


Abstract
Radiopharmaceuticals that can target the random metastatic dissemination of melanoma tumors may present opportunities for imaging and staging the disease as well as potential radiotherapeutic applications. A novel molecule, 2-(2-(4-(4-123I-iodobenzyl)piperazin-1-yl)-2-oxoethyl)isoindoline-1,3-dione (MEL037), was synthesized, labeled with 123I, and evaluated for application in melanoma tumor scintigraphy and radiotherapy. Methods: The tumor imaging potential of 123I-MEL037 was studied in vivo in C57BL/6J female mice bearing the B16F0 murine melanoma tumor and in BALB/c nude mice bearing the A375 human amelanotic melanoma tumor by biodistribution, competition studies, and SPECT. Results: 123I-MEL037 exhibited high and rapid uptake in the B16F0 melanoma tumor at 1 h (13 %ID/g [percentage injected dose per gram]), increasing with time to reach 25 %ID/g at 6 h. A significant uptake was also observed in the eyes (2 %ID, at 3–6 h after injection) of black mice. No uptake was observed in the tumor or in the eyes of nude mice bearing the A375 tumor. Because of high uptake and long retention in the tumor and rapid body clearance, the mean contrast ratios (MCR) of 123I-MEL037 were 30 and 60, at 24 and 48 h after injection, respectively. At 24 h after injection of mice bearing the B16 melanoma, SPECT indicated that the radioactivity was located predominately in the tumor followed by the eyes, whereas no specific localization of the radioactivity was noted in mice bearing the A375 human amelanotic tumor. In competition experiments, uptake of 123I-MEL037 in brain, lung, heart, and kidney—organs known to contain σ-receptors—was not significantly different in haloperidol-treated animals compared with control animals. Therefore, reduction of uptake in tumor and eyes of the pigmented mice bearing the B16F0 tumor suggested that the mechanism of tumor uptake was likely due to an interaction with melanin. Conclusion: These findings suggested that 123I-MEL037, which displays a rapid and very high tumor uptake, appeared to be a promising imaging agent for detection of most melanoma tumors with the potential for development as a therapeutic agent in melanoma tumor proliferation.
Malignant melanoma is a very aggressive cancer with a high rate of metastasis and increasing incidence in developed countries. Despite the significance of this disease and compared with advances in other areas of cancer, there are still no effective treatments available, although early detection and improved diagnostic methods have decreased considerably the mortality rates over the last decade (1). A key feature of melanoma tumors is the extensive pigmentation present in most tumor cells, thus making it a very attractive target for both diagnosis and treatment. An advantage of this system is that uptake can be optimized to cells containing melanin, thus providing a selective mechanism by which a significant target-to-nontarget ratio could be achieved.
Several biochemical entities have been investigated as potential imaging and radiotherapeutic agents, including monoclonal antibodies (2), iodothiouracils (3), melanocortin-1 receptor-targeting peptides (4), chloroquine (5), methylene blue dye (6), and iodobenzamides (7).
Preclinical investigations with several melanin-targeting radiopharmaceuticals based on benzamides demonstrated selective uptake in melanoma tumor-bearing mice (8–10). Moreover, the iodobenzamides 123I-N-(2-diethylaminoethyl) 4-iodobenzamide (123I-BZA) (7), 123I-N-(2-diethylaminoethyl)-2-iodobenzamide (123I-BZA2) (11,12), and 123I-iodobenzamide (123I-IBZM) (13) and the iodobenzylamine N-[3-(4-morpholino)propyl]-N-methyl-2-hydroxy-5-iodo-3-methylbenzylamine (ERC9 (14) have been evaluated in melanoma patients, resulting in excellent detection of melanoma and its metastases with high sensitivity and selectivity. These studies confirmed the efficacy of these iodinated radiopharmaceuticals as useful imaging agents in patients with cutaneous and ocular melanoma on the basis of the selective high affinity binding to melanin-containing melanocytes. Other iodobenzamide or benzoate derivatives possessing a piperidine chain have been evaluated in preclinical studies for melanoma detection (15,16). Here tumor uptake was mediated through a σ-receptor–based mechanism, as demonstrated by compounds such as N-(N-benzylpiperidin-4-yl)-4-iodobenzamide (IBP) (17).
Our radiopharmaceutical optimization program aimed to deliver iodinated benzamide analogs that could display high tumor uptake and rapid clearance from the body suitable for scintigraphy and potential therapeutic applications. This involved the incorporation of a variety of stable functional groups into the benzamide-tertiary amine structures to maximize the uptake and clearance profiles of the resultant molecules. Part of the structure optimization included the substitution of the piperidine moiety for piperazine, a group readily used in the preparation of several σ-receptor ligands, and rigidification of the benzamide by replacing it with an isoindoline-1,3-dione. Another unique feature was the insertion of a methylene-carbonyl unit and the incorporation of the iodine on the benzylamine instead of the benzamide, as with IBP and related benzamides. Consequently, we report the synthesis of 2-(2-(4-(4-123I-iodobenzyl)piperazin-1-yl)-2-oxoethyl)isoindoline-1,3-dione, (123I-MEL037) (Fig. 1) and its biologic evaluation in melanoma tumor-bearing mice.
