


Abstract
The endothelium constitutes a functionally active organ critically involved in angiogenesis. Nitric oxide (NO) is an important regulator of vascular homeostasis and angiogenesis and stimulates glucose metabolism in certain cells. We thus investigated the effect of exogenous NO on 18F-FDG transport in human endothelial cells. Methods: Human umbilical vein endothelial cells (HUVECs) were treated with the NO donors sodium nitroprusside (SNP) or diethylenetriamine (DETA), in concentrations of 1 μmol/L–1 mmol/L for up to 24 h. 18F-FDG uptake levels corrected for protein content were determined by cellular radioactivity measured after 30-min incubation. Cells were evaluated for total hexokinase activity and plasma membrane glucose transporter 1 (GLUT1) levels, and involvement of potential signaling pathways was investigated by cotreatment with respective protein kinase inhibitors. Results: Both SNP and DETA stimulated HUVEC 18F-FDG uptake, which began at 16 h and peaked at 24 h. The increase in 18F-FDG uptake was dose dependent, reaching 464.0% ± 49.8% and 254.5% ± 10.8% of control levels at 24 h with 1 mmol/L SNP and DETA, respectively. Exposure of HUVECs to 1 mmol/L SNP resulted in a 3.5 ± 0.3-fold elevation in hexokinase activity (P < 0.01) and a significant increase in GLUT1 levels. SNP-stimulated 18F-FDG uptake was abolished by cotreatment with cycloheximide, the tyrosine kinase inhibitor genistein, the phosphatidylinositol-3 kinase (PI3K) inhibitor wortmannin, or the protein kinase C inhibitor staurosporine. Conclusion: NO stimulates 18F-FDG uptake in HUVECs through an increase in GLUT1 expression and hexokinase activity, which appears to involve both protein kinase C and PI3K pathways.
The vascular endothelium is an active, dynamic tissue that controls many important functions, including nutrient exchange, regulation of vascular tone and perfusion, and inflammatory responses (1). Endothelial cells are also responsible for promoting angiogenesis in tumor and ischemic lesions (2,3). To successfully induce angiogenesis, it is crucial for endothelial cells to metabolically adapt to adverse environments. This is dependent on a sufficient supply of energy, which, in endothelial cells, is predominantly generated by active metabolism of glucose (4,5). Therefore, elucidation of mechanisms governing the regulation of glucose transport in endothelial cells is necessary to better understand the process of angiogenesis. In addition, such studies are also important because endothelial glucose transport rates may contribute to the overall uptake of 18F-FDG in tumor or vascular lesions on PET images (6).
Nitric oxide (NO) is an important signaling molecule with critical roles in angiogenesis (7,8). NO has been shown to stimulate glucose transport in a variety of nonendothelial cells (9–12). However, the role of NO in regulating endothelial glucose metabolism has not yet been evaluated. Thus, the purpose of this study was to elucidate the effect of NO on human endothelial cell glucose transport as measured by 18F-FDG uptake and to further investigate the molecular mechanisms involved in such an effect.
MATERIALS AND METHODS
Cell Culture
Human umbilical vein endothelial cells (HUVEC; American Type Culture Collection) were maintained in endothelial cell basal medium (EBM; Clonetics, Cambrex Corp.) supplemented with epidermal growth factors, 12% fetal bovine serum, 2 g/L glucose, and 100 U/mL penicillin-streptomycin in a humidified atmosphere of 5% CO2 at 37°C. Culture media were changed every 3 d, and only cells from passage 4–5 were used for experiments. To each well of 12-well culture plates, 1 × 105 HUVECs were seeded with 1.5 mL of culture media, which reached 80% confluence 48 h later on the day of experiments.
Exposure to Nitric Oxide Donors
HUVECs were exposed to sodium nitroprusside (SNP; Sigma Chemical Co.) or diethylenetriamine (DETA; Sigma) in concentrations of 1 μmol/L to 1 mmol/L by their addition to the culture medium. Cells were then incubated in a humidified atmosphere of 5% CO2 at 37°C for 24 h. The concentration of NO released from the NO donors was measured by incubating 100 μL of culture media with an equal volume of Griess reagent containing 1% sulfanilamide, 0.1% N-(1-naphthyl)ethylenediamine dihydrochloride, and 2.5% phosphoric acid for 10 min. Absorbance at 540 nm was measured using a spectrophotometer, and results were expressed as concentration relative to unstimulated cells.
Potential cytotoxic effects of SNP in our experiments were evaluated by inspection of cell morphology, protein assays, and trypan blue assays. Trypan blue assays were performed by incubating SNP (0, 0.5, 1, or 5 mmol/L) treated cells with 0.4% trypan blue for 10 min, followed by counting the percentage of positive staining cells (6 samples per group).
Cellular 18F-FDG Uptake Measurements
Uptake measurements were performed in HUVECs treated with or without various concentrations of SNP or DETA by adding 370 kBq (10 μCi) of 18F-FDG to the culture medium of the cells and incubation in a humidified atmosphere of 5% CO2 at 37°C. Time course experiments showed that 18F-FDG uptake in HUVECs reached a relative plateau after 30 min incubation (data not shown). Hence, the remaining 18F-FDG uptake experiments were performed with 30-min incubation, when the adherent cells were washed twice with phosphate-buffered saline (PBS), then lysed in 1 mL distilled water before measurement of radioactive counts on a high-energy γ-counter (Wallac). 18F-FDG uptake levels of each sample were corrected for protein content as determined by the Bradford method and expressed as percentage of uptake relative to that of control cells.
