


Apoptosis is a physiologic form of cell death, which plays an important role in embryogenesis, cellular homeostasis, tissue atrophy, and removal of damaged and mutated cells (1). Caspases, which act as a molecular chain saw, are the central components of the apoptosis response. Caspases are cysteine proteases that cleave after an aspartate residue in their substrates. At least 7 of 14 known mammalian caspases have important roles in apoptosis (2). The caspases involved in mammalian apoptosis are divided into the following categories: the initiator caspases, which include caspases-2, -8, and -10; and the effector or executioner caspases, which include caspases-3, -6, and -7. An initiator casapse is characterized by an extended N-terminal region, which is important for its functions and comprises one or more adaptor domains (CARD or DED). An effector caspase usually contains 20–30 residues in its prodomain sequence. All caspases are produced as single-chain inactive zymogens and must undergo proteolytic activation during apoptosis. The effector caspases—both zymogen and activated enzyme—exist as constitutive homodimers. The activation of effector caspases is induced by initiator caspases through cleavage at a specific Asp residue, resulting in separate large and small subunits, which subsequently associate with each other. In contrast, initiator caspases are autoactivated without proteolytic cleavage and may be present predominantly as a monomer (e.g., caspase-9) or in equilibrium between monomers and dimers (e.g., caspase-8).
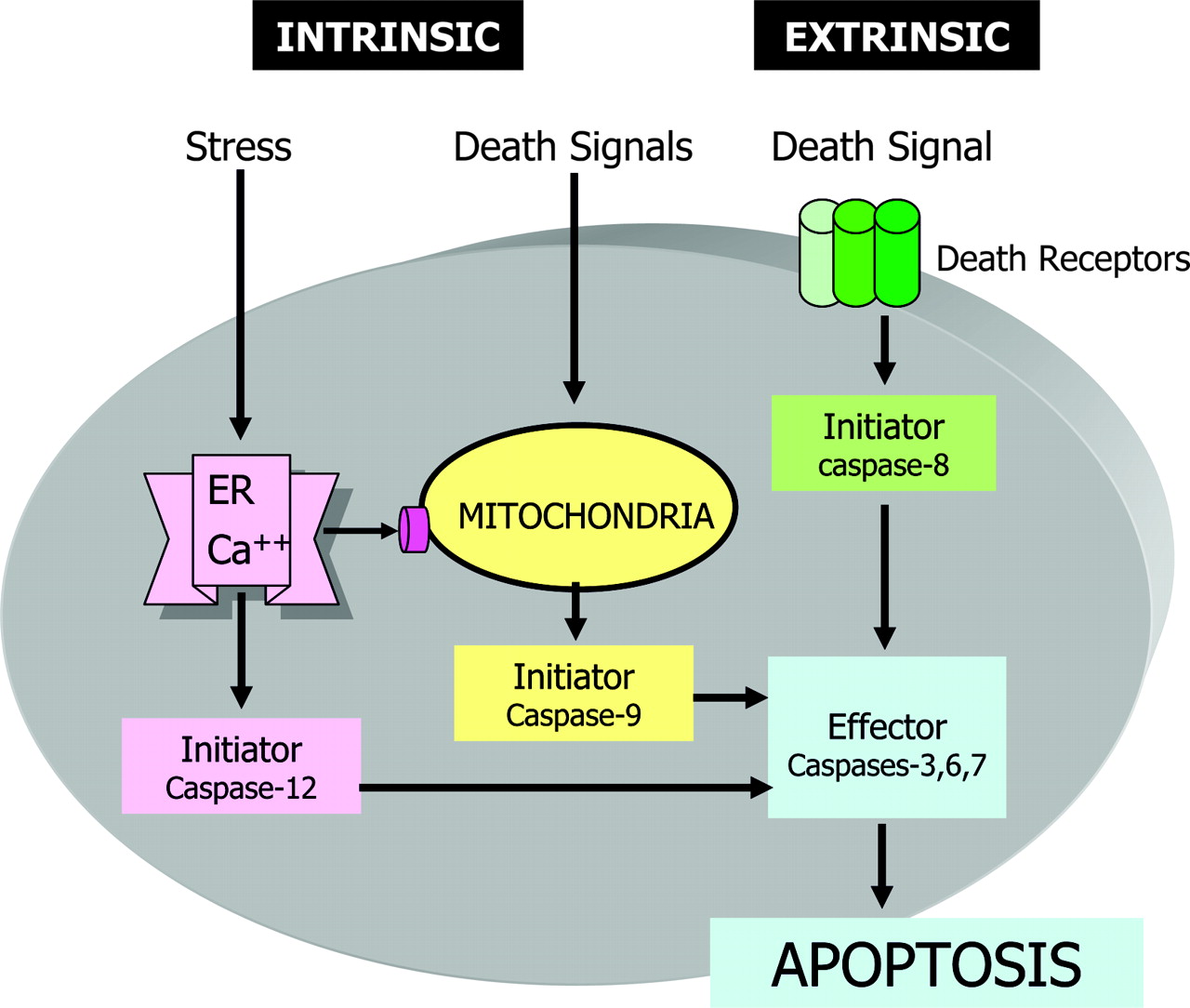
There are 3 major signaling pathways of apoptosis in which caspases are activated (Fig. 1): the death receptor pathway (3,4), the mitochondrial pathway (5,6), and a recently recognized endoplasmic reticulum (ER) stress pathway (7,8). The death receptor pathway, or so-called extrinsic death pathway, is initiated by binding of an extracellular death ligand (such as CD95L and tumor necrosis factor-α [TNF-α]) to its cell-surface death receptor (such as CD95 and TNF-α receptors), resulting in the formation of a homotrimeric ligand-receptor complex. The trimerization of the cytoplasmic death domain of death receptors results in the recruitment of adapter proteins (e.g., Fas-associated death domain, TNF-α receptor-associated death domain) and initiator procaspase to form a death-inducing signaling complex (DISC), which forms a platform for activation of initiator caspases and turning on executioner or effector caspases to complete the apoptotic process (Fig. 2). The mitochondrial pathway is activated in response to several stimuli, including ischemic or oxidative stress, and ultraviolet radiation or chemotherapeutic agents. The