


Abstract
For gene therapy to be efficacious in the treatment of cancer, therapeutic transgenes must be limited in their expression to tumor cells and must be expressed at sufficiently high transcriptional levels. Moreover, the inadequacy of gene delivery must be overcome by induction of toxicity to neighboring nontargeted cells. Combining targeted radionuclide therapy with gene therapy using human telomerase promoters has shown promise in these respects, and the efficacy of this scheme has been assessed in vitro using transfectant mosaic tumor spheroids. To enable the evaluation of targeted radiotherapy combined with gene transfer in vivo, we have developed a transfectant mosaic xenograft (TMX) model. Methods: Human telomerase promoters were used to drive expression of the noradrenaline transporter (NAT) transgene in 2 human cell lines (UVW and EJ138). Promoter activity was assessed in xenografts in nude mice by determination of the uptake of the radiopharmaceutical 131I-metaiodobenzylguanidine (131I-MIBG) and by measurement of tumor growth. The efficacy of 131I-MIBG treatment was also assessed in TMXs to determine the delay in growth of tumors composed of various proportions of NAT-expressing cells—a likely clinical scenario after gene delivery in vivo. Results: In terms of induction of the capacity for active uptake of 131I-MIBG and the resultant inhibition of tumor growth in vivo, both telomerase promoters (hTR and hTERT) were similar in potency to the CMV (cytomegalovirus) promoter as controlling elements for the expression of the NAT transgene. In TMXs derived from UVW and EJ138 cells, 131I-MIBG uptake was proportional to NAT gene expression (rs = 0.910, P < 0.001 for UVW; rs = 0.971, P < 0.001 for EJ138). Inhibition of the growth of these tumors correlated with the fraction of NAT-transfected cells (rs = 0.910, P < 0.001 for UVW; rs = 0.971, P < 0.001 for EJ138), and substantial tumor growth delay was observed when 5% of the xenograft was composed of NAT-positive cells. Conclusion: TMXs constitute a suitable model to measure the efficacy of cancer gene therapy strategies when <100% of the tumor mass can be targeted to express the therapeutic transgene.
The identification of genetic peculiarities of tumor cells has led to the development of gene-based strategies for the treatment of cancer. Two significant barriers to the success of this therapeutic strategy are inadequate tumor specificity of expression of transgenes and low efficiency of gene transfer into targeted tumors. However, experimental and clinical data signify promise for the amalgamation of gene transfer with radiotherapy (1), and our recent investigations reveal that these obstacles may be overcome by the integration of gene therapy with radiopharmaceutical targeting (2–5).
131I-Metaiodobenzylguanidine (131I-MIBG)—an analog of adrenergic neuron blockers (6)—is actively accumulated by neuroectodermal tumors that express the noradrenaline transporter (NAT) (7–9). 123I-MIBG or 131I-MIBG is currently used for diagnostic imaging, and 131I-MIBG is an effective agent for the treatment of neuroblastoma (10–14). Regrettably, the uptake of the drug in malignant sites is heterogeneous, suggesting that this therapy alone is unlikely to cure disease, and a growing body of experimental and clinical evidence indicates that it is advantageous to use 131I-MIBG in combination with other therapies (15–18).
We introduced the NAT transgene into non-NAT–expressing cells derived from various tumor types, thereby creating the capacity for active uptake of 131I-MIBG and resulting in dose-dependent cytotoxicity (2,19). This demonstrated, to our knowledge, for the first time, the possibility of 131I-MIBG treatment of tumors other than those derived from the neural crest. Subsequently, we refined this therapeutic scheme by using the tumor-specific telomerase promoters (hTR and hTERT) to control NAT expression (3). Unlike most mammalian control elements, these were strong inducers of expression in vitro, similar in potency to that of the ubiquitous CMV promoter (4,20,21).
An attractive characteristic of therapeutic strategies that combine gene manipulation with the administration of radiopharmaceuticals is an inherent, radiologic bystander effect derived from the transfer of radiation-induced toxic factors to, and cross-fire irradiation of, nontransfected cells (4). These phenomena are due to biologic processing of the physical radiation insult and radioactive-decay particle bombardment of neighboring cells, respectively. The powerful bystander effect generated by irradiation has the potential to overcome heterogeneity with respect to transgene expression and concentration of radiopharmaceutical in tumors (5). We have assessed the usefulness of this effect using our recently developed transfectant mosaic spheroid (TMS) model, composed of mixtures of NAT-transfected and untransfected cells (22). This enabled our demonstration that the magnitude of cell kill by radiolabeled MIBG was greater than the NAT-transfected fraction in TMS, indicative of a bystander effect in this 3-dimensional culture system. Furthermore, we recently reported that intracellularly concentrated 131I-MIBG induced greater toxicity to bystander cells than external beam γ-irradiation (5).
Here we describe the development of a transfectant mosaic xenograft (TMX) model, the in vivo equivalent of TMS, for the evaluation of gene-radionuclide therapy that combines telomerase-dependent, tumor-specific expression of the NAT transgene with radiopharmaceutical induction of bystander effects.
MATERIALS AND METHODS
Synthesis of 131I-MIBG
Chemicals were purchased from Aldrich Chemical Co. High-performance liquid chromatography (HPLC)–grade solvents were obtained from Rathburn Chemicals. Carrier-free sodium 131I-iodide was purchased from GE Healthcare. No-carrier-added 131I-MIBG was prepared using a solid-phase system in which the precursor of MIBG was attached to an insoluble polymer via the tin-aryl bond (23). The reaction conditions, HPLC purification procedure, and radiochemical yield were as described previously (24).
Plasmids
The bovine NAT (bNAT) complementary DNA (cDNA) was kindly provided by Dr. Michael Bruss and Prof. Heinz Bonisch (University of Bonn, Germany). Recombinant plasmids containing the bNAT cDNA under control of the CMV, hTR, or hTERT promoter were constructed as previously described (2,3,21).
Cell Culture and Transfection of NAT Gene
Media and supplements were obtained from Invitrogen. The transgene hosts were the human glioma cell line (UVW) previously described (4) and the human bladder carcinoma cell line EJ138 (5). UVW cells were maintained in Eagle's minimum essential medium (MEM) with 10% (v/v) fetal bovine serum, penicillin/streptomycin (100 U/mL), fungizone (2 μg/mL), and glutamine (200 mmol/L). The EJ138 cells were cultured in the same MEM medium as described for UVW cells with the addition of 0.1 nmol/L nonessential amino acids. The cells were cultured at 37°C in an atmosphere of 5% CO2.