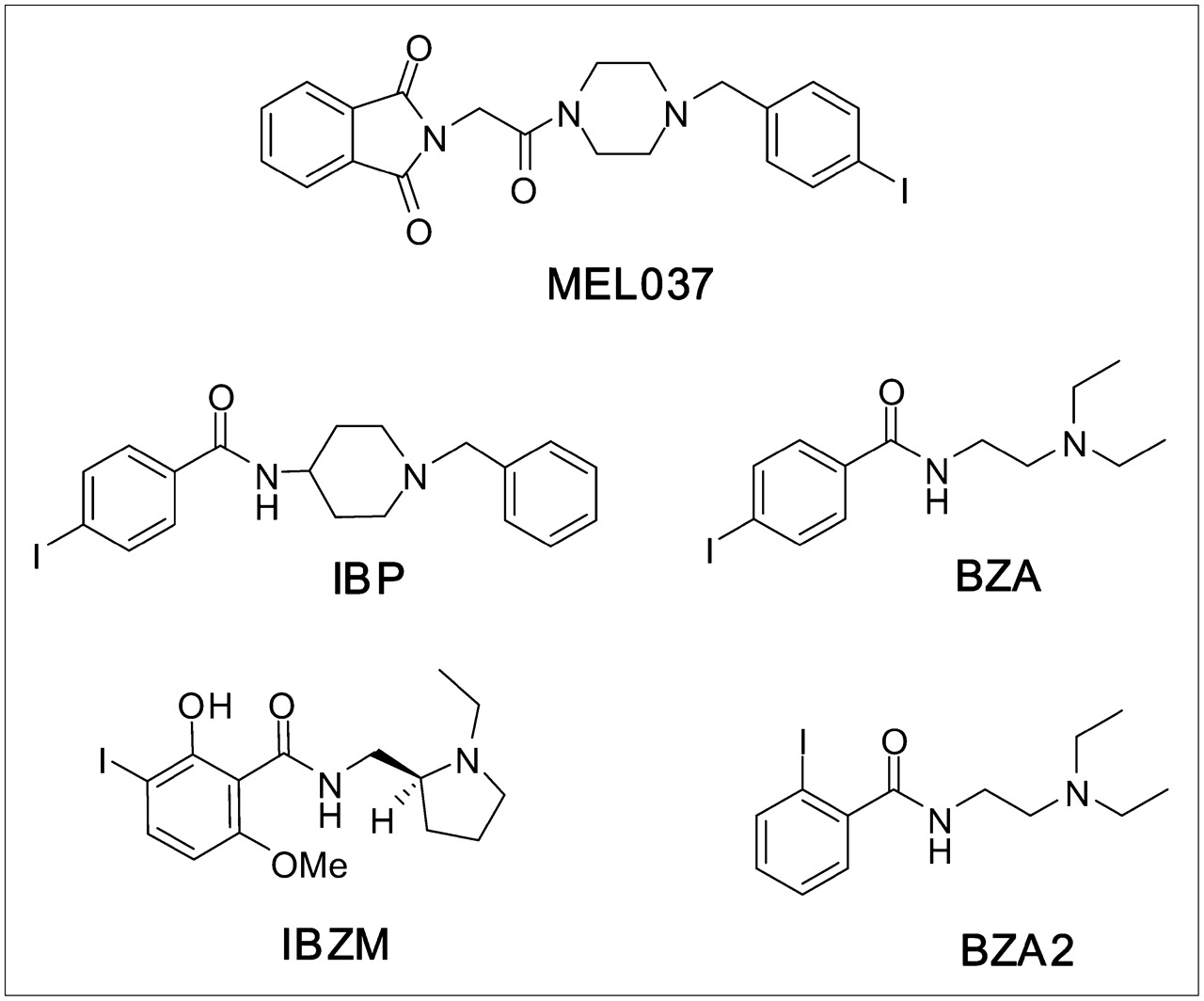
Chemical structures of MEL037, IBP, BZA, IBZM, and BZA2.
MATERIALS AND METHODS
General
1-(4-Iodobenzyl)piperazine (2a) was purchased from ACB Blocks Ltd. All other chemicals were purchased from Sigma-Aldrich and used without further purification. 123I was produced by the National Medical Cyclotron (Sydney, Australia), using the 124Xe (p, 2n) reaction and was delivered as 123I-NaI in 0.02 mol/L NaOH at a concentration of 37 GBq/mL. 3H-1,3-Di-O-tolylguanidine (3H-DTG) (2.2 TBq/mmol) and 3H-(+)-pentazocine (2.2 TBq/mmol) were purchased from Perkin Elmer Life Sciences Australia. Melting points were determined in open capillary tubes using a Gallenkamp melting point apparatus and are uncorrected. Nuclear magnetic resonance (NMR) spectra were performed on a Bruker Avance DPX 400 operating at 400 MHz for 1H NMR spectra and 100 MHz for 13C NMR spectra. Elemental analysis was done at The Campbell Microanalytical Laboratory (Department of Chemistry, University of Otago, Otago, New Zealand). Low-resolution mass spectrometry (LRMS) was performed on a Micromass ZMD Quadrupole Mass Spectrometer whereas high-resolution mass spectrometry (HRMS) was performed using a Bruker Daltonics BioApex-II 7T FTICR mass spectrometer equipped with an off-axis Analytic electrospray (ES) ionization source. Preparative high-performance liquid chromatography (HPLC) was performed on a Waters Empower system, equipped with a 600 gradient pump and a 486 UV(ultraviolet) detector using a C18 Alltech Econosphere column (10 μm, 22 × 250 mm). After radiolabeling, 123I-MEL037 was purified by an HPLC system equipped with linear variable UV, set at 254 nm, and a γ-detector using a reverse-phase C18 Phenomenex Bondclone column (10 μm, 7.8 × 300 mm) eluting with acetonitrile/0.1 mol/L ammonium acetate (70:30) at 1.5 mL/min. For radiochemical purity and stability measurements, the radioiodinated tracer solution was ascertained on an analytic HPLC system using a C18 Phenomenex Luna column (5 μm, 4.6 × 150 mm) eluting with acetonitrile/0.1 mol/L ammonium acetate (45:55) at 1 mL/min. Measurement of unmetabolized radiotracer was performed by thin-layer chromatography (TLC) analysis on silica gel plates with a preconcentration zone (Merck). The TLC plates were developed with ethyl acetate/methanol/concentrated NH4OH (85:15:0.2), and the radioactivity distribution was measured using a static radiochromatogram analyzer (Berthold Co).
Chemistry
2-(2-(4-(4-Iodobenzyl)Piperazin-1-yl)-2-Oxoethyl)isoindoline-1,3-Dione (MEL037).