activation of mitochondria results in collapse of the mitochondrial membrane potential and release from the intermitochondrial membrane space of some of the well-characterized proteins into the cytoplasm. Among them, the most intriguing one is cytochrome c, which binds to Apaf-1 and in the presence of dATP or ATP (deoxyadenosine triphosphate or adenosine triphosphate) induces conformational changes in Apaf-1 to form an apoptosome. The apoptososme, similar to DISC in the extrinsic pathway, serves as a platform to recruit and activate initiator caspase-9. Activated caspse-9 then activates effector caspases to induce apoptosis. In the ER stress pathway, which may be induced by misfolded proteins and changes in ER calcium, apoptosis is mediated via activation of yet another initiator caspase (caspase 12). Active caspase-12 then activates common effector caspases to induce apoptosis. ER stress may also induce apoptosis via the mitochondrial pathway (8).
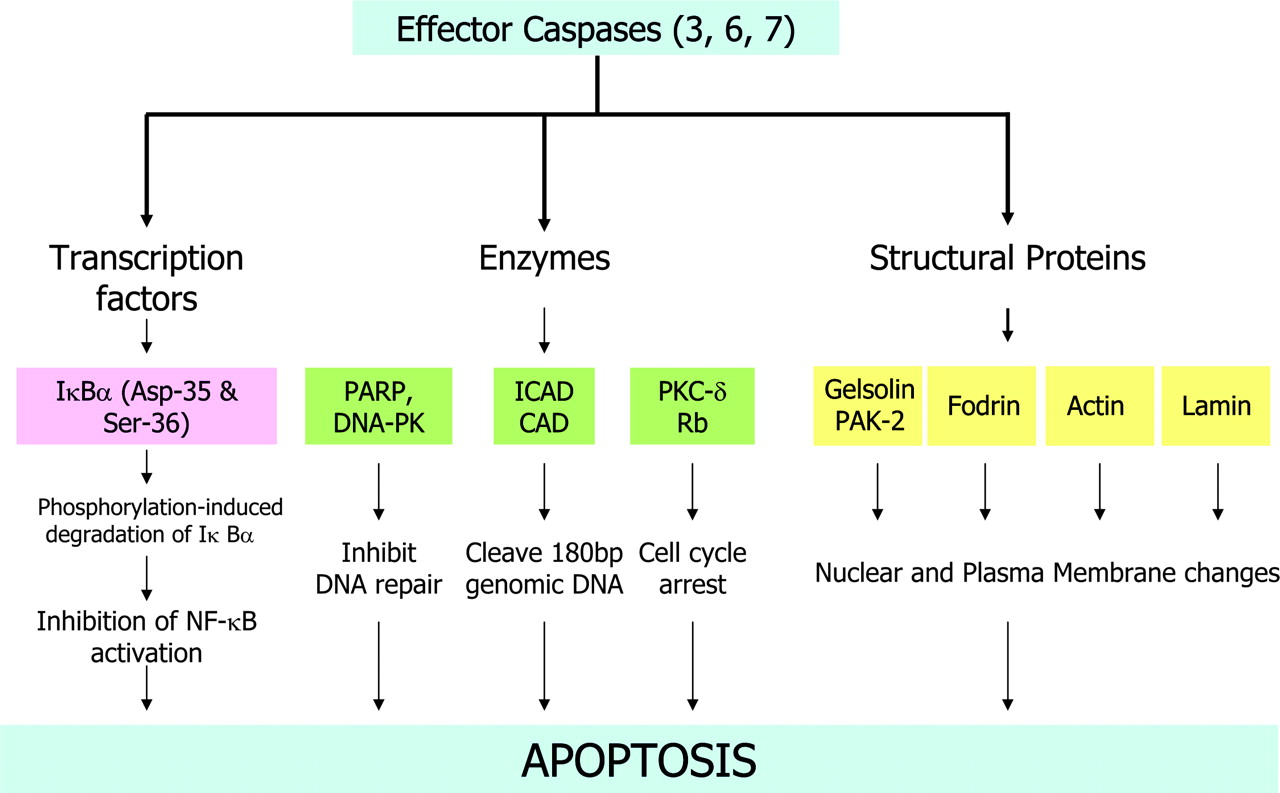
Once activated, effector caspases (caspases-3, -6, and -7) proteolytically cleave several cytoplasmic and nuclear target substrates (Fig. 3), resulting in characteristic biochemical and morphologic changes of apoptosis (9). The effector caspases recognize at least 4 continguous amino acids—namely, P4-P3-P2-P1—and cleave after the C-terminal residue (P1), usually an Asp (10). Although initially, the P1 residue was considered to be exclusively Asp, it has been demonstrated that some caspases can also cleave after Glu (11,12); for mammalian caspases, the preferred P3 position is invariably Glu (13). Therefore, the preferred specificity of cleavage of caspases has been suggested as X-Glu-X-Asp. There are apparently >400 caspase substrates. Caspase-6 or caspase-7 cannot compensate for the absence of caspase-3. Caspases-3 and -7 are redundant with respect to the cleavage of PARP, which is a substrate for caspase-6. The optimum sequence of substrate specificity for caspase-6 (VEHD), which cleaves PARP, is distinct from those for caspases-3 and -7 (DEVD). Therefore, the substrates with DEVD sequence used by Bauer et al. (14), in the study reported on pages 1066–1074 of this issue of The Journal of Nuclear Medicine, may delineate mostly caspase-3–and caspase-7–mediated apoptosis and may not delineate target substrates for caspase-6.