Measurement of Total Hexokinase Activity
Total cellular hexokinase activity was measured using the method of Vinuela et al. as modified by Waki et al. (13). Briefly, cells were homogenized in a buffer containing 50 mmol/L triethanolamine and 5 mmol/L MgCl2 (pH 7.6), and supernatants containing free and mitochondria-bound hexokinase were obtained by centrifugation at 1,000g at 4°C for 5 min. A buffer comprised of the homogenization buffer containing 0.5 mmol/L glucose, 5 mmol/L adenosine triphosphate, 0.25 mmol/L reduced nicotineamide adenine dinucleotide phosphate, and 6 units glucose-6-phosphate dehydrogenase was preincubated at 20°C for 15 min. Samples were then added, and the reaction mixture was measured for absorbance. Hexokinase activity was determined from a standard curve, with 1 unit defined as the enzyme activity that phosphorylates 1 μmol/L of glucose per minute at 20°C. Aliquots were removed to measure the protein content, and the enzyme activity was expressed as mU/mg protein.
Western Blot Analysis for GLUT1 Expression
Two 150-mm plates of 80% confluent HUVECs that were either untreated or treated with 1 mmol/L SNP were used to obtain membrane proteins for Western blot. After culture media were aspirated, the attached cells were washed twice with PBS, then solubilized with 500 μL of lysis buffer containing 0.0856 g/mL sucrose, 10 mmol/L/mL N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES), 25 μmol/L/mL ethylenediaminetetraacetic acid, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mmol/L phenylmethylsulfonylfluoride and centrifuged at 1,000g for 20 min. The supernatants from the 2 plates were combined and mixed with 1.5 mL of lysis buffer containing 0.0856 g/mL sucrose, 10 mmol/L/mL HEPES, and 10 mmol/L/mL MgCl2, incubated at 4°C for 1 h, and centrifuged at 45,000 rpm for 60 min, and the pellet was dissolved with a minimum volume of PBS. After measurement of protein content using Bradford assays, 25 μg of membrane protein, which amounts to protein obtained from approximately 1 × 107 HUVECs, were loaded on each lane. The proteins were separated on a 10% polyacrylamide gel, electroblotted to a hydrobond enhanced chemiluminescence nitrocellulose membrane (Amersham Biosciences), and reacted with a polyclonal antibody against human glucose transporter 1 (GLUT1; 1:1,000 dilution; Calbiochem). GLUT1 protein was visualized using a horseradish peroxidase-conjugated secondary antirabbit IgG antibody (1:1,000 dilution; Caltag) and a high-performance chemiluminescence film.
SNU-C5 human colon cancer cells that are known to express very high levels of GLUT1 were used as positive controls. As a negative control, proteins from SNP-treated cells were loaded and underwent procedures as described but without the primary antibody.
Effect of Protein Synthesis Inhibitors and Protein Kinase Inhibitors
To elucidate the cellular signaling pathways involved in the stimulatory effect of NO, we measured 18F-FDG uptake in HUVECs treated with 1 mmol/L SNP for 24 h in the presence or absence of inhibitors of candidate protein kinase pathways. These included 4 μmol/L staurosporine, a protein kinase C (PKC) inhibitor; 200 nmol/L wortmannin, a specific phosphatidylinositol 3-kinase (PI3K) inhibitor, 500 μmol/L genistein, a tyrosine kinase inhibitor; and 100 ng/mL cycloheximide, a protein synthesis inhibitor. The inhibitors were added to cells 1 h before treatment with SNP.
Statistical Analysis
All experiments were repeated at least 3 separate times, and the mean ± SD of triplicate samples from a single representative experiment was presented. The range of results obtained from repeated experiments was also presented. Student t test was used to evaluate the statistical significance between groups, and P values <.05 were considered significant.