UVW or EJ138 parental cell lines were stably transfected with the recombinant plasmids using Effectene transfection reagent (Qiagen) as previously described (2–5,21). The transfectants were maintained by addition of geneticin (G418) at every passage, and stable transfectants were not further selected into clones derived from single transfected cells. This provided a model representative of the heterogeneity of transgene expression expected after in vivo gene delivery. Stably transfected cell populations were obtained approximately 4 wk after transfection. The growth characteristics of the transfectants in terms of plating efficiency, doubling time, and radiosensitivity were indistinguishable from those of the nontransfected parent line.
131I-MIBG Uptake In Vitro
Cells were assessed for 131I-MIBG uptake as previously described (2). Briefly, UVW and EJ138 cells were seeded in 6-well plates at an initial density of 0.5 × 105 cells per well and cultured for 48 h. 131I-MIBG incorporation was measured by incubating the cells for 2 h at 37°C in complete medium with 7 kBq of 131I-MIBG. Nonspecific accumulation was measured in the presence of 1.5 mM desmethylimipramine (DMI) (Sigma-Aldrich), a specific and potent NAT inhibitor. After incubation, medium was removed, the cells were washed with phosphate-buffered saline, and radioactivity was extracted using 2 aliquots of 10% (w/v) trichloroacetic acid. The activities of the extracts were measured in a γ-well counter (Packard Biosciences Ltd.). Specific uptake, expressed as counts/minute × 103 per 106 cells, was calculated by subtracting values obtained in the presence of DMI from total uptake.
Experimental Animals
Six-week-old female, congenitally athymic nude mice of strain MF1 nu/nu were obtained from Charles River plc. In vivo experiments were performed in accordance with the U.K. Coordinating Committee for Cancer Research guidelines on experimental neoplasia in animals (25).
Xenografts
Xenografts were established in nude mice by subcutaneous injection of 3 × 106 UVW cells, EJ138 cells, or the transfected derivatives freshly harvested at 60%–70% confluence to ensure that their growth was in logarithmic phase. Mosaic xenografts were established by injection of 3 × 106 cells comprising various proportions (0%–100%) of NAT-transfected to parental cells. NAT-transfected cells were grown in the absence of geneticin during the passage before injection into the murine flanks to avoid geneticin toxicity to the mixed parental cells. Capacity for active uptake of MIBG was confirmed in cells in monolayer culture on the day of injection. 131I-MIBG uptake and therapy determinations were initiated 7 d (UVW cells) or 12 d (EJ138 cells) later, at which time tumor volume was approximately 60 mm3. To monitor potential toxicity, body weight was measured daily and experimental animals were scored for signs of distress using standard guidelines (26). Mice whose xenografts grew to 1,900 mm3 were euthanized.
Real-Time Reverse Transcriptase Polymerase Chain Reaction (RT-PCR)
Mice were sacrificed when mosaic tumors were approximately 250 mm3. The tumors were excised and added to 10 volumes of TRI reagent (Ambion). The tissue was homogenized and RNA was extracted according to the manufacturer's instructions. Preparation of cDNA and RT-PCR were performed using TaqMan RT-PCR methodology and reagents (Perkin-Elmer Applied Biosystems), as described previously (21,27). Primer and probe sequences were designed from published sequences for bNAT (accession no. U09198) using the ABI prism PrimerExpress TM version 1.0 software and were custom synthesized (MWG-Biotech). The sense primer corresponded to bases 1583–1602 of the bNAT sequence, and the antisense primer was complementary to bases 1637–1657. These primers generated a PCR product of 75 base pairs. The internal probe corresponded to bases 1612–1635 of the bNAT sequence. The probe was labeled with the fluorescent reporter dye 6-carboxyfluoroscein (FAM) at the 5′ end and the quencher molecule 6-carboxytetramethylrhodamine (TAMRA) at the 3′ end. The housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal standard for all real-time PCRs. To enable quantitation of the starting number of copies of the target sequence, standard curves were constructed for bNAT and GAPDH as previously described (27). After TaqMan PCR of standards and unknown samples, the Opticon Monitor (version 3.1) sequence detection software (Bio-Rad Laboratories Ltd.) was used to determine the initial amounts of unknown samples by comparison of their threshold cycle (Ct) values with those of known standards.
Tumor Uptake of 131I-MIBG
These experiments were performed on groups of 6 mice. The animals received, by intraperitoneal injection, 2 MBq 131I-MIBG. The precise activity administered to each mouse was measured using a Curiemonitor-2 ionization chamber (Radiation Components). After 48 h, the mice were euthanized. Tumors were excised and weighed, and the associated radioactivity was measured in an automated γ-counter (Packard Biosciences Ltd.). The γ-counter measurement of each sample, in counts/minute, was converted to an absolute value (in MBq) by comparison with the measurements obtained from standards of known activity. From this value, and knowledge of the weight of the tissue sample and activity of 131I-MIBG initially administered to the mouse, the activity in tumor was expressed as the percentage injected dose per gram of tissue (%ID/g). Correction was made for radioactive decay that had taken place since the time of injection.
131I-MIBG Experimental Therapy
When the tumors were approximately 60 mm3, mice were injected intraperitoneally with 12 MBq of no-carrier-added 131I-MIBG. Groups of 12 mice were randomized and used for each treatment group. Subcutaneous tumors were measured with calipers immediately before treatment and every 2 or 3 d thereafter. On the assumption of ellipsoidal geometry, diameter measurements were converted to an approximate volume by multiplying half of the longest diameter by the square of the mean of the 2 shorter diameters. Mice whose xenograft volume reached 1,900 mm3 were euthanized. For each treatment group, mean relative tumor volumes (volume at any time point divided by volume immediately before treatment) were plotted against time, and the areas under the individual time–activity curves were determined by trapezoidal approximation to allow comparison among treatment groups.
