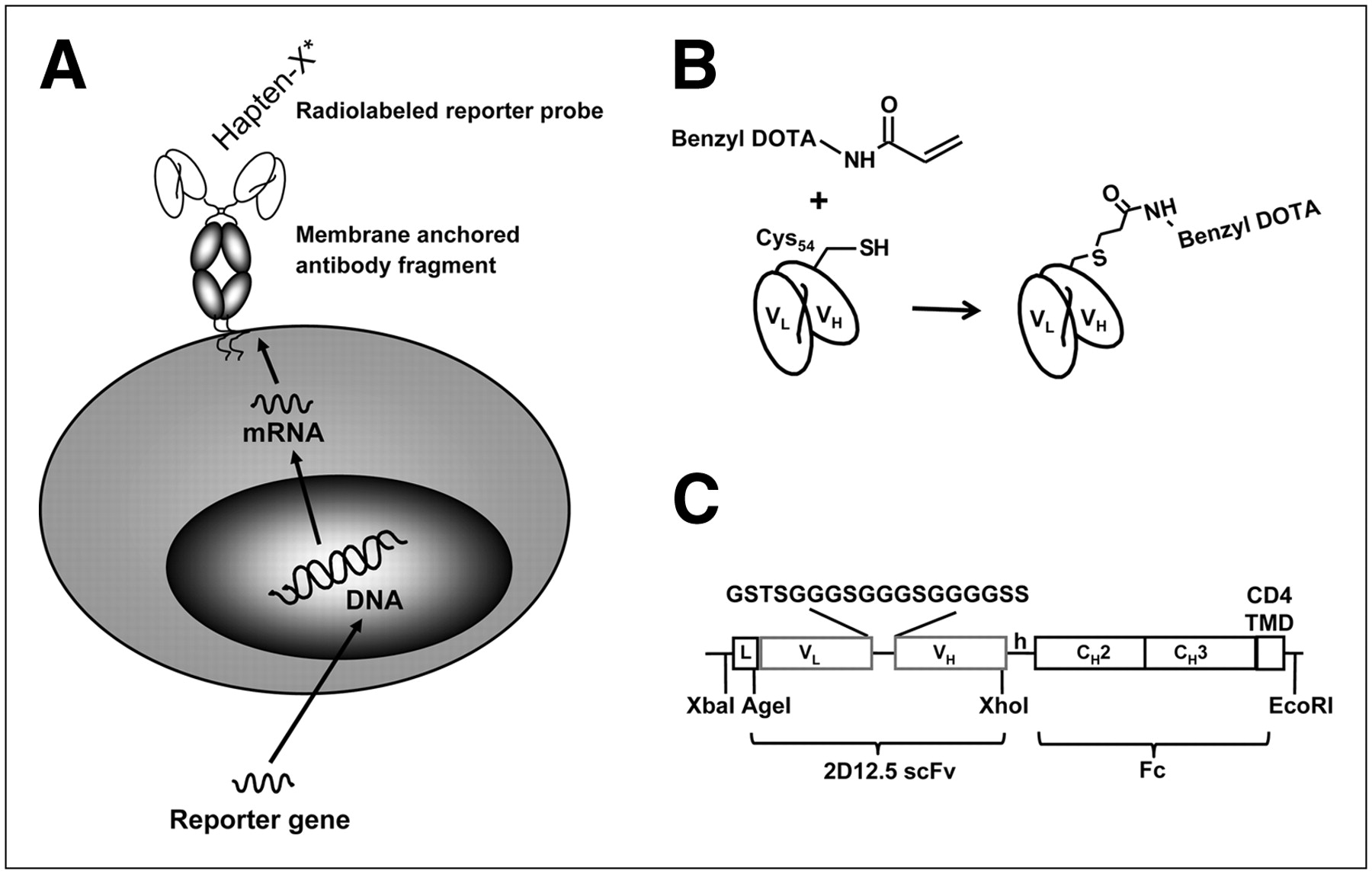
(A) Use of antibody fragments as reporter genes. Cells are transfected with gene-encoding antibody fragment with membrane-anchoring domain. Antibody fragment is expressed on cell surface and acts as artificial receptor for binding small radiolabeled haptens. (B) Irreversible binding of AABD to 2D12.5/G54C. Covalent bond is formed between cysteine 54 in variable heavy chain of antibody and acryl group of AABD. (C) Schematic presentation of DAbR1 fusion gene. DAbR1 reporter gene consists of scFv fragment of 2D12.5/G54C fused to human Fc (CH2-CH3) fragment and human T cell CD4 transmembrane domain, which anchors DAbR1 in plasma membrane. VH = variable heavy; VL = variable light.