N-Phthaloylglycine (1) (0.37 g, 1.81 mmol), 1-(4-iodobenzyl)piperazine (2a) (0.48 g, 1.81 mmol), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) (0.42 g, 2.18 mmol), N-hydroxybenzotriazole (HOBT) (0.30 g, 2.18 mmol), and N-methylmorpholine (NMM) (0.73 g, 7.25 mmol) were dissolved in 10 mL N,N-dimethylformamide (DMF) and the reaction mixture was stirred at room temperature for 24 h. DMF was removed in vacuo and the resulting residue was partitioned between ethyl acetate and water. The organic layer was concentrated, and the remaining residue was purified by column chromatography on silica (methanol/dichloromethane, 10:90) to give a light-yellow solid. Recrystallization from ethyl acetate and dichloromethane produced a white crystalline solid (0.38 g, 43%), m.p. (melting point) 152°C–153°C. 1H NMR (CDCl3) δ 2.43 (t, J = 5.0 Hz, 2H, CH2), 2.52 (t, J = 5.0 Hz, 2H, CH2), 3.48 (s, 2H, CH2), 3.52 (t, J = 5.0 Hz, 2H, CH2), 3.61 (t, J = 5.0 Hz, 2H, CH2), 4.48 (s, 2H, CH2CO), 7.08 (d, J = 8.2 Hz, 2H, ArCH), 7.45 (dd, J = 3.0, 5.5 Hz, 2H, ArCH), 7.66 (d, J = 8.2 Hz, 2H, ArCH), 7.72 (dd, J = 3.0, 5.5 Hz, 2H, ArCH). 13C NMR (CDCl3) δ 39.1 (CH2), 42.3, 44.8, 52.5, 52.7 (CH2), 62.2 (CH2), 92.7 (ArC-I), 123.5 (ArCH), 131.0 (ArCH), 132.3 (ArC), 134.0 (ArCH), 137.5 (ArCH), 163.8 (C=O), 168.1 (C=O). MS (mass spectrometry): ES(+ve) m/z 490 (M+1). Elemental Analysis: Anal. (C21H20N3O3I) C, H, N; Calcd 51.55, 4.12, 8.59; found, 51.74, 4.35, 8.48.
2-(2-(4-(4-Bromobenzyl)Piperazin-1-yl)-2-Oxoethyl)isoindoline-1,3-Dione (Br-MEL037).
The bromo analog of MEL037 was prepared from 1-(4-bromobenzyl)piperazine (2b) (1.24 g, 4.87 mmol) using the same procedure as the synthesis of MEL037. Recrystallization from ethyl acetate and dichloromethane produced a white crystalline solid (1.49 g, 69%), m.p. 142°C–144°C. 1H NMR (CDCl3) δ 2.44 (t, J = 4.8 Hz, 2H, CH2), 2.51 (t, J = 4.8 Hz, 2H, CH2), 3.49 (s, 2H, CH2), 3.53 (t, J = 4.8 Hz, 2H, CH2), 3.61 (t, J = 4.8 Hz, 2H, CH2), 4.48 (s, 2H, CH2CO), 7.21 (m, 2H, ArCH), 7.45 (m, 2H, ArCH), 7.72 (m, 2H, ArCH), 7.87 (m, 2H, ArCH). 13C NMR (CDCl3) δ 38.9 (CH2), 42.1, 44.6, 52.3, 52.5 (CH2), 61.8 (CH2), 120.9 (ArC), 123.3 (ArCH), 130.5 (ArCH), 131.3 (ArCH), 132.1 (ArC), 133.9 (ArCH), 136.6 (ArC), 163.7 (C=O), 167.8 (C=O). MS: ES(+ve) m/z 442 (M[Br79] + 1) (10%), 464 (M[Br79] + Na + 1) (100%). Elemental Analysis: Anal. (C21H20N3O3Br) C, H, N; Calcd 57.02, 4.56, 9.50; found, 57.25, 4.66, 9.38.
2-(2-(4-(4-(Trimethylstannyl)Benzyl)Piperazin-1-yl)-2-Oxoethyl)isoindoline-1,3-Dione (3).
To Br-MEL037 (0.4 g, 0.90 mmol) in anhydrous toluene (18 mL) was added hexamethylditin (0.44 g, 1.36 mmol) and palladium tetrakistriphenylphosphine (30 mg, 0.02 mmol). The reaction mixture was heated to reflux for 48 h with further addition of palladium tetrakistriphenylphosphine (30 mg) every 8 h before it was cooled, filtered, and then evaporated to dryness. The resulting residue was purified by column chromatography (ethyl acetate/petroleum spirit, 50%–100%) to give a white solid (175 mg, 37%), m.p. 172°C–174°C. 1H NMR (CDCl3) δ 0.30 (s, 9H, Sn(CH3)3), 2.50 (m, broad, 4H, CH2), 3.60 (m, broad, 6H, CH2 and CH2CO), 4.48 (s, 2H, CH2), 7.33 (d, J = 7.2 Hz, 2H, ArCH), 7.34 (dd, J = 3.1, 5.4 Hz, 2H, ArCH), 7.48 (d, J = 7.2 Hz, 2H, ArCH), 7.88 (dd, J = 3.1, 5.4 Hz, 2H, ArCH). 13C NMR (CDCl3) δ −9.6 (Sn(CH3)3), 39.0 (CH2CO), 52.0 (broad, CH2), 62.5 (CH2), 123.5 (ArCH×4), 132.3 (ArCH×2 and ArC×2), 134.0 (ArCH×2, ArC), 136.5 (ArC-Sn), 164.2 (C=O), 168.0 (2×C=O). MS: ES(+ve) m/z 550 (M+Na). HRMS (high-resolution MS): CI(+ve) Calcd for C24H30N3O3Sn (M+Na+) 550.1138; found, 550.1135.
Lipophilicity
The lipophilicity of MEL037 and its bromo analog was assessed using HPLC by determining the logP7.5 value using a Waters C18 column (X-Terra; 5 μm, 4.6 × 150 mm) eluting with a mobile phase consisting of methanol and phosphate buffer (0.1 mol/L, pH 7.5), 50:50, at 1 mL/min at pH 7.5 representing physiologic conditions. The logP7.5 of the studied compounds was estimated by a comparison of its retention time with that of standards of known log P values (18).