RESULTS
NO Concentration in Media of SNP-Treated HUVECs
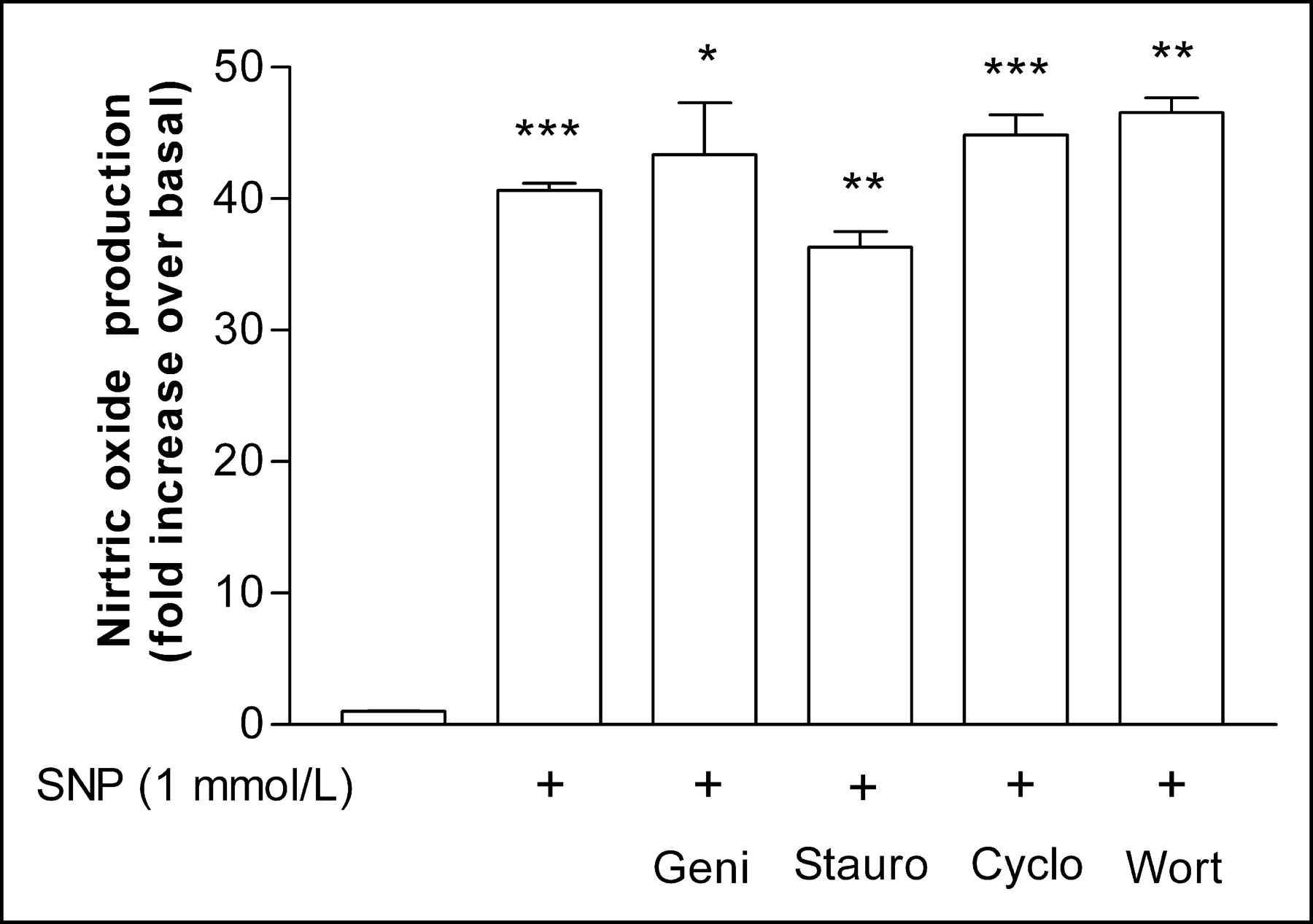
Treatment of HUVECs with 1 mmol/L SNP for 24 h led to a significant increase in NO levels released in culture media to 40.7 ± 0.8-fold over that for controls (Fig. 1). The experiment was repeated 2 more times, which showed 32.8 ± 1.7- and 43.3 ± 1.7-fold increases in NO levels (interassay mean, 38.9 ± 5.5-fold increase). The increase in NO by SNP was not affected by the presence of the inhibitors genistein, staurosporine, cycloheximide, or wortmannin, which respectively showed 40.6 ± 0.67-, 36.3 ± 1.8-, 44.8 ± 2.2-, and 46.5 ± 1.6-fold higher NO levels compared with controls (Fig. 1). The interassay averages of SNP-induced NO increases in the presence of the inhibitors were 38.9 ± 6.4-, 40.8 ± 5.7-, 43.0 ± 5.0-, and 32.7 ± 5.1-fold, respectively.

Nitric oxide generation levels by SNP. Concentrations were measured in culture media after incubation with 1 mmol/L SNP for 24 h. Data are mean ± SD of a representative experiment with triplicate samples expressed as relative increase compared with controls. SD of control group (first bar on left) is too small to discern. Geni = genistein; Stauro = staurosporine; Cyclo = cycloheximide; Wort = wortmannin; *P < 0.05, **P < 0.01, ***P < 0.005.
Effect of NO Treatment on HUVEC 18F-FDG Uptake
There was no change in cell morphology or protein content in HUVECs treated with 0.5 or 1 mmol/L SNP for 24 h compared with controls. Trypan blue assays confirmed viability after treatment with 0.5 mmol/L and 1 mmol/L SNP (94.6 ± 1.1 and 94.3% ± 2.3%, respectively) to be comparable with that of untreated controls (94.5% ± 2.0%). In contrast, however, 24-h exposure to 5 mmol/L SNP significantly decreased HUVEC viability to 55.8% ± 2.0% (P < 0.0001).