To demonstrate the feasibility of antibody-based reporter genes, we have used the engineered antibody 2D12.5/G54C as a reporter gene and yttrium-(S)-2-(4-acrylamidobenzyl)-DOTA (*Y-AABD) as a reporter probe. The 2D12.5/G54C antibody binds yttrium-DOTA and also other rare earth–DOTA metal complexes. Binding of *Y-AABD to 2D12.5/G54C brings the acryloyl group of AABD near an engineered cysteine side chain at position 54 of the heavy chain of the antibody (Fig. 1B), which results in covalent binding of AABD via a Michael reaction (22). To achieve cell surface expression of 2D12.5/G54C, we fused the single-chain Fv fragment (scFv) of 2D12.5/G54C with a human immunoglobulin Fc region and the CD4 transmembrane domain. The resulting protein DOTA antibody reporter 1 (DAbR1) was expressed in the human glioblastoma cell line U-87. Cell culture and biodistribution studies and small-animal PET imaging of tumor-bearing mice demonstrated high and specific binding of *Y-AABD to tumor cells expressing DAbR1. These data indicate that antibody-based reporter genes that irreversibly capture probe molecules represent a promising new platform for the development of new reporter probe–reporter gene combinations.
MATERIALS AND METHODS
Design and Gene Assembly of 2D12.5/G54C scFv-Fc-CD4 Construct (DAbR1 Gene)
The variable light and heavy genes from the murine 2D12.5 monoclonal antibody containing the G54C mutation (22) were amplified and assembled into scFv by polymerase chain reaction (PCR). In separate PCRs, genes encoding human IgG4 Fc (hinge-CH2-CH3; gift from Dr. Mirela Claude Jensen) and CD4 transmembrane domain and truncated intracellular domain of the CD3 complex ζ-chain (23) were amplified and assembled into 1 gene (Fc-CD4). The final 2 PCR products were assembled in pUC (New England Biolabs) already containing a 5′ leader peptide sequence using the restriction enzyme sites shown in Figure 1C. The complete complementary DNA construct encoding the leader and scFv-Fc-CD4 was transferred using XbaI and EcoRI sites into the mammalian expression vector pcDNA3.1 (−) under the control of the cytomegalovirus promoter (Invitrogen).
Cell Line, Culture Conditions, and Transfection Procedure
The U-87 human glioma cell line, obtained from American Type Culture Collection, was maintained at 37°C in complete Dulbecco's modified Eagle's medium containing 10% (v/v) fetal bovine serum. The DAbR1–pcDNA vector was transfected into U-87 cells by electroporation (Amaxa) according to the manufacturer's instructions. Stably transfected cells were selected with the antibiotic G418 (500 μg/mL; Invitrogen) in complete Dulbecco's modified Eagle's medium. After about 3 wk, G418-resistant clones were isolated and analyzed individually for the expression of human Fc by Western blots and flow cytometry (fluorescence-activated cell sorter [FACS]). These experiments were performed when the cell density was about 10 million cells/75-cm2 tissue culture flask.
Western Blot Analysis
Samples (20 μg of total proteins from whole-cell lysates) were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (10%) and transferred to a nitrocellulose membrane. After 1 h at room temperature in blocking buffer (Licor Bioscience), the membranes were probed with rabbit antihuman Fc fragment (1:1,000; Jackson ImmunoResearch Lab). The membranes were washed with phosphate-buffered saline (PBS) with polysorbate 20 and incubated with IRDye 800–conjugated goat antirabbit antibody (infrared dye, 1:10,000; Licor Bioscience). Membranes were washed 4 times with Tris-buffered saline with polysorbate (TBST), and then rinsed with PBS before being scanned using an Odyssey infrared imaging system according to the manufacturer's instructions (Licor Bioscience).
Flow Cytometry
Either DAbR1-expressing or wild-type U-87 cells (1 × 106) were incubated with rabbit antihuman IgG antibody (Fc-specific) for 20 min in PBS containing 1% fetal bovine serum (FACS buffer). Cells were washed twice with formaldehyde buffer and incubated with Alexa Fluor 488–conjugated goat antirabbit IgG (Invitrogen). After 2 washes, cells were analyzed by a FACScan flow cytometer (Becton Dickinson) using the CellQuest 3.1 software for acquisition and the ModFit LT 2.0 software (Verity) for analysis.