Radiolabeling
To a solution of the trimethylstannyl precursor (3) (0.25 mg, 0.5 μmol) in ethanol (200 μL), 123I-NaI (1 GBq, 30 μL), chloramine-T (100 μg, 0.45 μmol in 100 μL H2O), and HCl (100 μL, 1 mol/L) were successively added. After 5 min at room temperature, Na2S2O5 (5 mg, 30 μmol in 100 μL H2O), NaHCO3 (5 mg, 60 μmol in 100 μL H2O), and 350 μL of HPLC mobile phase were added. The resulting mixture was purified on the semipreparative HPLC system. The radiolabeled compound (retention time = 15 min) was collected and dried in vacuo. The radioiodinated tracer was recovered with 100 μL ethanol and then formulated in 10 mL saline for biologic evaluation.
Ligand-Binding Assays
The percentage inhibition of unlabeled MEL037 to σ1- and σ2-receptors was determined in in vitro competitive binding assays at 10−6 and 10−5 mol/L concentrations. The assays were performed according to literature methods with minor modification (19). Briefly, the percentage of inhibition was determined by incubating, in triplicate, aliquots of diluted rat brain membrane (300 μg of protein) with a 10−5 mol/L concentration of MEL037 in 50 mmol/L Tris-HCl (pH 8.0) with 3H-(+)-pentazocine (3 nmol/L) at 37°C for 2.5 h for σ1-receptors or with 3H-DTG (10 nmol/L) and (+)-pentazocine (1 μmol/L) at 25°C for 1.5 h for σ2-receptors. In both cases nonspecific binding was determined in the presence of haloperidol (10 μmol/L). After incubation, the reaction was terminated by rapid filtration using a Brandel 48-well cell harvester (Brandel) over Whatman GF/B glass fiber filters that were soaked in a solution of 0.5% polyethyleneimine at 4°C for at least 2 h before use. Filters were washed 3 times with 5 mL of ice-cold wash buffer (50 mmol/L Tris-HCl, pH 7.4). The filters were collected and the amount of bound radioactivity was measured using a β-scintillation counter. The percentage inhibition of unlabeled MEL037, at a 10−5 mol/L concentration, was also determined for other receptors using specific radioligands and tissue membranes (Novascreen Biosciences) according to literature methods (Table 1). The 50% inhibitory concentration (IC50) values were then converted to apparent Ki (inhibition constant) values using the Cheng–Prussof equation and radioligand Kd (dissociation constant) values (3H-(+)-pentazocine Kd σ1 = 2.5 nmol/L; 3H-DTG Kd σ2 = 77 nmol/L).
Experimental Conditions for In Vitro Assays and Percentage of Inhibition of MEL037 for Selected Neurotransmitter Systems
In Vivo Studies
Animal experiments were performed in compliance with the National Health and Medical Research Council Australian Code of Practice for the care and use of animals for scientific purposes. Female C57BL/6J black and BALB/c nude albino mice (15–18 g) of 5 wk age were obtained from the Animal Resources Centre, Western Australia. B16F0 murine melanoma cells were originally obtained from the European Collection of Cell Cultures and A375 human amelanotic melanoma cells were originally obtained from the American Type Culture Collection. Before transplantation, B16F0 and A375 melanoma cells were maintained as a monolayer in RPMI culture medium supplemented with 10% fetal calf serum and antibiotics and passaged with trypsinization. Early passages were frozen and stored in liquid nitrogen. Cells were passaged to P = 10 and then discarded. Frozen aliquots were grown in a monolayer culture to between 80% and 95% confluence and for transplantation were trypsinized and washed with Ca2+- and Mg2+-free phosphate-buffered saline (PBS). For inoculation, B16F0 melanoma cells were resuspended in Ca2+- and Mg2+-free PBS at 3 or 5 × 106 viable cells per milliliter, and 0.1 mL was subcutaneously injected at the left flank of 6- to 7-wk-old C57BL/6J mice. Eleven days later, tumors could be palpated in >98% of inoculated animals. A375 human melanoma cells were resuspended at 1 × 107 viable cells per milliliter, and 0.1 mL was injected subcutaneously at the left flank of 6-wk-old BALB/c nude mice; 25–26 d later tumors could be palpated in ∼60% of inoculated animals developing tumors.
Biodistribution
Eleven days (B16F0 melanoma) and 25 d (A375 human melanoma) after tumor transplantation, 123I-MEL037 (0.37–0.74 MBq, 100 μL) was injected intravenously into the mice via the tail vein. At defined times after injection (1, 3, 6, 24, and 48 h), groups of mice (n = 5) were sacrificed by CO2 administration, which was followed by cervical dislocation, weighing, and dissection. Selected organs were weighed, and their radioactivity was measured with a γ-counter (Wallac model 1480). The remaining activity in the carcass was also determined to obtain the total activity in the mouse at a defined time point. The fraction of injected activity (%ID) in the organs was calculated by comparison with suitable dilutions of the injected dose. The radioactivity concentration in the organ and the mean concentration in the mouse (%ID/g) were found by dividing the %ID for each organ by the weight of the organ. A mean contrast ratio in the tumor at time of sacrifice (MCRt) was calculated for each animal by dividing the tumor concentration by the radioactive concentration in the mouse excluding the tumor according to the following formula: MCRt = (activity/tissueg)/(remaining activity at time t/animal weightg).
Competition Studies
In competition experiments, groups of mice bearing B16F0 murine melanoma (n = 5) were injected intravenously with 1 mg/kg haloperidol 5 min before the radiotracer (0.37–0.74 MBq) dose. The mice were sacrificed 1 or 24 h after injection, and tissues were handled as described for the biodistribution study. Radioactive concentrations in organs and tumor of treated animals were compared with that of control animals. Statistical significance was evaluated using the 1-way ANOVA test. The criteria for significance were P < 0.05 and P < 0.01.