The currently available target for molecular imaging of the apoptotic death comprises cell-surface externalization of phosphatidylserine (PS) (15–17). The success of this target lies not only in the relevance of apoptosis in a large number of pathologies but also the availability of the recombinant human protein annexin A5, which binds to PS with a high specificity in the complex tissue environment. Apoptosis, besides PS, offers several other potential targets, such as the activated caspases. Of these activated (executioner) caspases, caspase-3 is the bearer of the greatest promise. In contrast to exteriorized PS on the cell surface, activated caspase-3 resides predominantly in the cytosol (and mitochondria) protected from the extracellular milieu by the selectively permeable plasma membrane. This imposes a technical constraint on the design of suitable probes for in vivo targeting. The specificity of the probe is a lesser problem since the peptide motif within caspase-3 substrates is well recognized and has been applied successfully in a variety of assays to measure caspase-3 activity in vitro. The real challenge is to facilitate entry of the probe into the cell and ensure its retention in an apoptotic cell (with activated caspase-3).
Bauer et al. (14) have deftly engineered the activated caspase-3 binding peptide motif DEVD to a peptide motif borrowed from the repertoire of HIV—the Tat-sequence. The latter motif increases the uptake of radiolabeled DEVD-peptide into the cell in vitro. The radiolabel probe is selectively retained in the apoptotic cell (compared with the viable cells) due to cleavage of the chimeric peptide by activated caspase-3. This strategy offers a very interesting and intriguing concept, wherein the specific uptake of the radiolabeled probe is turned on by activated caspase-3. Cleavage of chimeric peptide by activated caspase-3 was demonstrated in the additional set of experiments conducted in a cell-free system. However, authors have not considered other explanations of retention than “turn me on;” for instance, the difference in cellular uptake could be attributed to a variable membrane dynamics between apoptotic and living cells. Experiments that use a Tat-chimera with a noncleavable radiolabeled peptide motif can obviously provide some clarification. Another concern arises from the finding that the probe uptake gradually erodes over a 20-min period, as evident in Figure 5 of the article by Bauer et al. The specific uptake may not be turned on for long enough to shine above background levels for successful molecular imaging of apoptosis in vivo. Furthermore, although the caspase substrates used by Bauer et al. are expected to be applicable in most cases of apoptotic cell death, which is predominantly caspase dependent, it may not delineate caspase-independent apoptosis. Finally, no caspase activation is involved in another form of programmed cell death—autophagy (18)—that may not be identified by the proposed technique. In vivo experimental animal studies should determine the ultimate utility and potential clinical applicability.