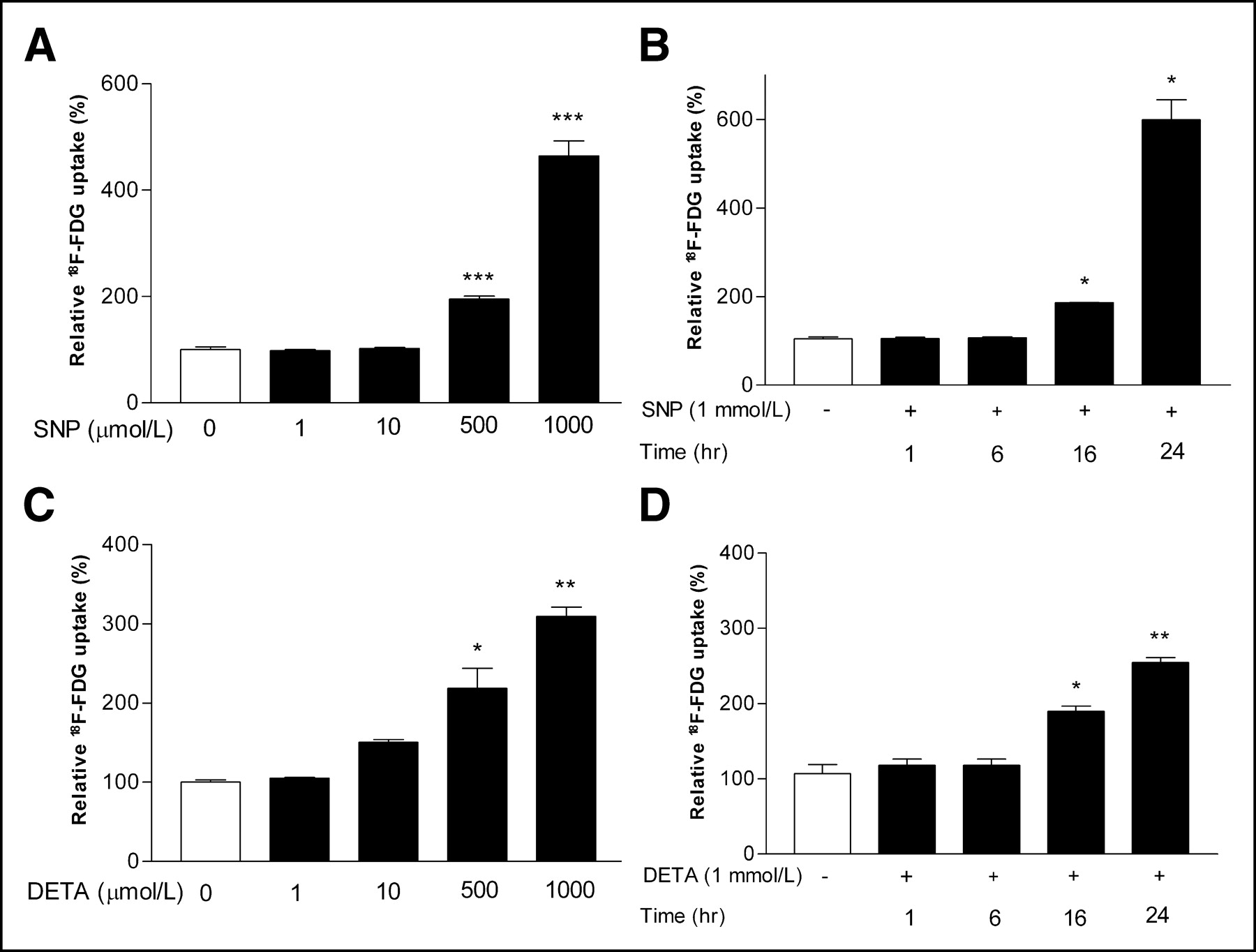
Exposure of HUVECs to either SNP or DETA resulted in a significant enhancement in 18F-FDG uptake in a dose-dependent manner. Treatment with 0.5 and 1 mmol/L SNP for 24 h increased 18F-FDG uptake to 206.3% ± 2.7% and 464.0% ± 49.8%, respectively, of control levels (both P < 0.001; Fig. 2A), and the same concentrations of DETA increased 18F-FDG uptake to 218.9% ± 35.9% (P < 0.05) and 309.7% ± 16.1%, respectively (P < 0.005; Fig. 2C). The range of 18F-FDG increase stimulated by 24 h of 1 mmol/L SNP observed in 7 separate experiments was between 368.5% ± 26.5% (P < 0.001) and 599.3% ± 63.0% (P < 0.05) of control levels (interassay mean, 499.3% ± 89.9%). Time course studies using 1 mmol/L SNP or DETA revealed an increase in 18F-FDG uptake at 16 h of exposure (176.5% ± 18.3% and 166.8% ± 8.7%, respectively; both P < 0.05; Fig. 2B), with a maximum uptake at 24 h (599.5% ± 63.1%; P < 0.01; and 223.9% ± 7.7%; P < 0.005; Fig. 2D).

Relative 18F-FDG uptake levels in HUVECs. Dose dependence of 18F-FDG uptake levels after 24-h incubation with various concentrations of SNP (A) and DETA (C). Time course of increase in 18F-FDG uptake levels in cells treated with 1 mmol/L of SNP (B) and DETA (D). Data are mean ± SD of percentage uptake relative to controls from single experiment representative of 3 (SNP) or 2 (DETA) separate experiments. *P < 0.01, **P < 0.005, ***P < 0.001.
Hexokinase Activity and GLUT1 Expression Levels
Because 18F-FDG is both transported and phosphorylated within the cell, we investigated hexokinase activity and GLUT1 expression levels of HUVECs. After exposure to 1 mmol/L SNP for 24 h, total cellular hexokinase activity was significantly increased to levels 3.5 ± 0.3-fold of those for controls (P < 0.01; Fig. 3). In 2 more repeated experiments, hexokinase activity was increased to 2.3 ± 0.2-fold (P < 0.05) and 2.5 ± 0.4-fold (P < 0.001) of control levels.