Preparation of 90Y-AABD and 86Y-AABD
The bifunctional chelating agent AABD was prepared following the method of Corneillie et al. (24). 90Y in 0.05 M hydrochloric acid (specific activity, 18.5 TBq/mg) was purchased from PerkinElmer, and 86Y in 0.1 M hydrochloric acid (specific activity, 1 TBq/mg) was purchased from Isotrace Technologies (25). A total of 37 MBq (1 mCi) of 90Y or 86Y was incubated at 37°C for 1 h with 1.2 mM AABD in 0.5 M triethylammonium acetate buffer (pH, 5.5). At the end of incubation, diethylenetriamine pentaacetate (DTPA) was added to the final concentration of 10 mM and incubated at room temperature for 5 min to scavenge any unchelated 90Y or 86Y.
In Vitro Antibody Binding Assay
One million DAbR1-expressing and wild-type U-87 cells were plated in 6-well plates in triplicate the day before the experiment. On the day of the experiment, cells were incubated with 37 kBq (1 μCi) of 90Y-AABD in 1 mL of RPMI medium containing 0.5% fetal bovine serum, with rocking at room temperature for 2 h. The cells were then washed twice with cold PBS and lysed with 0.1% sodium dodecyl sulfate. The lysates were transferred to scintillation liquid and counted in a β-counter (5600; Packard) to determine the amount of radioactivity bound to the cells. Duplicate 6-well plates without radioactivity were used to determine the number of cells per well.
Xenograft Models
Severe combined immunodeficient mice were purchased from the Jackson Laboratory. All animal manipulations were conducted with sterile techniques using the guidelines of the UCLA Animal Research Committee. Wild-type and DAbR1-expressing U-87 cells were used to generate xenografts in mice. Cells growing exponentially in culture were suspended in 50% PBS and 50% Matrigel (BD Biosciences) and injected subcutaneously at the right shoulder (1–3 × 106 cells/mouse). To evaluate whether the accumulation of 90Y-AABD correlated with DAbR1 expression levels, DAbR1-expressing U-87 cells were diluted with wild-type U-87 cells to obtain a mixed population containing 0%, 10%, 25%, 50%, and 100% of DAbR1-expressing cells. Animals were used for biodistribution studies or underwent small-animal PET/CT scans after tumor volumes had reached 50–100 mm3 (1–3 wk after injection).
Biodistribution Studies in Tumor-Bearing Animals
Mice bearing U-87 wild-type or U-87-DAbR1 xenografts were injected via the tail vein with 1,480 kBq (40 μCi) of 90Y-AABD in 100 μL of saline. Groups of 3 mice each were sacrificed at 1 and 24 h after injection of 90Y-AABD. Blood, organs, and tumors were removed and weighed, and the amount of radioactivity was determined using a β-counter (5600; Packard). Radioactivity determinations were normalized by the weight of the tissue and amount of radioactivity injected to obtain the percentage injected dose per gram (%ID/g) of tissue.
Small-Animal PET/CT Imaging
Small-animal PET/CT scans were performed using the microPET FOCUS 220 (26) (Siemens Preclinical Solutions) and MicroCAT II scanners (Siemens Preclinical Solutions). For PET/CT, mice were anesthetized using 1.5%−2% isoflurane and positioned in a plastic imaging chamber that minimized positioning errors between PET and CT (27). After an injection of 86Y-AABD (3,700 kBq [100 μCi]) via a tail vein catheter, a 1-h dynamic PET scan was acquired. Fourteen hours after injection, the same mice underwent a second, 10-min static scan. Images were reconstructed by filtered backprojection, using a ramp filter with a cutoff frequency of 0.5 Nyquist. Image counts per pixel per second were calibrated to activity concentrations (Bq/mL) by measuring a 3.5-cm cylinder phantom filled with a known concentration of 86Y. Immediately after each PET scan, mice underwent a 7-min micro-CT scan, using routine image acquisition parameters (27). For quantification, activity concentrations were expressed as percentage of the decay-corrected injected activity per gram of tissue by using the AMIDE software (28). Spheric regions of interest were drawn around the tumor and various normal organs. Regions of interest were defined on fused PET/CT images generated by the AMIDE software to ensure accurate anatomic positioning.
Statistical Analysis
Results were expressed as mean ± 1 SD. Statistical comparisons were made by 2-tailed t tests or ANOVA as appropriate. Linear regression analysis was used to study the association between quantitative parameters. A P value of less than 0.05 was considered statistically significant.
RESULTS
Expression of DAbR1 in Human U-87 Glioma Cells and In Vitro Binding of 90Y-AABD
Expression of the DAbR1 protein by transfected U-87 clones was confirmed by Western blots (Fig. 2A). Next, we evaluated whether the DAbR1 protein traffics to the cell surface. FACS analysis (performed on nonpermeabilized cells) demonstrated binding of the anti-Fc antibody to U-87-DAbR1 cells but not to U-87 wild-type cells (Figs. 2B and 2C). Because the anti-Fc antibody cannot cross the cell membrane, specific binding to U-87-DAbR1 cells proves cell surface expression of DAbR1.