Metabolite Studies
For metabolite studies, animals were injected with 3–5 MBq of 123I-MEL037. Unmetabolized radiotracer in the plasma, brain, and tumors was determined at 0.25, 1, 3, and 24 h after injection by radio-TLC analysis. Samples of the brain and tumor (100–200 mg minced in small pieces) and plasma (0.2 mL) were added to unlabeled MEL037 (20 nmol, 10 μL of 1 mg/mL acetonitrile solution) and acetonitrile (1 mL), exposed for 2 min to an ultrasonic probe designed for cell disruption (Sonicator; Misonix Inc., USA), and then centrifuged. The radioactivity of the precipitate was measured to quantify the acetonitrile extraction efficiency. If necessary, a second extraction with acetonitrile (1 mL) was performed to ensure maximum recovery of the radioactivity. The supernatant was evaporated to dryness, redissolved in 50 μL methanol, and applied to TLC plates.
SPECT
SPECT was performed using a high-resolution γ-camera (X-SPECT; Gamma Medica Inc.), designed for laboratory animals, equipped with an array of discrete 2 × 2 × 6 mm NaI(Tl) crystals optically isolated from each other and a high-resolution, parallel-hole collimator that has a 12.5 × 12.5 cm field of view. The mouse was anesthetized via inhalant isoflurane (Forthane; Abbott) in 200 mL/min oxygen using a nose cone fitted to the animal bed and imaged for 10–40 min at 1, 24, and 48 h after intravenous injection of 7–10 MBq 123I-MEL037.
RESULTS
Chemistry and Radiochemistry
Compound MEL037 was synthesized from commercially available 1 and 2a using EDC and HOBT with NMM in DMF (yield = 43%) (Fig. 2). Similarly, condensation of piperazine 2b with the acid 1 by the same route afforded Br-MEL037 (yield = 69%). Subsequent treatment of the bromo precursor Br-MEL037 with hexamethylditin in the presence of catalytic palladium tetrakistriphenylphosphine in refluxing toluene yielded the stannane 3 (yield = 37%). The log P values of MEL037 and Br-MEL037 were 3.6 and 3.3, respectively. The regioselectivity of the labeling of MEL037 in the 4 position on the benzene ring was achieved by electrophilic substitution of a trimethyltin leaving group using chloramine-T as oxidant. The radiochemical yield of 123I-MEL037 preparation was 65% ± 9% (n = 12). Radiochemical purity as assessed by analytic HPLC was >99% and the specific activity was >2 GBq/nmol based on the limit of detection of the HPLC system. The radiochemical purity was still >98% when 123I-MEL037 was stored at −20°C for 24 h and 97% when kept in 1% ethanol saline solution at room temperature for 4 h.
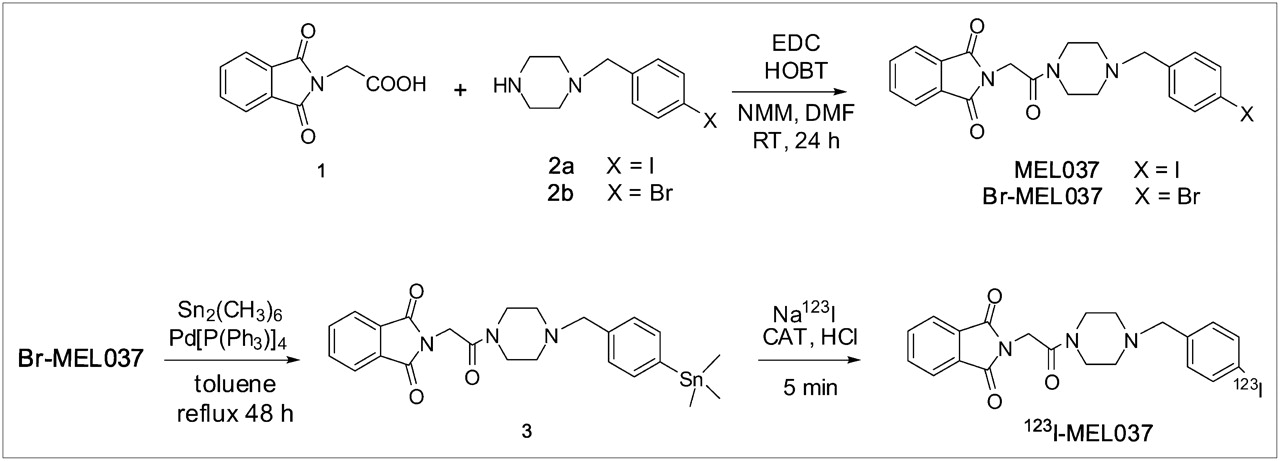
Preparation of MEL037 and radiolabeling of 123I-MEL037. RT = room temperature; CAT = chloramine-T.
Ligand-Binding Assays
The potency of unlabeled MEL037 to inhibit the specific binding of 3H-(+)-pentazocine to σ1-receptors and of 3H-DTG to σ2-receptors from rat brain was determined in competitive binding assays at 10−6 and 10−5 mol/L concentration of unlabeled compound. In spite of structural similarities with IBP, very low affinities for σ1- and σ2-receptors were obtained for MEL037. At 10−5 and 10−6 mol/L concentrations, MEL037 was found to inhibit 95% and 54%, respectively, of σ1-binding of 3H-(+)-pentazocine (Ki σ1 = 0.5 μmol/L) and at 10−5 mol/L, 60% of σ2-binding of 3H-DTG (Ki σ2 = 10 μmol/L) (Table 1). Similarly, the bromo analog Br-MEL037 did not present any σ-receptor potency. As the studied compound is not an inhibitor for σ-receptors, its potency on other receptor systems was assayed. MEL037 did not display any affinity for other receptors studied.