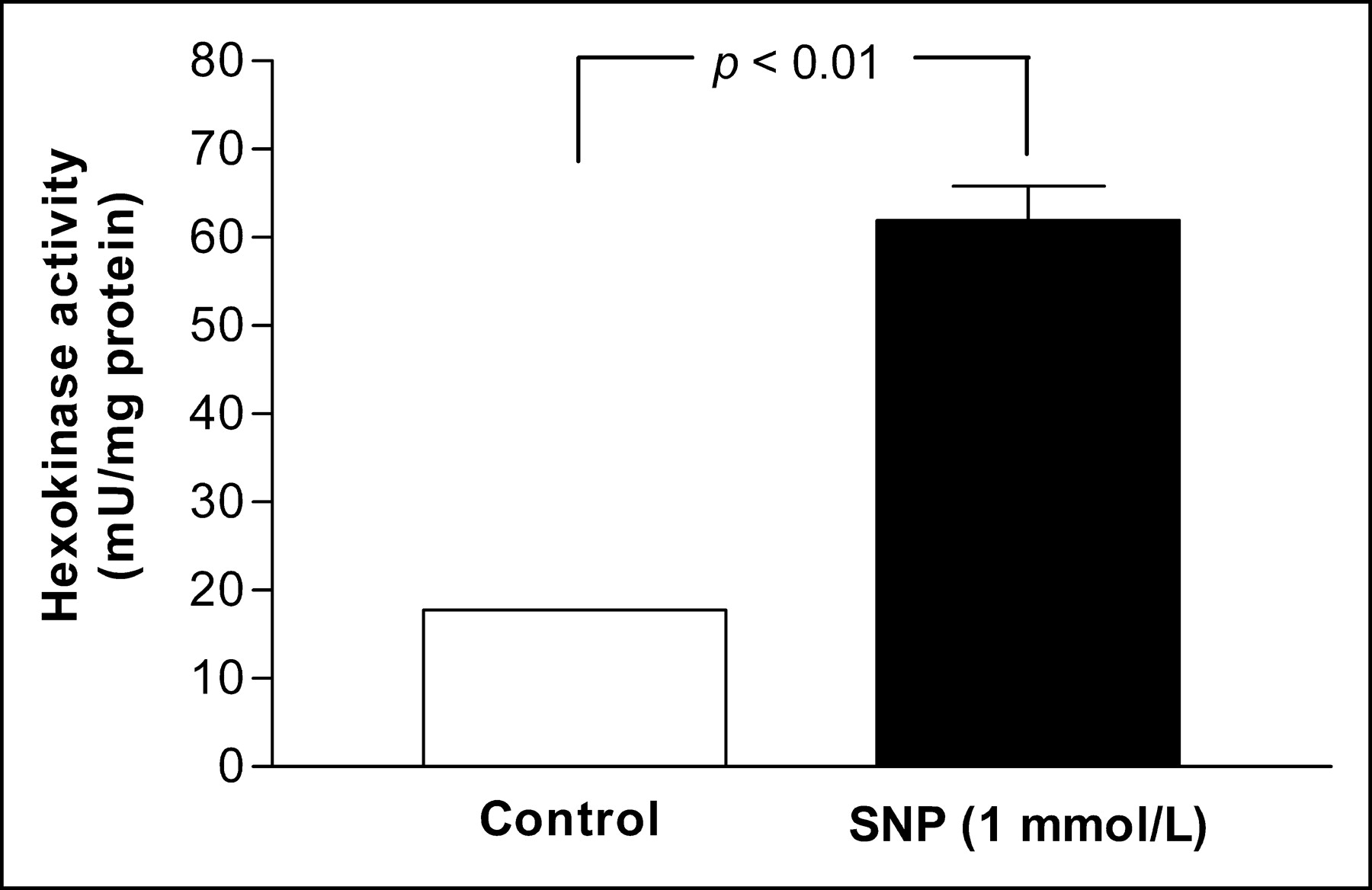
Total hexokinase activity. Black bar indicates HUVECs incubated with 1 mmol/L SNP for 24 h. Results are expressed as mean ± SD of mU/mg protein of triplicate samples obtained from single experiment representative of 3 separate experiments.
Western blot analysis of plasma membrane GLUT1 protein also revealed a significant increase in expression levels in HUVECs treated with 1 mmol/L SNP for 24 h compared with unstimulated controls (Fig. 4). Absence of visible bands in the negative control lane indicates specificity of the Western blot results.

Representative Western blot for plasma membrane GLUT1. SNU-C5, human colon cancer cells; SNP, HUVECs treated with 1 mmol/L SNP; control, untreated HUVECs; negative, HUVECs treated with 1 mmol/L SNP but without application of primary antibody.