(A) Western blots of wild-type and DAbR1-expressing U-87 cell lysates using antihuman Fc antibody for detection. FACS analyses of wild-type (B) and DAbR1-expressing U-87 (C) cells with antihuman Fc antibody. x-axis shows fluorescence due to antihuman Fc antibody binding, and y-axis shows side scatter; both are in arbitrary units, as common in flow cytometry. (D) In vitro binding of 90Y-AABD to wild-type and DAbR1-expressing U-87 cells. cpm = counts per minute; SSC = side scatter; WT = wild type.
After 2 h of incubation with 90Y-AABD, the binding by U-87-DAbR1 cells was 24,484 (±158) cpm/million cells, whereas it was only 253 (±1.4) cpm/million cells for U-87 wild-type cells. Thus, transfection with DAbR1 led to an almost 100-fold increase (ratio, 96.8) in the binding of the reporter probe 90Y-AABD (Fig. 2C). The U-87-DAbR1 cells expressed membrane-bound DAbR1 protein that was able to specifically bind high amounts of the reporter probe 90Y-AABD in vitro.
Biodistribution of 90Y-AABD in Tumor-Bearing Mice
At 1 h after injection, tumor binding of 90Y-AABD in DAbR1-positive tumors was 6.23 (±1.32) %ID/g, and the tumor-to-plasma ratio was 45:1. At 24 h after injection, the binding of 90Y-AABD in DAbR1-positive tumors was 4.89 (±0.62) %ID/g, with a tumor-to-plasma ratio of 428:1 (Tables 1 and 2). At both time points, tumor uptake of 90Y-AABD was significantly higher than that of all other sampled tissues (P < 0.05). Uptake of 90Y-AABD in U-87 wild-type tumors was 0.16 (±0.02) %ID/g at 1 h after injection, which decreased to 0.05 (±0.03) %ID/g at 24 h after injection (Table 1). Thus, the uptake of 90Y-AABD by U-87-DAbR1 was 39-fold and 100-fold higher than that of the wild-type tumors at 1 and 24 h after injection, respectively (P < 0.001). Table 1 summarizes the uptake of 90Y-AABD in the various tissues sampled at 1 and 24 h after injection. Table 2 shows the corresponding tumor-to-organ ratios. Of all normal tissues sampled, the liver showed the highest radioactivity uptake, with approximately 3 %ID/g at 1 h after injection and 0.9 %ID/g at 24 h. For DAbR1 tumors, the tumor-to-liver ratios at 1 and 24 h after injection were 2.36 (±0.44) and 6.30 (±1.32), respectively. The tumor-to-kidney ratios were 7.67 (±2.30) at 1 h after injection and 33.46 (±6.12) at 24 h after injection. The excreted radioactivity, which might contain metabolic products of *Y-AABD, was not investigated further. In mice injected with mixtures of U-87 wild-type and U-87-DAbR1 cells, the accumulation of 90Y-AABD at 24 h after injection correlated with the number of DAbR1-positive U-87 cells (r = 0.89, P < 0.001 for linear fit; Fig. 3).

90Y-AABD accumulation in tumors consisting of mixed populations of DAbR1-positive and -negative U-87 cells. Each data point shows average and SEM for 3 mice. Statistically significant linear relationship between percentage of DAbR1-positive tumor cells and uptake of 90Y-AABD (r = 0.89, P < 0.001) is demonstrated.