Statistical Analysis
One-way ANOVA was used to test the differences in the strength of viral and telomerase promoters and differences in radiopharmaceutical uptake and tumor growth delay for different proportions of NAT-expressing cells in tumors. Post hoc testing used the Bonferroni correction for multiple comparisons. Associations between radiopharmaceutical uptake, NAT expression, and the proportion of NAT-transfected cells in TMX were evaluated by the Spearman rank correlation (2-tailed). P values < 0.05 were considered significant.
RESULTS
Cellular Uptake of 131I-MIBG In Vitro
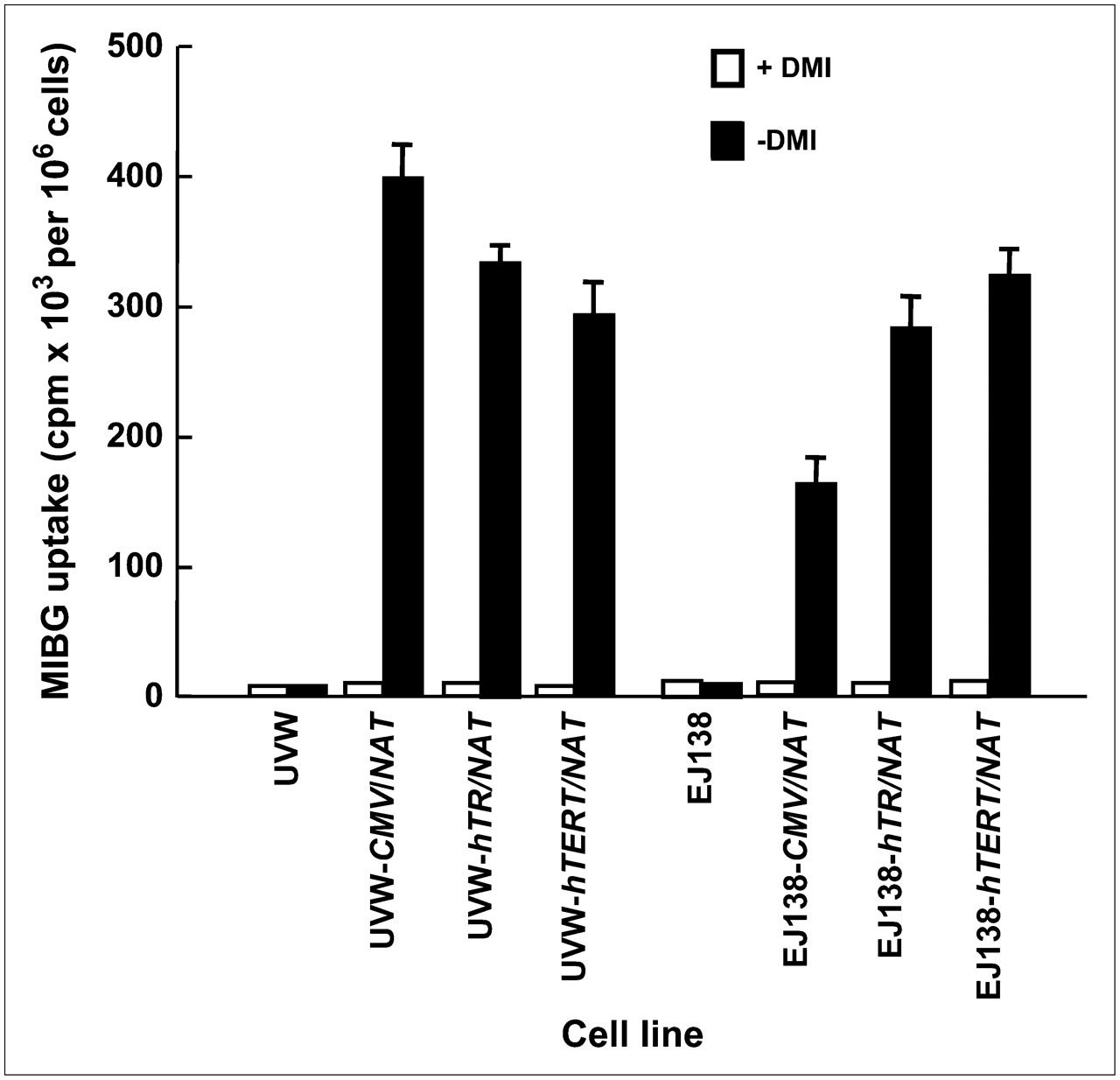
Untransfected UVW and EJ138 cells had negligible active accumulation of 131I-MIBG (Fig. 1). UVW cells transfected with the NAT gene, controlled by the CMV, hTR, or hTERT promoter, showed, respectively, 39-, 32-, or 30-fold higher 131I-MIBG uptake relative to the same cells treated with the specific monoamine uptake inhibitor DMI. EJ138 cells transfected with the NAT gene, controlled by the CMV, hTR, or hTERT promoter, showed, respectively, 16-, 28-, or 31-fold higher 131I-MIBG uptake relative to DMI-treated cells (Fig. 1). There was a significant difference in the accumulation of 131I-MIBG between CMV- and hTR-controlled cells (P < 0.05 for UVW; P < 0.01 for EJ138). However, the difference in 131I-MIBG concentration between cells transfected with either of the telomerase promoters was not significant.