Effect of Inhibitors on NO-Stimulated 18F-FDG Uptake
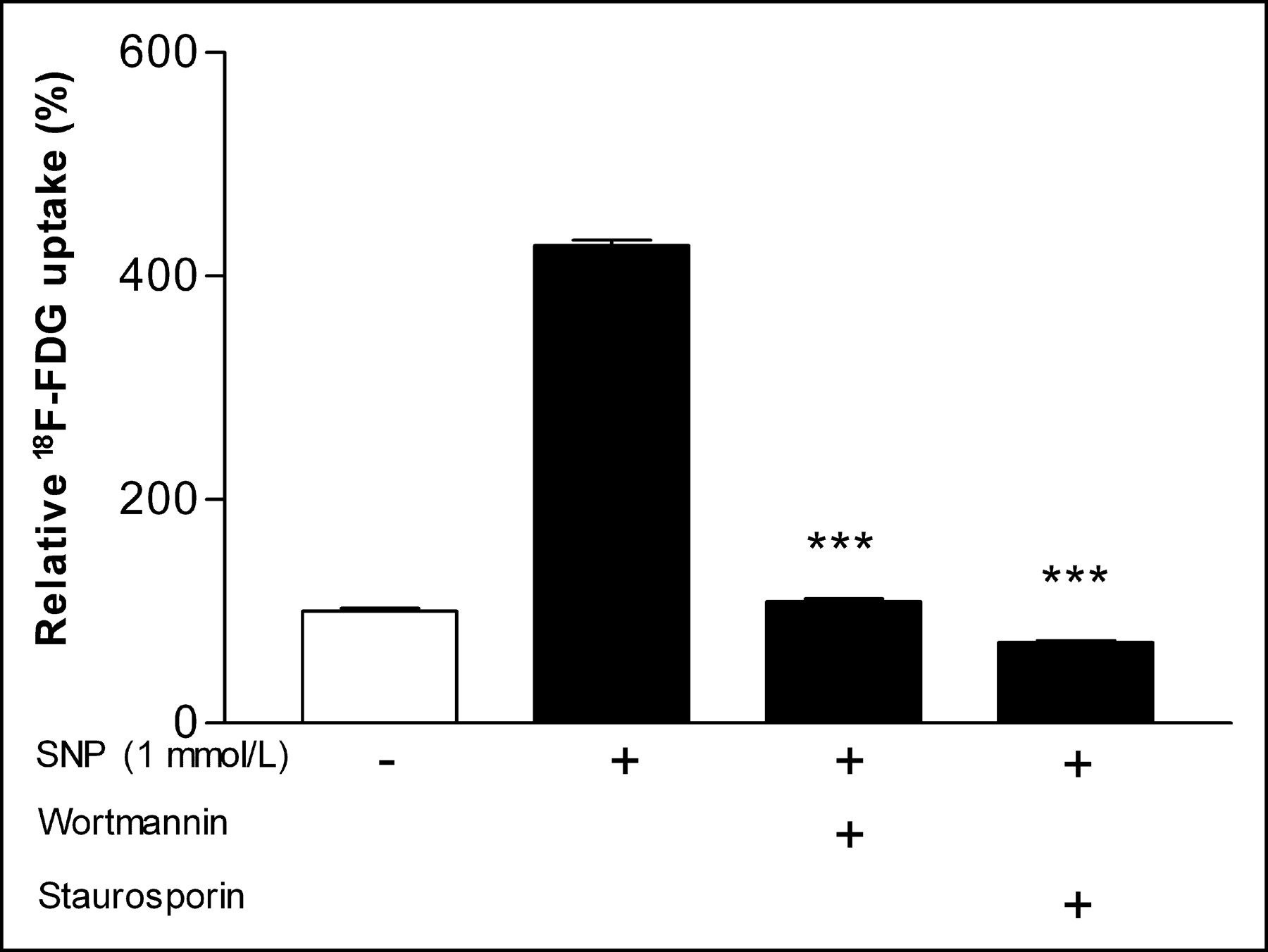
Cycloheximide completely abolished SNP-stimulated 18F-FDG uptake, resulting in uptake levels of 65.1% ± 2.6% of unstimulated control cells (P < 0.001; Fig. 5). In the presence of genistein, 18F-FDG uptake decreased to 133.6% ± 9.0% of control levels compared with 427.0% ± 8.8% in its absence (P < 0.001; Fig. 5). Wortmannin significantly attenuated the SNP effect and resulted in 18F-FDG uptake that was only 109.0% ± 4.5% of control levels (P < 0.001 compared with SNP-treated cells without wortmannin; Fig. 6). Staurosporine completely blocked the SNP effect, resulting in 18F-FDG uptake levels even lower than those of unstimulated control cells (72.2% ± 2.8%; P < 0.001 compared with SNP-treated cells without staurosporine; Fig. 6).

Effect of cycloheximide and genistein on SNP-stimulated 18F-FDG uptake. Inhibitors were added 1 h before 24-h stimulation with 1 mmol/L SNP. Results are expressed as percentage of uptake relative to mean of controls. Data are mean ± SD of triplicate samples from single experiment representative of 3 separate experiments. ***P < 0.001.