Biodistribution of 90Y-AABD (n = 3)
Tumor-to-Blood and Tumor-to-Organ Ratios
Imaging of DAbR1-Positive U-87 Tumors with High Contrast by Small-Animal PET
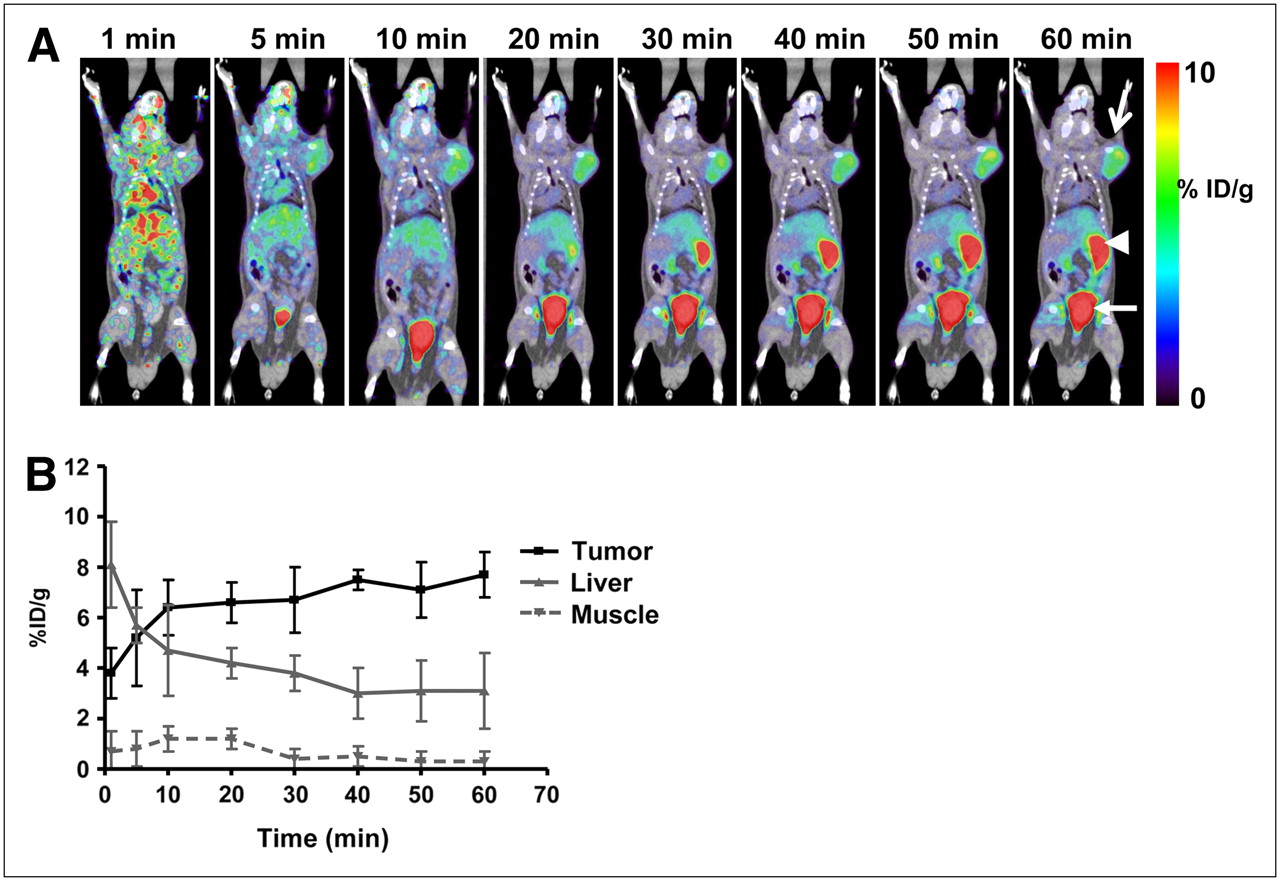
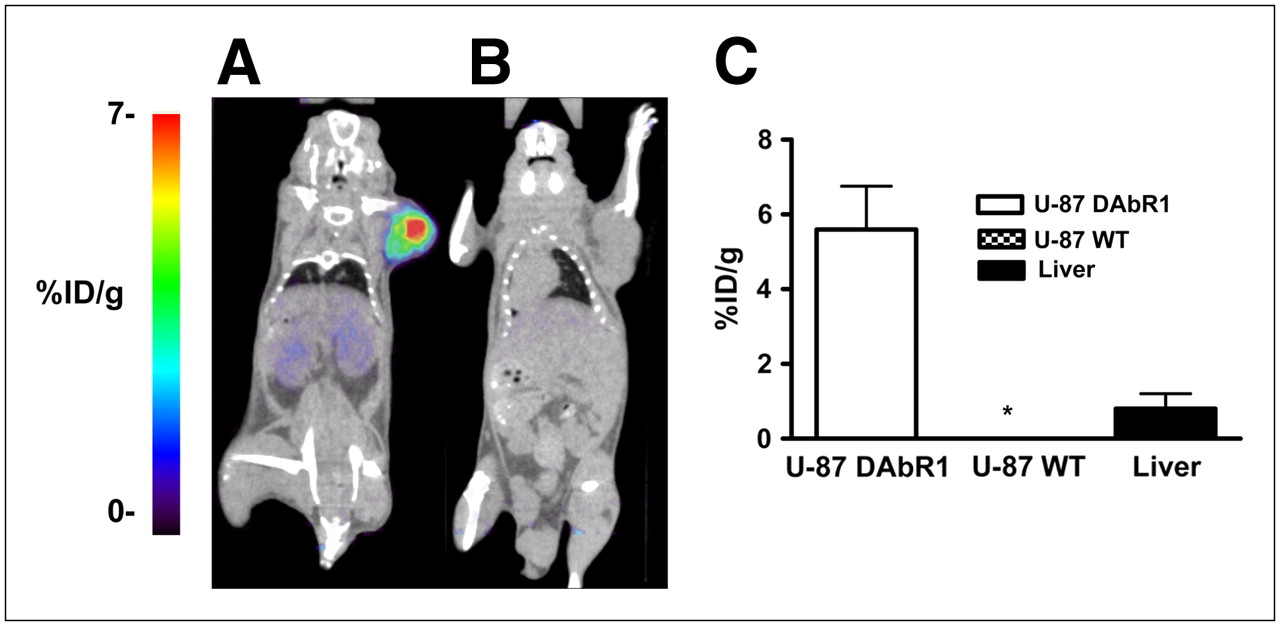
Figure 4A shows a series of dynamic images (coronal sections) from 1 to 60 min after injection in a mouse carrying a U-87-DAbR1 xenograft. Images demonstrate 86Y-AABD accumulation in the tumor and rapid renal and hepatobiliary clearance of 86Y-AABD. Corresponding time–activity curves for tumor tissue, skeletal muscle, and liver are shown in Figure 4B (average of 3 scans). At 1 h after injection, the radioactivity uptake for DAbR1 tumors was 7.7 (±0.9) %ID/g, liver uptake was 3.1 (±1.5) %ID/g, and muscle activity was 0.3 (±0.4) %ID/g. U-87 wild-type xenograft tumors showed background activity levels at 1 h after injection (0.28 [±0.2] %ID/g) with 86Y-AABD. In PET scans acquired at 14 h after injection, the U-87-DAbR1 tumors demonstrated intense focal tracer uptake, whereas radioactivity had cleared from the kidneys, liver, and gastrointestinal tract (Fig. 5A). In wild-type