The work of Bauer et al. (14) also provides an impetus for engineering the probes that target the intracellular cascade of apoptosis. In extending the proposal of Bauer et al., it may become possible to use substrate binding sites of active effector caspases for labeling. Recently, Ni et al. (19), using the crystal structure of the unoccupied substrate binding sites of caspase-3, demonstrated the critical side chain movement in a hydrophobic pocket in the active site. The side chain of Tyr-204 is rotated so that the phenol group occupies the S2 subsite in the active site. The rotation of this side chain is critical for the binding of substrates. Barring some limitations, the labeling of caspase substrates with a common amino acid sequence is an important contribution in the area of molecular imaging for apoptosis.

Three different roads but a common destination.
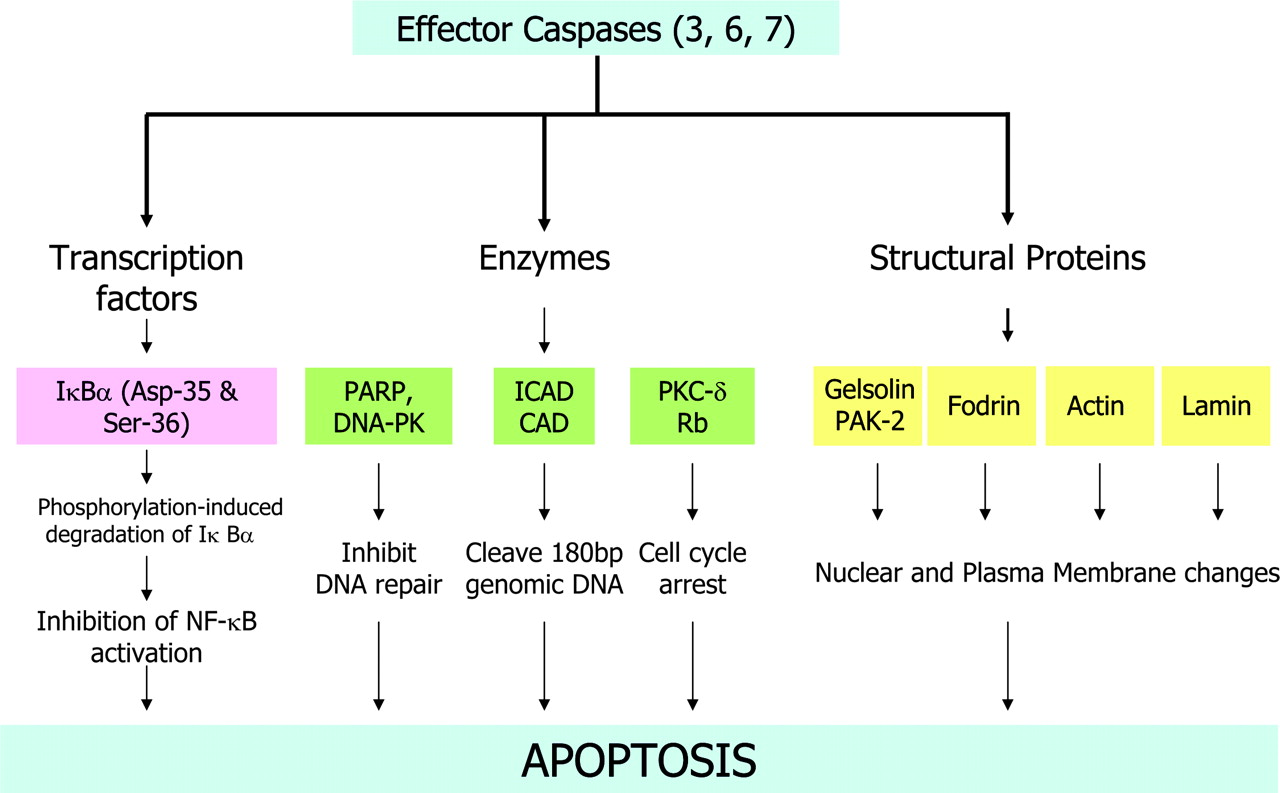
Target substrates of effector caspases. PARP = poly(ADP-ribose) polymerase; DNA-PK = DNA-dependent protein kinase; ICAD = inhibitor of caspase-activated deoxyribonuclease; CAD = caspase-activated deoxyribonuclease; PKC-δ = protein kinase C δ; Rb = retinoblastoma protein; PAK-2 = p21-activated kinase-2; NF = nuclear factor.