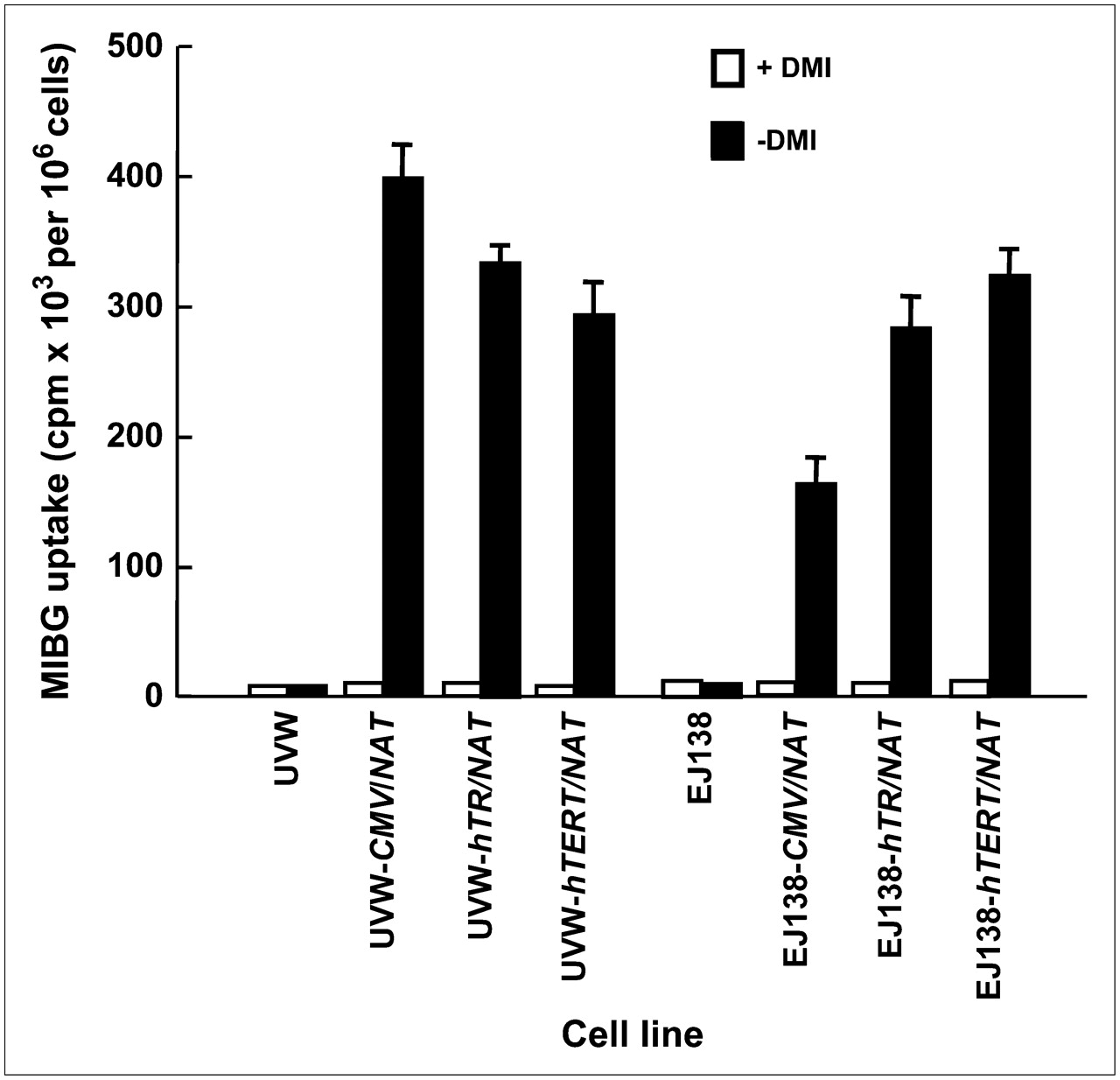
131I-MIBG uptake by UVW and EJ138 cells—untransfected or transfected with the NAT gene controlled by the CMV, hTR, or hTERT promoter. Evaluation of uptake was performed in the presence or absence of DMI, a specific inhibitor of NAT-mediated uptake. Results are mean ± SD of 3 determinations performed in triplicate.
Effect of Transgene Promoter on Tumor Uptake of 131I-MIBG In Vivo
After intraperitoneal administration of 131I-MIBG, toxicity was not observed in any of the animals. Initially, we compared the retained fraction of the injected dose of radiopharmaceutical per gram of tumor, at 48 h after injection, in xenografts composed entirely of NAT transfectants of UVW or EJ138 cells. One-way ANOVA revealed no significant difference in the accumulation of radioactivity between xenografts containing the NAT transgene driven by the ubiquitous CMV promoter or either of the telomerase promoters in either cell line (Table 1).
131I-MIBG Concentration in Tumor Xenografts
Effect of Transgene Promoter on 131I-MIBG-Induced Inhibition of Tumor Growth In Vivo
Figure 2 shows the effect of 131I-MIBG treatment on the growth of UVW and EJ138 xenografts transfected with the NAT transgene under control of the CMV, hTR, or hTERT promoter. Untreated control xenografts, derived from UVW and EJ138 and expressing NAT under control of the CMV promoter, grew with kinetics similar to that of the tumors that had no capacity for active accumulation of 131I-MIBG. For the sake of clarity, these data are omitted from the figure.
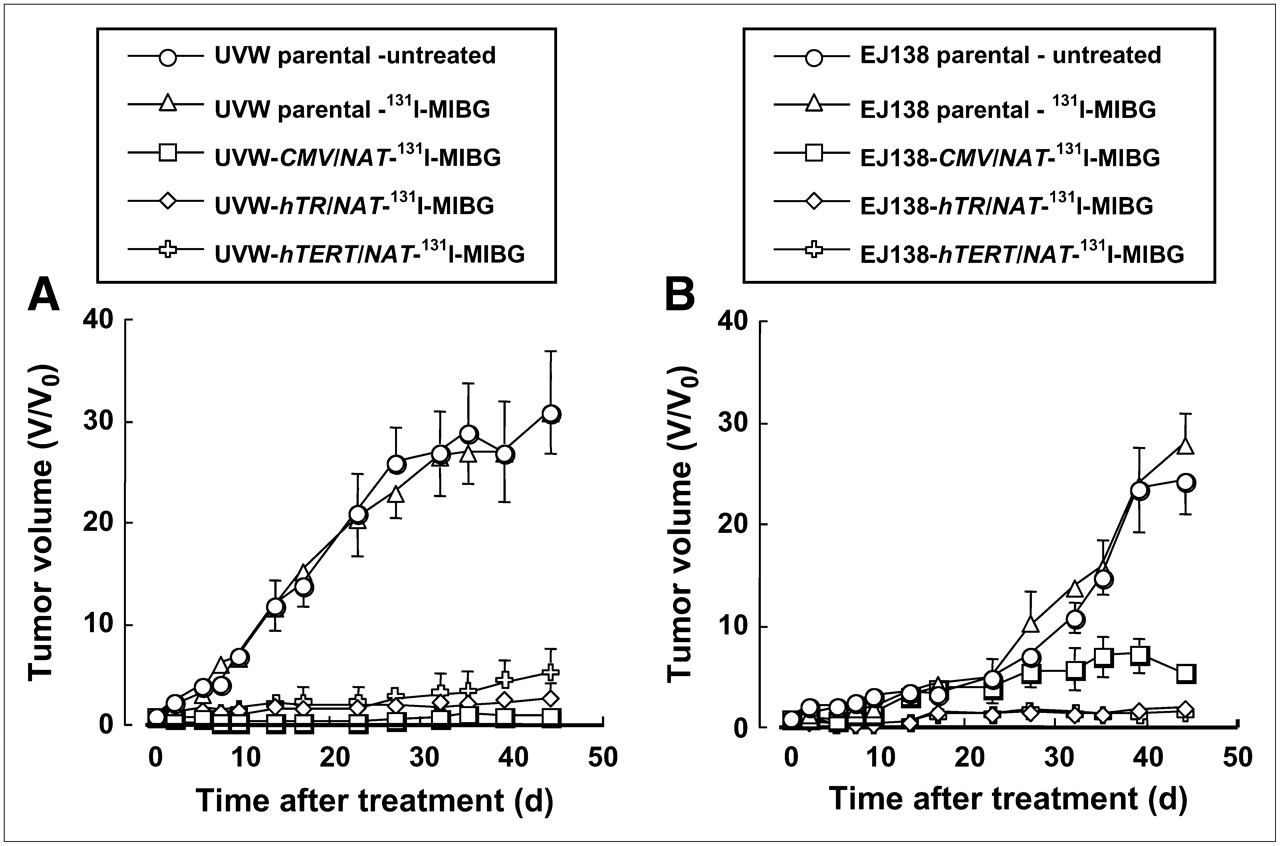
Effect of 131I-MIBG (12 MBq) treatment of xenografts composed of UVW cells (A) or EJ138 cells (B). Cells were untransfected (parental) or transfected with the NAT gene under control of the strong ubiquitous viral promoter (CMV) or either of the human telomerase (hTR and hTERT) promoters. Each treatment group consisted of 12 mice. Results are mean ± SD.
UVW Tumors.
Overall differences in the effectiveness of 131I-MIBG treatment—as measured by the area under the growth curves of tumors derived from cells whose NAT expression was controlled by various promoters—were highly significant (ANOVA, P < 0.001). Post hoc testing demonstrated significantly greater growth inhibition in the NAT transfectants (UVW-CMV/NAT, UVW-hTR/NAT, and UVW-hTERT/NAT) relative to that of nontransfectants (UVW parental) (P < 0.001) but no significant difference between telomerase (either UVW-hTR/NAT or UVW-hTERT/NAT) and the ubiquitous CMV promoter-controlled transfectants (UVW-CMV/NAT) or between untreated and 131I-MIBG–treated nontransfectants (UVW parental).