Biodistribution
To evaluate 123I-MEL037 as a tumor marker, the biodistribution of the radiotracer was studied in black mice bearing the B16F0 murine melanoma tumors and in nude mice bearing the A375 human amelanotic melanoma. Six hours after injection of the radiotracer, the highest uptake (20–30 %ID) was found in the gastrointestinal tract (Table 2). At 24 h after injection, <3% of injected dose was found in the intestine. In the liver, the uptake amounted to 15 %ID that cleared with a biologic half-life of 5 h to reach a value of 0.7–1.3 %ID at 24 h according to the animal species. In kidney, lung, spleen, and heart, the uptake values of 123I-MEL037 were <2 %ID at 1 h and 0.2 %ID at 24 h after injection. In the spleen of C57BL/6J black mice, large variations of the 123I-MEL037 uptake values were observed in groups of animals at each time point, which correlated with the extent of pigmentation in the organ. In the BALB/c nude mice, the uptake values in this organ were homogeneous for each group. The thyroid uptake of the 123I-MEL037 was particularly low and increased slightly during the study from 0.05 %ID at 1 h to ∼0.2 %ID from 6 to 24 h. In C57BL/6J black mice, a significant uptake of 123I-MEL037 was observed in the eyes (2.3 %ID). In BALB/c nude mice, the eye uptake was 100 times lower than that in black mice. As a consequence of the simultaneous low organ uptake and rapid clearance, the amount of radioactivity that remained in the black and nude mouse body at 24 and 48 h after injection was less than 5 and 2 %ID, respectively.
Biodistribution of 123I-MEL037 in Mice: Uptake (%ID) in Organ (Decay-Corrected)
The values of tumor and organ concentration, expressed in %ID/g, are summarized in Table 3. In the B16 melanoma tumor, 123I-MEL037 exhibited a high uptake at 1 h (13 %ID/g) that increased with time and reached 25 %ID/g at 6 h. The tumor uptake was still 10 %ID/g at 24 h. In the A375 amelanotic tumor, the uptake was very low, decreasing from 1.8 to 0.4 %ID/g between 1 and 24 h after injection. For each of the 2 mice species, the uptake values in heart, lung, and kidney ranged from 3 to 9 %ID/g at 1 h after injection to values <1 %ID/g at 24 h. In the brain, at 1 and 24 h, the uptake values of 123I-MEL037 were 2 and 0.2 %ID/g, respectively. The tumor-to-body contrast of 123I-MEL037 was ascertained by calculating the MCRt. These values are reported in Table 3. Because of the reduction of the radioactive concentration in the body and the slower decrease in target tissues such as tumor and the eyes, MCR24 was 35 and MCR48 was 60 in black mice. The corresponding MCR24 was <1 in nude mice.
Biodistribution of 123I-MEL037 in Mice: Uptake (%ID/g) and MCRt (Decay-Corrected)
Competition Studies
The blocking effect of haloperidol on tracer uptake in vivo was examined over 24 h in selected organs and tumor tissue to assess the uptake mechanism in the B16 tumor. As seen in Table 4, at 1 h after injection, the radioactive concentration in the tumor was significantly reduced in treated animals compared with that in control animals (−23%; P < 0.05). Simultaneously, significant increases of 10%–15% in lung, heart, blood, and brain uptakes were observed. At 24 h after injection, significant decreases in uptake values were observed in the tumor (−50%; P < 0.01), thyroid (−40%; P < 0.05), eyes, and liver (−16%, P < 0.05). At this time, the uptake in lung, heart, blood, and brain was found to be unchanged.
Effect of Haloperidol on Organ Uptake of 123I-MEL037 in Black Mice Bearing B16 Tumor (Decay-Corrected)
Metabolite Studies
After ultrasonic disruption and protein elimination, >96% of the radioactivity in the brain and plasma was recovered in acetonitrile. In the tumor, the recovery was 85%, 84%, and 75% at 15 min, 1 h, and 3 h, respectively. In plasma, at 1 h after injection, 60% of the radioactivity was related to unmetabolized 123I-MEL037. The remaining 40% of the radioactivity represented polar radioactive metabolites. In the brain and tumor at 3 h, the metabolite study indicated that >90% of the recovered radioactivity represented unmetabolized 123I-MEL037 (Fig. 3). Metabolite measurements were unreliable at 24 h because of the very low activity present in the brain and plasma, whereas in the tumor only a recovery of 37% recovery was possible.

Fraction of unmetabolized 123I-MEL037 in plasma, brain, and tumor at 0.25, 1, and 3 h after injection.
SPECT Studies
The whole-body distribution of 123I-MEL037 in mice bearing the B16F0 melanotic and the A375 amelanotic melanoma was monitored over 48 h using SPECT. Figure 4 shows typical SPECT images of mice acquired 24 h after the radiotracer injection. In the image of the mouse bearing the B16F0 melanoma tumor, the highest radioactivity concentration was observed in the tumor. Lower concentrations of radioactivity were seen in the eyes and in the abdomen. No tumor or eye uptake was observed in the mouse bearing A375 amelanotic melanoma tumors. These observations confirmed the quantitative results obtained in the biodistribution studies.