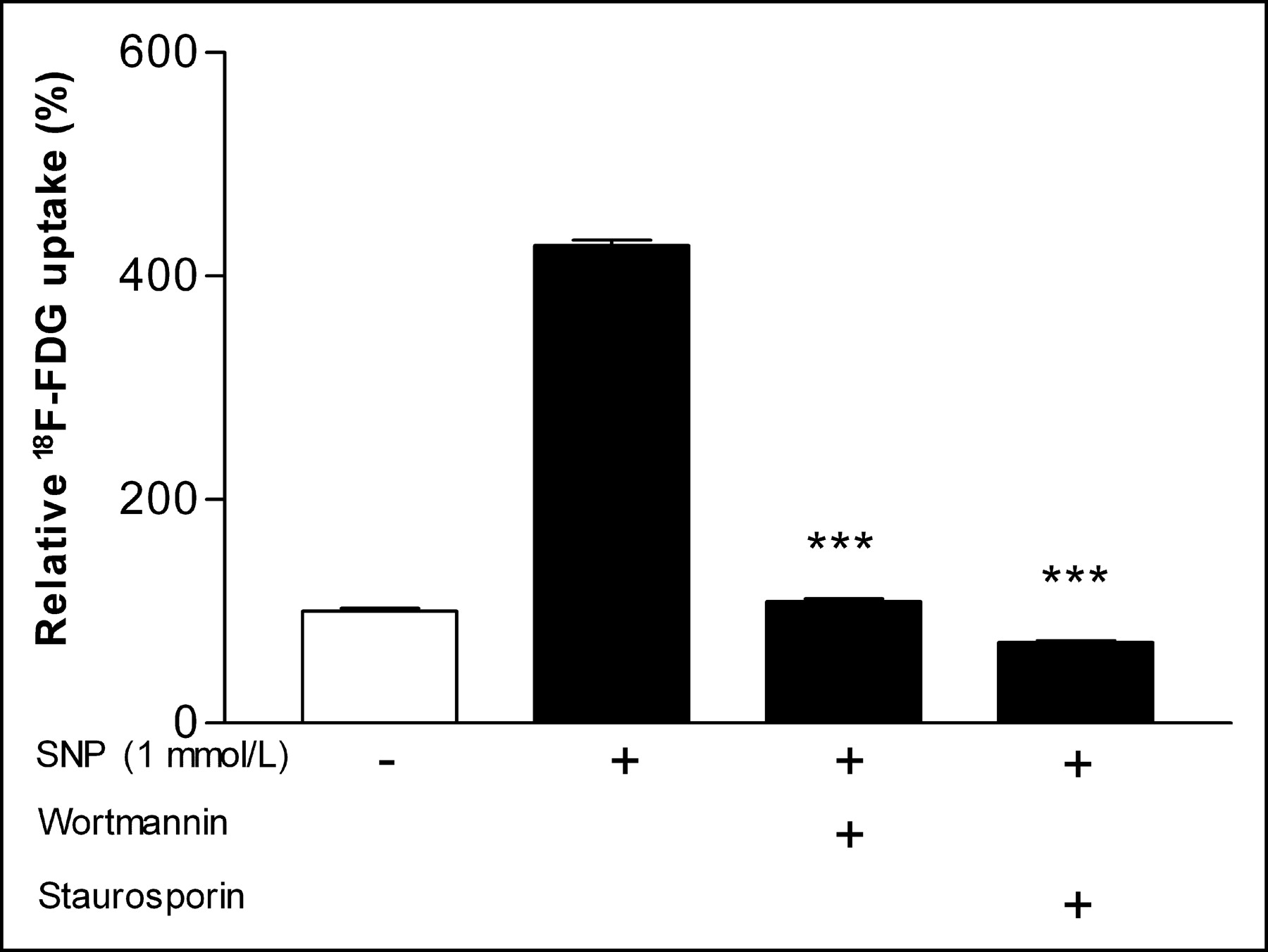
Effect of wortmannin and staurosporine on SNP-stimulated 18F-FDG uptake. Inhibtitors were added 1 h before 24-h stimulation with 1 mmol/L SNP. Results are expressed as percentage of uptake relative to mean of controls. Data are mean ± SD of triplicate samples from single experiment representative of 3 separate experiments. ***P < 0.001.
In 3 separate experiments, the interassay means of SNP-induced 18F-FDG uptake in the presence of genistein, cycloheximide, wortmannin, and staurosporin were 191.5% ± 54.2%, 59.4% ± 19.2%, 116.5% ± 47.0%, and 35.1% ± 6.6% of control levels, respectively.
DISCUSSION
Our study showed that exogenous NO significantly stimulates 18F-FDG uptake in HUVECs and is accompanied by an increase in cellular hexokinase activity and GLUT1 expression levels.
NO plays an important role in normal and pathologic vascular biology and is a critical mediator of angiogenesis (7). Angiogenic factors induce endothelial release of NO, which in turn mediates a multiplicity of angiogenic processes, such as endothelial cell survival, proliferation, and migration, whereas derangements in NO synthesis impair angiogenesis (14,15).
NO donors are also known to have dose-dependent cytotoxic effects on various cell types. Ioannidis et al. (16) reported damage of rat sinusoidal endothelial cells treated with 5 mmol/L SNP, and Gooch et al. (17) observed cytotoxic effects in bovine aortic endothelial cells treated with 1 mmol/L SNP after 24-h incubation but not in HUVECs treated with 0.7 mmol/L SNP. Under our experiment conditions, however, we confirmed that treatment for 24 h with SNP concentrations of up to 1 mmol/L did not cause significant cytotoxic effects, although cytotoxicity was observed with 5 mmol/L SNP.
This study revealed that, in addition to proangiogenic effects, NO also has a role in augmenting endothelial cell glucose transport. Endothelial cells are the principal effector cells of angiogenesis, and their survival and function are dependent on the availability and utilization of energy substrates. Endothelial cells are now recognized to rapidly transport and actively metabolize glucose (18). Endothelial glucose metabolism is upregulated in response to such stimuli as exposure to hypoxia, glucose deprivation, or treatment with growth factors (6,19–21). These stimuli have generally been shown to increase glucose transport activity after 12–24 h of treatment, which is similar to the SNP exposure time required for maximum 18F-FDG uptake in our study (19,21).