EJ138 Tumors.
One-way ANOVA again indicated an overall statistically significant difference in growth inhibition between the various cell types (P < 0.001), and the Bonferroni posttest indicated significantly greater growth inhibition in the transfectants (EJ138-CMV/NAT, EJ138-hTR/NAT, and EJ138-hTERT/NAT) relative to that of nontransfectants (EJ138 parental) (P < 0.001). There was also greater growth inhibition for hTR promoter-driven NAT-expressing tumors (EJ138-hTR/NAT) and for hTERT promoter-driven NAT-expressing tumors (EJ138-hTERT/NAT) compared with CMV promoter-driven NAT-expressing tumors (EJ138-CMV/NAT) (P < 0.05). However, there was no statistically significant difference in growth between tumors in which NAT was expressed under control of the hTR or hTERT promoter (EJ138-hTR/NAT and EJ138-hTERT/NAT, respectively).
These observations indicated that both telomerase promoters were suitable for the regulation of NAT transgene expression and that, unlike most mammalian regulatory elements, their potency was similar (UVW cells) or superior (EJ138 cells) to that of the strong CMV promoter. Subsequent evaluation of the effect of efficiency of NAT gene transfection on the efficacy of 131I-MIBG targeted radiotherapy was conducted using the hTR promoter to control the expression of the NAT transgene.
Validation of TMX Model
To determine the suitability of the TMX model for examination of the efficacy of 131I-MIBG in tumors that are heterogeneous with respect to NAT transgene expression, radiopharmaceutical accumulation and NAT gene transcription levels were determined in xenografts composed of various proportions of NAT-expressing cells.
Seven groups of UVW- and EJ138-derived TMXs were prepared. These contained the following percentages of NAT expressers: 0%, 5%, 10%, 25%, 50%, 75%, and 100%. When the tumors had grown to approximately 60 mm3, 6 mice from each TMX group of 12 were sacrificed, and RNA was extracted for RT-PCR determination of NAT expression within the tumor. The remaining 6 mice in each group received intraperitoneal administration of 2 MBq of 131I-MIBG to examine tumor uptake of the radiopharmaceutical.