tumors, no radioactivity uptake above background was detected (Fig. 5B). Quantitative analysis of tracer uptake at 14 h after injection (Fig. 5C) demonstrated that radioactivity uptake in DAbR1 tumors (5.2 [±1.7] %ID/g) was 6.5 times that in the liver (0.8 [±0.4] %ID/g), the normal organ with the highest tracer uptake.

(A) Serial small-animal PET/CT images from 1-h dynamic scan of mouse bearing DAbR1-expressing tumor on right shoulder (U-87-DAbR1 xenograft, open arrow; tracer excretion to small bowel, arrowhead; urinary bladder, closed arrow). (B) Corresponding time–activity curves for tumor, liver, and skeletal muscle (average and SD, n = 3).
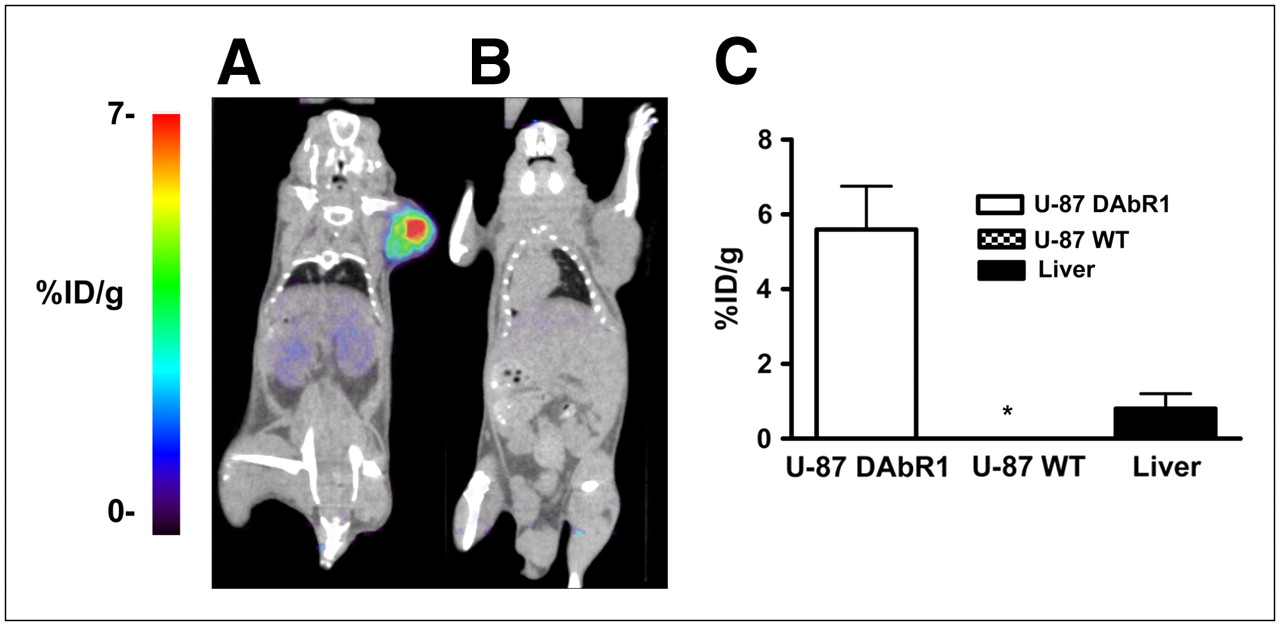
Small-animal PET/CT images of mice scanned 14 h after injection of 86Y-AABD, with mouse bearing DAbR1-positive U-87 tumor (A) and mouse bearing wild-type U-87 tumor (B). Radiotracer accumulation in wild-type tumors (n = 3) and DAbR1-expressing tumors (n = 3) is demonstrated, and radiotracer uptake in liver is shown for comparison (C). *Radiotracer uptake by wild-type U-87 tumors and muscle is less than 0.1 %ID/g. WT = wild type.