To our knowledge, the effect of NO on endothelial cell glucose metabolism has not been previously reported. On the other hand, NO has previously been observed to augment glucose metabolism in such cells as skeletal and cardiac myocytes, vascular smooth muscle cells, and adipocytes (9–12). The key steps that control cellular utilization of glucose are its translocation across plasma membrane through its transporters and subsequent phosphorylation by hexokinase. For insulin-responsive cells, such as adipocytes or myocytes, NO is known to enhance glucose transport via an increase in GLUT4 levels (11,12).
For endothelial cells, GLUT1 is the predominant and most abundant glucose transporter isoform (18). Endothelial GLUT1 expression is upregulated in response to hypoxia or glucose deprivation and by treatment with vascular endothelial growth factor (VEGF) (19–22). Our results show that NO is yet another stimulant that upregulates GLUT1 levels in endothelial cells. In addition to GLUT1 expression, we also found a significant increase in hexokinase activity for HUVECs treated with SNP. Taken together, our results indicate that both membrane expression of GLUT1 and hexokinase activity contribute to the observed augmented 18F-FDG uptake induced by NO.
The molecular mechanisms that govern cellular glucose metabolism are diverse, and multiple signaling cascades might be involved in its regulation for different cells. In our study, the protein synthesis inhibitor cycloheximide completely abolished the increase in 18F-FDG uptake, indicating that the SNP effect may require new protein synthesis. In addition to inhibition of protein synthesis, however, other effects of cycloheximide have been described for both increased and decreased glucose transport, which may be related to interference of cycloheximide with intrinsic activity or with GLUT translocation and retranslocation (23).
We also found that the SNP effect was significantly attenuated in the presence of genistein. Genistein is a potent tyrosine kinase inhibitor and has been shown to limit angiogenesis and inhibit insulin-stimulated cardiomyocyte glucose transport (24,25).
NO activates soluble guanylyl cyclase, which results in an increased intracellular synthesis of cyclic guanosine 3′,5′-cyclic monophosphate (cGMP). cGMP activates cGMP-dependent protein kinases that mediate localized and global signaling related to major NO effects (26). PI3K-Akt is activated by many angiogenesis promoting growth factors, and serves as a key mediator of such angiogenic processes as endothelial cell survival, proliferation, and migration (27). The PI3K-Akt pathway has been shown to mediate NO-induced endothelial cell migration and angiogenesis (8). PI3K-Akt also has a well-known central role in increasing glucose metabolism in insulin-responsive cells (28). In our study, the specific PI3K inhibitor wortmannin showed potent blocking of the SNP effect, indicating that it is partly mediated through activation of the PI3K-Akt pathway.
The PKC inhibitor staurosporine was a potent suppresser of the SNP effect, and cotreatment of HUVECs resulted in 18F-FDG uptake levels that were even lower than those in unstimulated controls. The PKC pathway has been reported to mediate VEGF-induced activation of endothelial Akt and stimulate angiogenesis (29). It has also been shown that PKC is essential for VEGF- and insulin-like growth factor-1-stimulated endothelial glucose transport (21,30). Notably, both PI3K and PKC were required for the stimulatory effect of insulin-like growth factor-1, which is similar to what we observed for the NO effect.
Taken together, the results of our inhibition experiments suggest that the actions of NO in stimulating HUVEC glucose transport involve interactions between multiple protein kinase pathways that require both PI3K and PKC. Because inhibition of PI3K and that of PKC each resulted in potent blocking of the SNP effect, activation of these pathways appears to occur in a sequential manner rather than in parallel.
CONCLUSION
NO enhances 18F-FDG uptake in human HUVECs together with an increase in membrane GLUT1 expression and hexokinase activity via pathways that involve both PI3K and PKC. These findings suggest an important role for NO in modulating glucose transport of endothelial cells.
Acknowledgments
The authors thank Jong-Oh Park, MS, for technical assistance. This work was supported in part by the national mid- and long-term nuclear research and development program grant M20243010001-04A0701-00110 from the Korean Ministry of Science and Technology. Presented in part at the 51st Annual Meeting of the Society of Nuclear Medicine (Philadelphia, PA), June 19–23, 2004.
Footnotes
Received June 8, 2004; revision accepted Sep. 24, 2004.
For correspondence or reprints contact: Kyung-Han Lee, MD, Department of Nuclear Medicine, Samsung Medical Center, 50 Ilwondong, Kangnamgu, Seoul, Korea.
E-mail: khnm.lee{at}samsung.com
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Fructose bisphosphatase 2 overexpression increases glucose uptake in skeletal muscle
- The Evolving Role of Succinate in Tumor Metabolism: An 18F-FDG-Based Study
- Principles of targeting endothelial cell metabolism to treat angiogenesis and endothelial cell dysfunction in disease
- Endothelial cell oxidative stress in diabetes: a key driver of cardiovascular complications?
- Fibronectin Stimulates Endothelial Cell 18F-FDG Uptake Through Focal Adhesion Kinase-Mediated Phosphatidylinositol 3-Kinase/Akt Signaling
- In Vitro Studies on the Signal Transduction of Thyroidal Uptake of 18F-FDG and 131I-Iodide