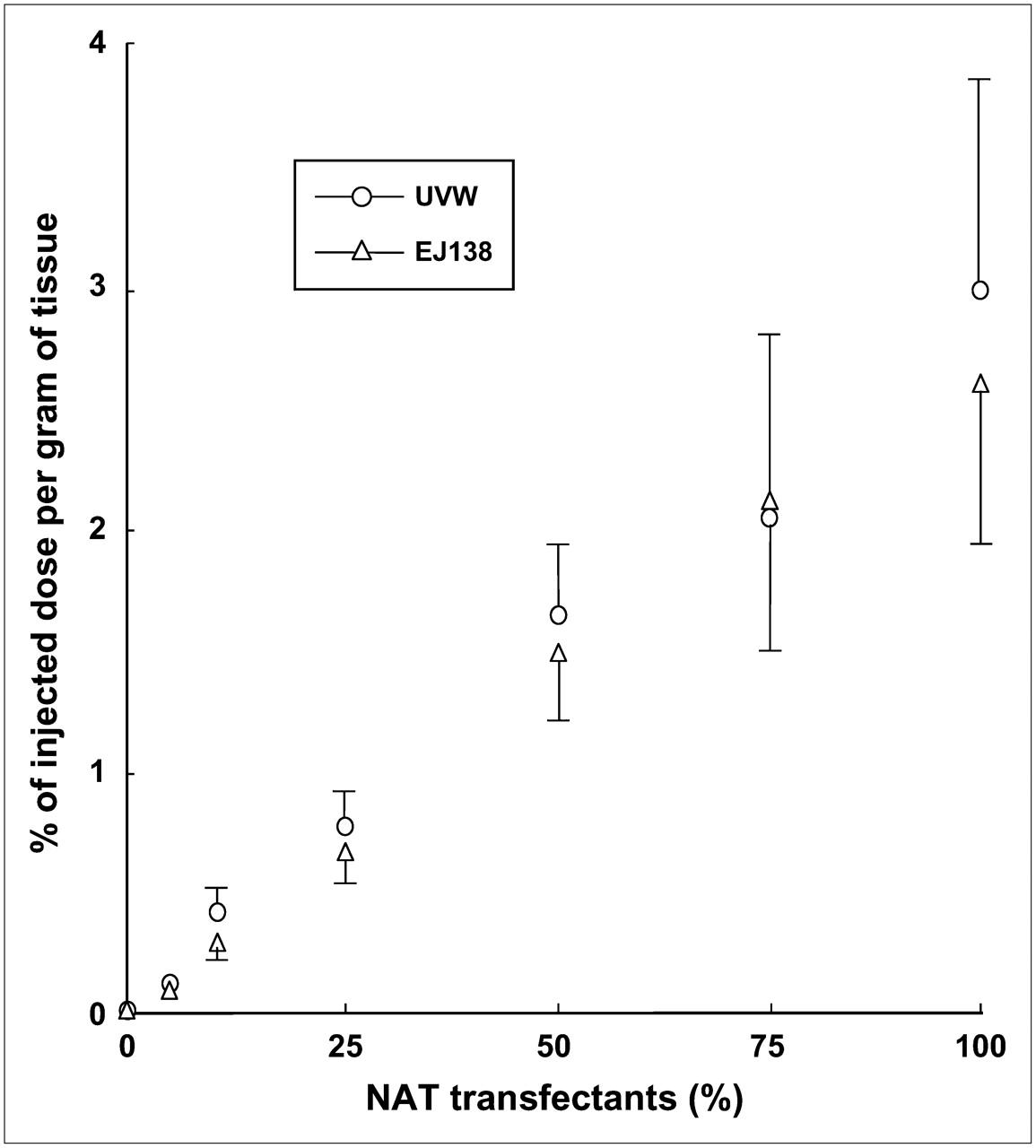
In TMXs derived from UVW and EJ138 cells, the percentage of NAT gene-transfected cells correlated significantly (P < 0.001) with the transcription level of the NAT transgene as a function of GAPDH expression (rs = 0.910 for UVW; rs = 0.971 for EJ138) (Fig. 3) and with the uptake of 131I-MIBG (rs = 0.966 for UVW; rs = 0.970 for EJ138) (Fig. 4).

Relationship between production of NAT messenger RNA and percentage of NAT gene transfectants in TMXs derived from UVW or EJ138 cells. Results are mean ± SD of 6 determinations. Significant correlations were established in UVW xenografts (rs = 0.910; P < 0.001) and EJ138 xenografts (rs = 0.971; P < 0.001).
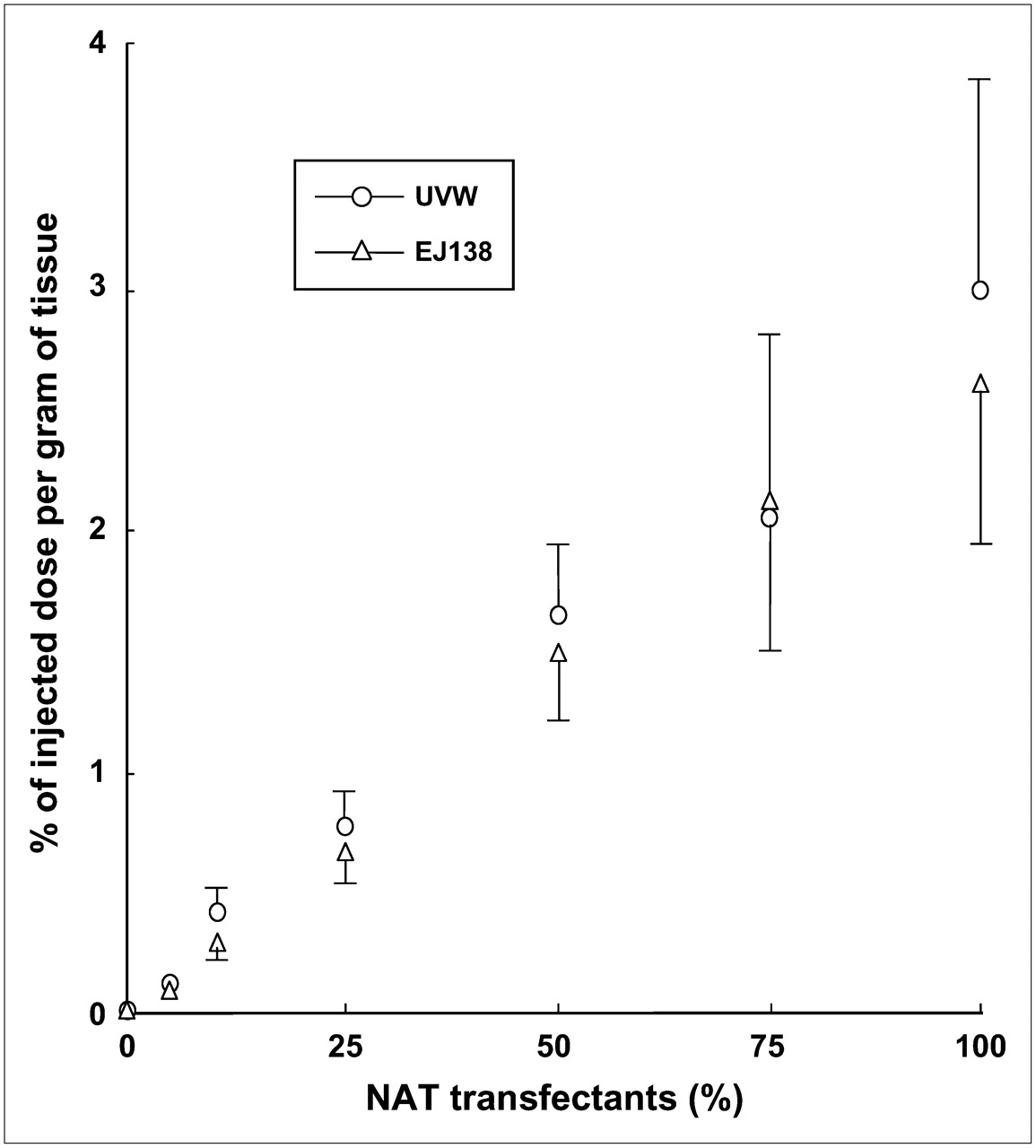
131I-MIBG uptake by TMXs, derived from UVW or EJ138 cells, as a function of proportional content of NAT gene transfectants. Results are mean ± SD of 6 determinations. Significant correlations were established in UVW xenografts (rs = 0.966; P < 0.001) and EJ138 xenografts (rs = 0.970; P < 0.001).