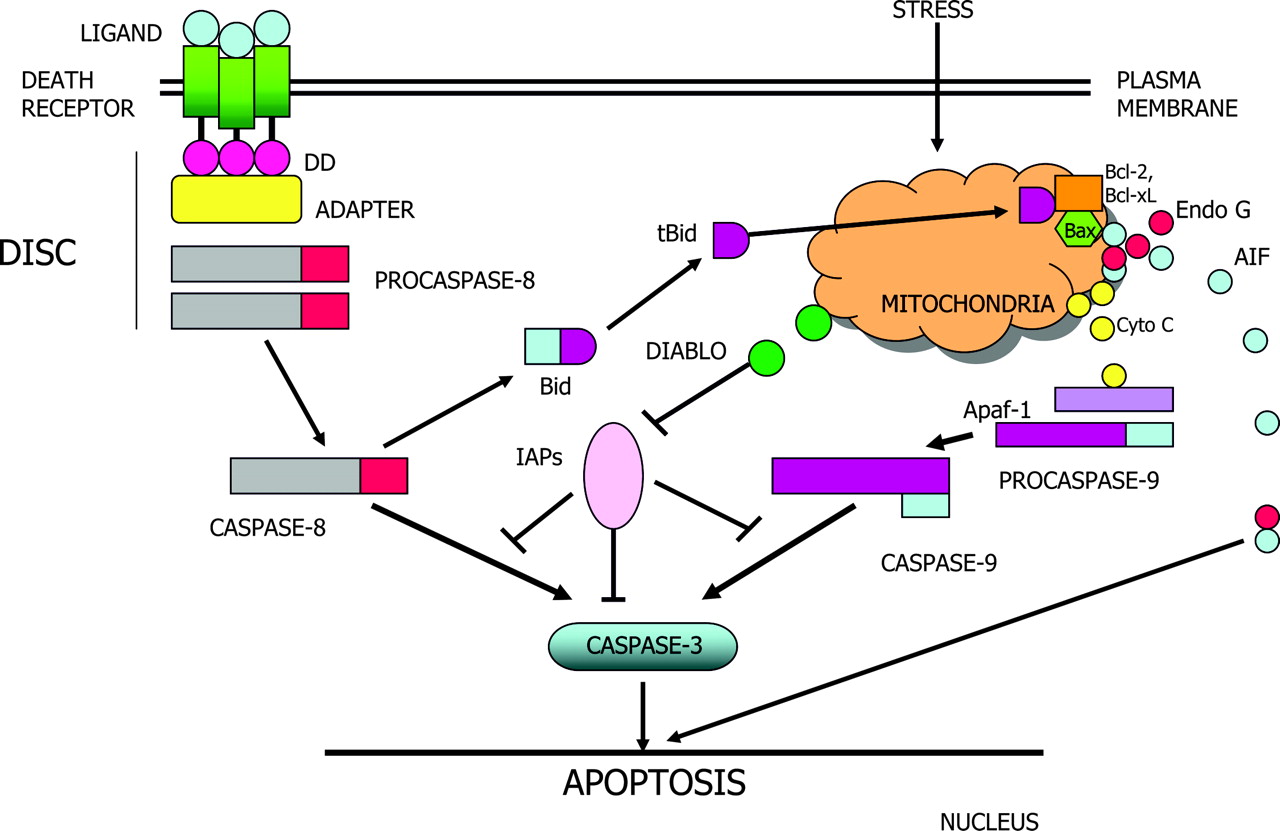
Death receptor and mitochondrial pathways of apoptosis. DD = death domain; AIF = apoptosis-inducing factor; Cyto C = cytochrome c; IAP = inhibitor of apoptosis protein.
Footnotes
Received Apr. 5, 2005; revision accepted Apr. 6, 2005.
For correspondence or reprints contact: Jagat Narula MD, PhD, University of California, Irvine, School of Medicine, 101 The City Dr., Bldg. 53, Mail Route 81, Orange, CA 92868-4080.
E-mail: narula{at}uci.edu





{kind=link}
{kind=link}
{kind=link}