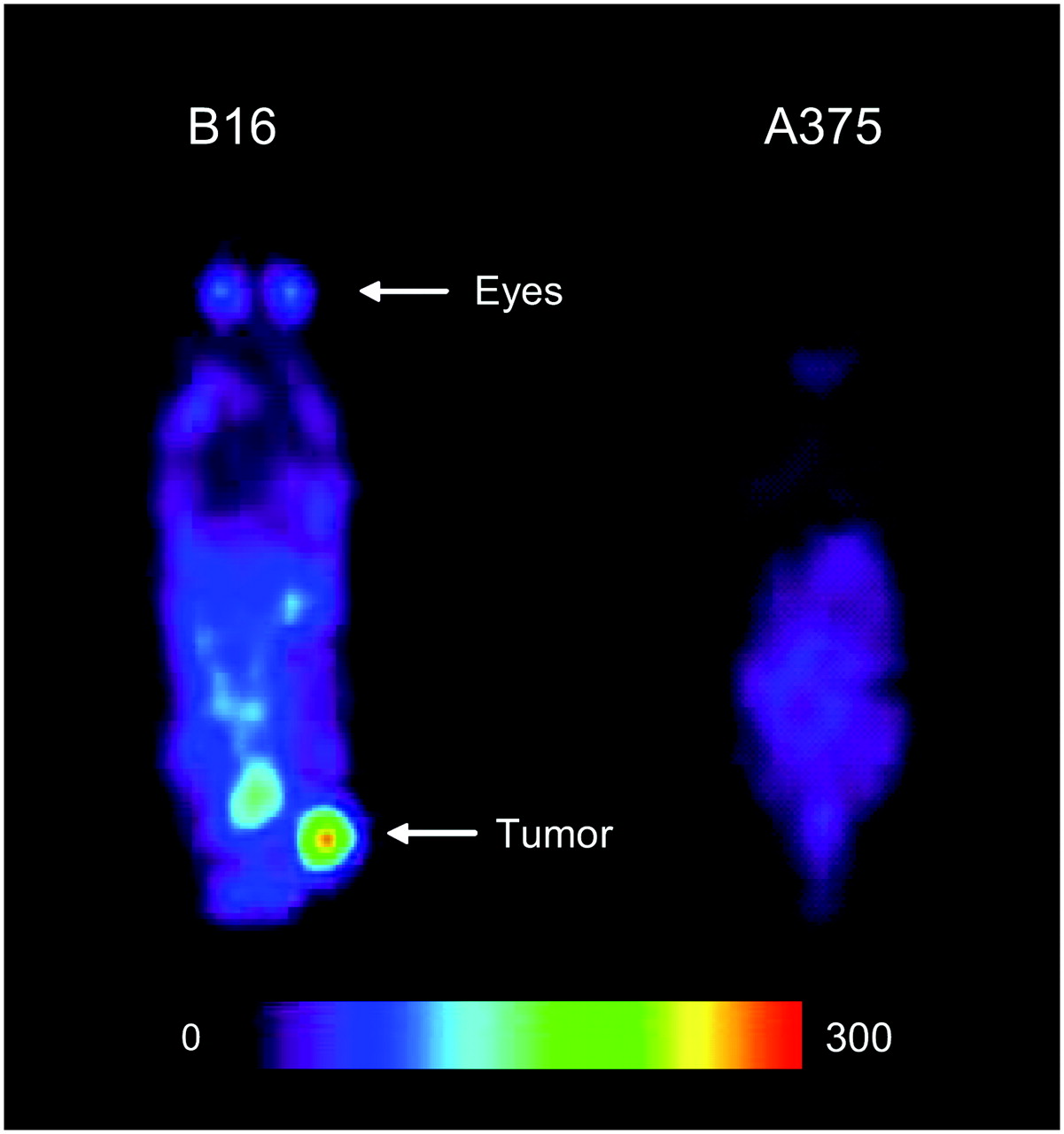
SPECT planar images obtained 24 h after injection of 10 MBq 123I-MEL037 in C57BL/6J mouse bearing B16F0 murine melanoma tumor and in BALB/c nude mouse bearing A375 human amelanotic melanoma tumor. Scale indicates the highest value in counts/pixel in image.
DISCUSSION
The aim of this study was to investigate the potential use of a new iodobenzylpiperazine, MEL037, as an imaging agent for melanoma tumor detection. MEL037 has biologic activity that may result from 3 potential mechanisms—that is, melanin binding (20), melanin synthesis (21), or affinity to the σ-receptors that are highly expressed in melanoma cells (22) and tumor tissues (23). Depending on the molecular structure, one of these mechanisms or a combination of mechanisms can take place. To understand the exact mode of action, unlabeled MEL037 was used in in vitro binding studies for σ1- and σ2-receptor affinity and 123I-MEL037 was used for in vivo biodistribution and competition experiments in 2 tumor models of melanoma.
Numerous iodobenzamides have already been prepared, and a clearer picture of the structure–activity relationships on cellular uptake and retention of activity in melanoma tumors is now emerging. In previous studies a relationship between the lipophilicity of iodobenzamides and tumor uptake was suggested (24,25). Hence, the measured lipophilicity of MEL037 (logP7.5 = 3.6) favors high uptake in the B16 tumors. The uptake mechanism of aminoalkyl-iodobenzamides, into melanoma cells, is believed to occur through the melanin biosynthesis pathway via an addition reaction to pheomelanin, a sulfur-containing intermediate found only in melanoma cells (21). In vitro studies of aminoalkyl-iodobenzamides indicated that the uptake is related to the melanin content of cells, and pharmacologic studies demonstrated that it is not σ-receptor dependent (8,24,26). Experiments in cell culture showed that uptake of 123I-BZA was rapid and high in B16 or M4Beu melanotic cells and was low in M3Dau amelanotic cells (20). Using the secondary ion mass spectrometry technique, the localization of BZA was demonstrated to be intracytoplasmic in tumoral melanocytes and in the pigmented structures of the skin and the eye and was unambiguously proven to be within melanosomes excluding a specific binding to membrane σ-receptors (27).
Conversely, the uptake in various tumors, including melanoma, of piperidinyl-benzamide derivatives and analogs, has been considered to be σ-receptor related (10,15,17). In vitro studies have demonstrated that these compounds possess high affinity for σ-receptors despite their nonselectivity for σ1–σ2 subtypes. Moreover, the specific localization of the radioiodinated piperidinyl-benzamides in the central nervous system as well as in tissues such as the liver, kidneys, and lungs—where σ-receptors are present in high concentration—strengthens the σ-hypothesis for the uptake of some benzamide derivatives in tumor tissues. In vitro binding assays with the σ-ligands 3H-(+)-pentazocine or with 3H-DTG demonstrated that MEL037 does not present a σ-receptor binding profile despite its molecular analogy with certain piperidine and piperazine derivatives. Furthermore, the competitive binding of MEL037 with specific radioligands for other receptor systems did not show any significant potency. To further support these results, the bromophenyl analog Br-MEL037 was used in in vitro competitive assays and presented the same pharmacologic profile.