Antitumor Effects of 131I-MIBG in TMXs
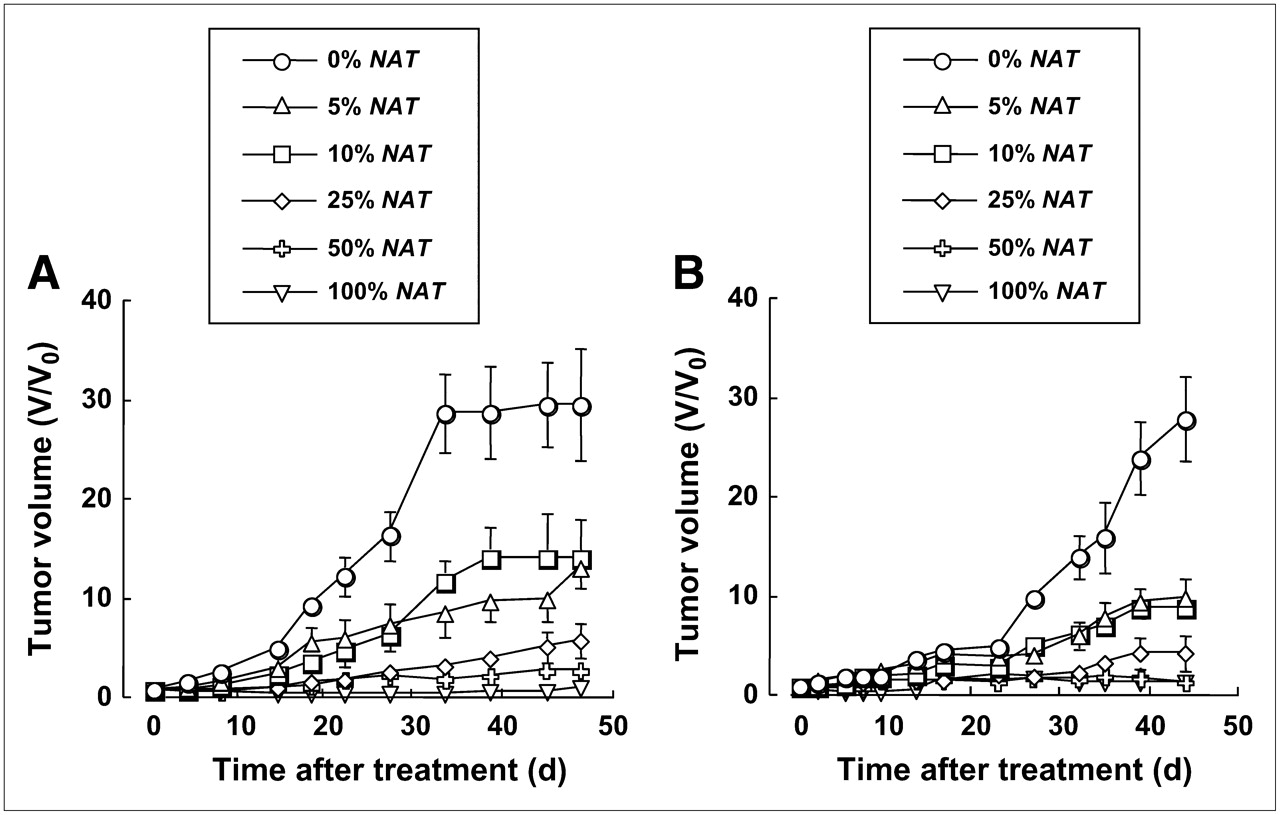
To assess the effect of 131I-MIBG on the growth of TMXs, animals with tumors composed of various proportions of NAT-expressing cells were intraperitoneally injected with 131I-MIBG (12 MBq/mL). The growth-inhibitory effect of 131I-MIBG on the various UVW and EJ138 TMXs is shown in Figure 5. Untreated control xenografts, derived from UVW and EJ138 and composed of 100% NAT-expressing cells, increased in size at the same rate as 131I-MIBG-treated tumors that contained no NAT-expressing cell. For the sake of clarity, these data were omitted from the figure.

Effect of 131I-MIBG (12 MBq) treatment of TMXs composed of various proportions of UVW cells (A) or EJ138 cells (B). Cells were untransfected (0%) or transfected with the NAT gene under control of the hTR promoter. Each treatment group consisted of 12 mice. Results are mean ± SD.
UVW Tumors.
There was a significant effect of the fraction of NAT-expressing cells on the susceptibility to inhibition of tumor growth by administration of 131I-MIBG (1-way ANOVA, P < 0.001). Post hoc analyses showed reduced growth rates of 5% and 10% NAT-positive tumor relative to non-NAT−expressing tumors (P < 0.001). Retardation of the rate of increase in tumor size was significantly greater in 25% compared with 5% and 10% NAT-expressing xenografts (P < 0.001). The growth rates of 100% NAT gene transfectants were slower than those of 50% transfectants (P < 0.05), and the increase in size of 50% NAT-positive tumors was slower than that of 25% transfectants (P < 0.05).
EJ138 Tumors.
As for UVW TMXs, EJ138 tumors displayed vulnerability to 131I-MIBG-induced growth impedance that was dependent on the percentage of NAT expression (1-way ANOVA, P < 0.001). Post tests revealed significant differences in the increase in tumor volume between 0% and 5%, 10% and 25%, and 25% and 50% NAT-positive TMXs (P < 0.001 for all comparisons). The growth rates of 50% and 100% NAT gene transfectants were statistically indistinguishable.