DISCUSSION
The present study indicates that antibody fragments that capture radiometal chelates are likely to be successful in the form of reporter genes for PET. Cell surface expression of an scFv fragment of the anti-DOTA antibody 2D12.5/G54C resulted in high, specific, persistent uptake of the reporter probe *Y-AABD. *Y-AABD was retained in the tumor tissue for 24 h, whereas it rapidly cleared from all normal organs, resulting in excellent tumor-to-background ratios and high-contrast PET images. In vivo binding of *Y-AABD by wild-type tumors was about 100 times lower than that of DAbR1-positive tumors and not different from background activity.
Biodistribution and tumor uptake of *Y-AABD compare favorably with data in the literature on other reporter gene–reporter probe combinations. Tumor uptake of *Y-AABD in tumors expressing DAbR1 appears to be at least as high as that reported for FHBG/HSV1-sr39tk, FIAU/HSV1-tk, and NET/metaiodobenzylguanidine (MIBG) (15,29–31) in stably transfected cell lines. Uptake of iodide in tumors transfected with NIS has been shown to be more than 30 %ID/g at 1 h after injection (32); however, this is followed by a rapid washout of radioactivity (0.4 %ID/g at 24 h after injection). The specificity of tracer uptake (ratio of tracer uptake by transfected vs. wild-type tumors) for *Y-AABD was almost 100 in our study. This is higher than the published literature data for FHBG (reported ratio, 11) (31) and FIAU (reported ratio, 65) (33). For NET/MIBG and NIS/iodide, ratios of 33 and 31 have been reported (15,32).
In addition to high and specific tumor uptake, *Y-AABD demonstrated a favorable biodistribution in mice bearing DAbR1-expressing tumors. Already at 1 h after injection, all tumor-to-organ ratios were greater than 2, and that was further improved at 24 h after injection (Table 2). Tumor-to-muscle and tumor-to-plasma ratios were more than 400 at 24 h, and the tumor-to-liver ratio was improved to 6 (Table 2). These tumor-to-organ ratios are higher than the ratios reported for FHBG, FIAU, MIBG, and radioiodine as reporter probes in stably transfected cells. For example, in rat glioma xenografts, the tumor-to-muscle and tumor-to-plasma ratios for FHBG at 24 h after injection were 124 and 25, respectively (33). The corresponding ratios for FHBG, MIBG, and radioiodine are on the same order or lower (15,32–35).
When comparing biodistribution and tumor uptake of other reporter gene–probe combinations with that of DAbR1–*Y-AABD found in this study, one needs to consider that different tumor cell lines, different modes of transfection, and different mouse strains were used. In addition, data were not normalized to the amount of protein or RNA synthesized by the transfected cells. Nevertheless, the high tumor-to-organ ratios obtained with DAbR1 and *Y-AABD relative to other reporter gene–reporter probe combinations indicates that DAbR1 and *Y-AABD represent a promising reporter gene–reporter probe combination.
Northrop et al. (18) have studied an anti-fluorescein isothiocyanate (FITC) scFv antibody fragment as a reporter gene for γ-camera imaging using 111In-DTPA-FITC as a reporter probe. However, 111In-DTPA-FITC demonstrated high kidney uptake and the tumor-to-kidney uptake ratio was less than 1 in this study (18). The maximum tumor uptake was 2.8 %ID/g, and the ratio of tumor uptake by cells expressing the anti-FITC antibody fragment and wild-type tumors was about 8. Thus, 111In-DTPA-FITC demonstrated a less favorable biodistribution, lower tumor uptake, and less specific tumor uptake than did the *Y-AABD used in our study. More recently, Roffler et al. (19) used an antidansyl (DNS) antibody as a reporter gene and the bivalent ligand DNS2–111In-DTPA as a reporter probe. At 48 h after injection, the radioactivity in anti-DNS–expressing tumors was about 2 %ID/g and 15 times higher than that of tumors transfected with a control gene. The tumor-to-blood, tumor-to-kidney, and tumor-to-liver ratios were approximately 72, 6, and 8, respectively (19). Imaging at earlier time points yielded less favorable tumor-to-organ ratios. When these results are compared with our data, the specificity of tumor uptake and the biodistribution in the *Y-AABD and DAbR1 system appear more favorable, enabling earlier imaging of tumors expressing the reporter gene.