The biodistribution of 123I-MEL037 was studied in 2 mice species—one bearing the B16F0 murine melanotic melanoma model and the other bearing the A375 human amelanotic melanoma model. These 2 models were used to assist in ascertaining the melanin versus nonmelanin mechanisms of uptake. In the B16 tumor, the uptake of 123I-MEL037 increased with time and reached 25 %ID/g at 6 h after injection. This uptake is 3 times higher than that for 125I-BZA2 under our experimental conditions and that reported in the literature (9) in the same tumor model. In the A375 tumor, the uptake was 20 times lower and diminished rapidly to 0.4 %ID/g at 24 h after injection. The frequency of expression of σ-receptors in melanoma tissue in vivo is not known. However, as the B16 mouse melanotic melanoma and A375 human amelanotic melanoma cell lines have been reported to express similar σ-receptor densities of 3 and 1.8 pmol/mg protein, respectively (22), it is likely that the accumulation of 123I-MEL037 within the B16 tumor is not due to σ-receptor interactions. Furthermore, it has been shown that the eye uptake of 123I-BZA in the C57BL/6J black mice was attributed to the accumulation of the radiotracer in the pigmented eye structures (28). In the present study, the significant uptake (2.3 %ID) of 123I-MEL037 in the eyes of the same animal species was 100 times higher than the eye uptake in BALB/c nude mice (nonpigmented animals), also supporting the hypothesis that 123I-MEL037 involves a specific interaction with melanin. A consequence of this is that 123I-MEL037 uptake values observed in the spleen of some C57BL/6J mice correlated well with the black coloration of the organ. The pigmentation, occurring in 30% of the young animals was attributed to the accumulation of melanin in melanocytes (29). In metabolite studies, the recovery of the radioactivity in the tumor dropped from 85% at 0.25 h after injection to 37% at 24 h after injection despite the 2 extractions with acetonitrile. A strong interaction of 123I-MEL037 with melanin within the cells and melanin insolubility could explain the poor recovery at later time points.
In each of the 2 mice species, the low thyroid uptake of the studied 123I-MEL037, <0.2 %ID at 24 h, is an indication that this tracer is stable to in vivo deiodination. In heart, lung, and kidney, the uptake values were lower than 1 %ID/g at 24 h. In the brain, the uptake values of 123I-MEL037 were 2 %ID/g at 1 h and had the same kinetics as in the blood. This nonspecific uptake is explained by the logP value (3.6), allowing the tracer to cross the blood–brain barrier. The tumor-to-body contrast as measured by MCR was 60 in C57BL/6J black mice bearing the B16 tumor and <1 in BALB/c nude mice bearing the A375 tumor, demonstrating that the amelanotic tumor was unable to concentrate the radiotracer.
It remained possible that the selective uptake of 123I-MEL037 by melanoma cells in the tumor is in part due to binding to σ-receptors perhaps associated with internalization of the compound. To confirm the melanin hypothesis versus the σ-receptor–mediated uptake, the blocking effect of haloperidol on the in vivo tracer uptake was examined over 24 h in B16 tumors and selected organs in which σ-receptors are present—that is, the brain, liver, heart, kidneys, and lungs. At 1 h after injection, no reduction of activity uptake in the central nervous system or the periphery was observed, but the 10%–15% increase was probably due to the higher radioactive concentration in the blood. Surprisingly, the radioactive concentration in the tumor decreased in treated animals by −23% and −50% at 1 and 24 h, respectively. At 24 h after injection, a significant decrease (−16 %ID/g) in activity in the eyes was observed, whereas the uptake in lung, heart, blood, and brain was found to be unchanged. Considering that the in vivo results showed a high uptake in B16 tumors and no uptake in A375 tumors that ordinarily would have the same amount of σ-binding sites, a decrease of activity in the tumor and eye uptake as a consequence of a σ-receptor interaction in a competitive challenge between the competitor and 123I-MEL037 is not in agreement with in vitro assays that indicated that MEL037 is not a potent σ-tracer.
Haloperidol, an antipsychotic drug with a σ-receptor pharmacologic profile, was found to have antiproliferative effects in tumor cell and B16 melanoma cell culture by inducing apoptosis (30,31). Phenothiazine derivatives have been demonstrated to have the same in vitro effect and to attenuate in vivo melanoma tumor growth (32). In contrast, the σ-receptor ligand DTG did not inhibit B16 cell growth (33). Moreover, chloroquine (34) and phenothiazine derivatives have been reported to have a high affinity for melanin and to accumulate in pigmented cells of eyes (35,36). Some in vitro studies were not conclusive in their ability to show the haloperidol inhibition of 125I-BZA binding to B16 pigmented cells (26). However, other studies have demonstrated a saturable binding of 3H-haloperidol to beef-eye melanin and to synthetic dopamine melanin, and in vivo autoradiography also revealed extensive 3H-haloperidol accumulation in melanin-containing tissues and, particularly, in eyes of pigmented mice (37). Therefore, the reduction by haloperidol of 123I-MEL037 uptake in the B16 tumor and in the eyes of C57BL/6J black mice, compared with that of untreated animals, could be reasonably explained by the competition between the drug and the radiotracer in the binding to melanin in these tissues. According to our in vitro data and our in vivo results, it is likely that MEL037 does not bind to σ-receptors within the B16 tumor but is subject to the same melanin interaction as the aminoalkyl-iodobenzamide BZA. As a consequence, MEL037 could also act as a type of prodrug interacting with melanin, which could subsequently exert an apoptotic effect on melanoma cells and could also be useful in enhancing the effect of cytotoxic agents (38).
CONCLUSION
We have prepared an iodobenzylpiperazine derivative in an attempt to develop new radiotracers to study melanoma. In spite of structural similarities with IBP, compounds MEL037 and Br-MEL037 did not have σ-receptor profiles. Biologic studies showed that 123I-MEL037 presented a very high uptake in melanotic tumor and that this uptake was related to melanin binding or biosynthesis. Further in vitro studies are warranted to understand the exact in vivo behavior of this benzylpiperazine and to fully understand the exact uptake mechanism in melanoma tumors. The radioiodinated benzylpiperazine, which displays a rapid and high tumor uptake, appeared to be a promising imaging agent for melanoma detection. As the studied compound displays a high melanoma-to-nontarget ratio and displays an interaction with melanin, it could also be suitable for development and evaluation as a potential therapeutic or radiotherapeutic agent.
Acknowledgments
The authors gratefully acknowledge Tim Jackson and Janette Chapman for technical assistance. We thank Dr. Marie-Claude Grégoire for helpful advice and image processing.
Footnotes
-
COPYRIGHT © 2007 by the Society of Nuclear Medicine, Inc.
References
- Received for publication March 12, 2007.
- Accepted for publication May 7, 2007.





{kind=link}
{kind=link}
{kind=link}
{kind=link}