The results of 131I-MIBG treatment of both xenografts suggest that low-percentage NAT positivity was associated with disproportionately greater efficacy than high-percentage NAT expression.
DISCUSSION
One of the main limitations to the success of cancer gene therapy is that gene transfer vectors do not achieve transfection of all cells in malignant tissue. Therefore, sterilization of tumors relies on the production by targeted cells of factors that are toxic to neighboring untransfected cells. Radioactive decay within tumor cells is lethal to vicinal cells due to cross-fire irradiation (28) and radiation-induced biologic bystander effects. The latter are produced by cellular processing of the physical radiation insult into factors that cause damage to adjacent unirradiated cells (5,29–34). Consequently, targeted radiotherapy has been proposed as an adjunct to gene therapy (3).
To evaluate the extent to which bystander effects may overcome the impediment to cure imposed by heterogeneous distribution of the radiopharmaceutical 131I-MIBG in tumors, we previously constructed TMSs composed of a range of proportions of NAT gene-transfected to nontransfected cells (4,19). These are representative of small tumors in which variable efficiency of gene transfer has been accomplished. In the present article, we describe a comparable model derived from tumor cells transfected with the NAT gene and grown as xenografts in the flanks of athymic mice. This allowed the investigation in vivo of the influence of gene transfer efficiency on the effectiveness of targeted 131I-MIBG therapy.
Although telomerase is generally undetectable in normal tissues, it is active in all types of human malignancy (35). Therefore, the use of telomerase promoters has been advocated as a means of restricting the expression of therapeutic transgenes to tumors (2–4). Generally mammalian promoters are characterized by low-expression activity compared with viral regulatory elements, resulting in inefficient activation of transcription and, consequently, low levels of production of therapeutic protein (36). However, we demonstrated previously that the hTR and hTERT promoters were comparable to the strong ubiquitous RSV and CMV promoters with respect to transgene expression in vitro (3,4,20,21).
The current study initially compared telomerase and CMV promoter activity in vivo. In terms of induction of the capacity for active uptake of 131I-MIBG and the resultant inhibition of tumor growth, both telomerase promoters (hTR and hTERT) were similar in potency to the CMV promoter as controlling elements for the expression of the NAT transgene. Therefore, our ensuing assessment of the dependence of NAT expression level in experimental tumors on the therapeutic potency of 131I-MIBG treatment was performed in TMXs wherein the hTR promoter was the regulatory element.
Xenografts, grown subcutaneously in the flanks of athymic mice and containing no NAT-expressing cell, showed negligible endogenous expression of NAT and negligible uptake of 131I-MIBG. In contrast, NAT gene expression and 131I-MIBG uptake in TMXs, composed of various proportions of hTR promoter-controlled, NAT-transfected and untransfected cells, increased in direct proportion to the fraction of NAT-transfected cells.
To assess growth inhibition in this model, representing variably transfected tumors in vivo, we measured the toxicity of 131I-MIBG to TMXs derived from 2 cell lines and composed of a range of proportions of NAT-expressing cells. In xenografts derived from both cell lines, retardation of tumor growth was observed, even when the tumor contained only 5% NAT positivity. Moreover, we observed that the inhibition of TMX growth by 131I-MIBG treatment was directly related to the proportion of NAT-expressing cells. In the absence of 131I-MIBG treatment, the level of NAT expression had no effect on the growth rate of xenografts.
Therefore, both NAT gene expression levels and 131I-MIBG uptake accurately reflected the percentage of NAT-positive cells in the tumors in vivo. This indicates the suitability of TMX for the examination of quantitative aspects of radionuclide treatment and gene therapy. For example, this model will allow the determination of the minimal NAT expression necessary for the imaging and sterilization by radiolabeled MIBG of tumors of a range of sizes that are 1–2 orders of magnitude greater than TMS. The TMX model will also facilitate the determination of optimal therapeutic dosage in relation to normal tissue toxicity.
Several in vitro studies have revealed that bystander effects are produced by irradiated cells and that these impact on neighboring unirradiated cells, causing death, mutation, or genomic instability (29–34,37,38). This phenomenon is prevalent at low radiation dose and low dose rate—both of which are characteristic of targeted radiotherapy (39). We recently showed that potent toxins are generated specifically by cells that concentrate radiohalogenated MIBG. These may be linear-energy-transfer–dependent and different from those generated by external beam γ-radiation (5).
To determine the potency of bystander effects in vivo elicited by cells that accumulated Auger electron–emitting radionuclides, Xue et al. (40) incorporated 5-125I-iodo-2′-deoxyuridine (125I-IUdR) into adenocarcinoma cells and mixed these with unlabeled cells before subcutaneous implantation in rodents. They reported inhibition of tumor growth as a consequence of factors released from the labeled cells. The TMX model described here is an alternative means of determining the importance of bystander effects in vivo. We expect that it will enable the quantitative analysis of aspects of targeted radionuclide therapy fundamental to its efficacy, such as the contribution by biologic bystander effects and physical radiation cross-fire as well as the significance of decay-particle range, half-life, and half-time of intratumoral residence of radionuclides.
Although this model can provide valuable information of therapeutic relevance, it may not exactly mimic effects that could be achieved clinically. For example, it is necessary to determine the effect of alternative routes of transgene delivery on the heterogeneity of distribution of transfected cells in TMXs. Moreover, it should also be possible to create mosaic xenografts composed of transfected peripheral rims or central regions. Such models could be used to represent transfections that are dependent on, for example, cellular proliferation or hypoxic promotion of expression, respectively.
CONCLUSION
We have characterized a novel TMX model with respect to the relationship between transcriptional activity of the NAT transgene, uptake of 131I-MIBG, and inhibition of growth. This will be a useful tool for the evaluation of gene therapy and the optimization of combination targeted radiotherapy.
Acknowledgments
This work was supported by grants from Cancer Research U.K. and the Neuroblastoma Society. We thank Heinz Bonisch and Michael Bruss, University of Bonn, for the kind gift of bNAT cDNA.
Footnotes
-
COPYRIGHT © 2007 by the Society of Nuclear Medicine, Inc.
References
- Received for publication March 31, 2007.
- Accepted for publication May 31, 2007.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.