In addition to reporter gene imaging, DAbR1–*Y-AABD could potentially be used for radiotherapy of malignant tumors after adenoviral gene transfer. Because *Y-AABD is rapidly cleared from the circulation and not accumulated in any normal tissue, the radiation exposure of normal tissues is expected to be small. In fact, the 2D12.5 antibody was initially developed for radioimmunotherapy with 90Y-DOTA complexes (36). Because 90Y emits high-energy β-radiation with a maximum tissue penetration of almost 1 cm, expression of the suicide gene by a fraction of the cancer cells within a tumor mass may be sufficient for achieving a therapeutic effect.
The following limitations of the study should be noted. We used 86Y for PET to obtain proof of principle that the *Y-AABD–DabR1 represents a promising reporter gene–reporter probe combination. However, the physical characteristics of 86Y limit its usefulness for PET. The positron abundance of 86Y is only 33%. Furthermore 86Y emits γ-rays with energies up to 3,000 keV, which are partly emitted simultaneously with the positron, so that a considerable amount of γ-coincidences occurs (37). These characteristics limit image quality of 86Y PET scans and make quantitative analysis of the PET signal challenging (37). AABD analogs can be labeled with other radionuclides more suitable for PET, such as 64Cu. The 2D12.5/G54C antibody binds a variety of DOTA radiometal complexes (36). Unfortunately, affinity is low for DOTA complexes with PET radionuclides such as 64Cu. U-87 cells expressing DAbR1 did not show significant binding of 64Cu-AABD in vitro (data not shown). However, the crystal structure of the 2D12.5/G54C-DOTA complex indicates that DOTA can be modified at position 2 without affecting 2D12.5/G54C binding (38). A bifunctional DOTA complex consisting of two 2-(bromoacetamidobenzyl)-DOTA molecules joined by a 1,4-dithio-butane linker has been shown to bind to 2D12.5/G54C with high affinity in vivo (39,40). Thus, 89Y-DOTA–64Cu-DOTA or similar constructs could potentially be used for PET.
The DAbR1 reporter gene was generated from the mouse monoclonal antibody 2D12.5/G54C. For clinical studies, a humanized version of 2D12.5 could be used to generate other DOTA-binding reporter genes. The potential of immunogenicity for such reporter genes is lower than that for viral proteins, such as HSV1-tk. However, the fusion of the antibody fragment to the transmembrane domain of CD4 may generate a potential site for immune reactions. Future studies are necessary to study the immunogenicity of DAbR1.
CONCLUSION
This study shows the feasibility of using an antibody fragment with irreversible binding to AABD as a reporter gene for PET. The high and prolonged tumor uptake of AABD, the rapid excretion of AABD from all normal tissues, and the low potential for immunogenicity make DAbR1–AABD a promising reporter gene–reporter probe combination for imaging transplanted cells and monitoring gene therapy.
Acknowledgments
We thank Gerald Toy, Jenny Shu, Dean Campbell, and Martin Walter for their help with the cell culture and animal experiments. We also thank Waldemar Ladno, Antonia Luu, Judy Edwards, and Dr. David Stout for excellent technical assistance with animal imaging. This work was supported by the UCLA ICMIC (NCI P50 CA086306) and the UCLA IMED (DE-FC03-87E60615) grants, and NIH Research grant CA016861.
Footnotes
-
COPYRIGHT © 2008 by the Society of Nuclear Medicine, Inc.
References
- Received for publication May 20, 2008.
- Accepted for publication August 11, 2008.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Molecular Imaging with Reporter Genes: Has Its Promise Been Delivered?
- Antibody with Infinite Affinity for In Vivo Tracking of Genetically Engineered Lymphocytes
- Thy1-Targeted Microbubbles for Ultrasound Molecular Imaging of Pancreatic Ductal Adenocarcinoma
- Development of a Radiolabeled Irreversible Peptide Ligand for PET Imaging of Vascular Endothelial Growth Factor
- Androgen-Independent Molecular Imaging Vectors to Detect Castration-Resistant and Metastatic Prostate Cancer
- Development of a Universal Anti-Polyethylene Glycol Reporter Gene for Noninvasive Imaging of PEGylated Probes