


Abstract
Detection of a new or recurrent breast cancer lesion relies on physical examination and imaging studies, primarily mammography, followed by histopathologic evaluation of biopsy tissue for morphologic confirmation. Approximately 66%–85% of detected lesions are not malignant. Therefore, biopsies are unnecessary for at least two thirds of patients. Human estrogen receptor-positive breast cancer cells typically display an elevated level of cyclin D1 protein because of the overexpression of CCND1 messenger RNA (mRNA) and an elevated level of insulin-like growth factor 1 (IGF1) receptor (IGF1R) because of the overexpression of IGF1R mRNA. We hypothesized that scintigraphic detection of CCND1 peptide nucleic acid (PNA) hybridization probes with a 99mTc-chelating peptide on the N terminus and an IGF1 peptide loop on the C terminus could detect CCND1 mRNA in human MCF7 breast cancer xenografts in nude mice from outside the body. Methods: We prepared the CCND1 probes as well as mismatched controls by solid-phase synthesis. We used fluorescence microscopy to detect the cellular uptake of fluoresceinyl probes and quantitative reverse transcription-polymerase chain reaction to detect the hybridization of probes to mRNA. We imaged 99mTc-probes in MCF7 xenografts scintigraphically and measured distribution by scintillation counting of dissected tissues. Results: IGF1R-overexpressing MCF7 cells internalized the fluorescein-chelator-CCND1 PNA-IGF1 peptide but not the mismatched control peptide. The chelator-CCND1 PNA-IGF1 peptide but not the control peptide lowered the level of cyclin D1 protein in IGF1R-overexpressing MCF7 xenografts in nude mice after intratumoral injection. IGF1R-overexpressing MCF7 xenografts in nude mice were visualized at 4, 12, and 24 h after tail vein administration of the 99mTc-CCND1 antisense probe but not the control probe. 99mTc-chimeras were distributed normally in the kidneys, liver, tumors, and other tissues. Conclusion: Cancer gene activity can be detected from outside the body by probing with radionuclide-chelator-PNA-peptide chimeras.
Mammography and physical examination, the only generally accepted screening tools available, miss up to 40% of early breast cancers, the most common noncutaneous cancers in U.S. women. Moreover, if an abnormality is found, an invasive diagnostic procedure still must be performed to determine whether the breast contains atypia or cancer, even though 66%–85% of abnormalities are benign (1). Recent advances in mammography, including 2 views rather than 1, spot compression, and digital images under the guidance of the Breast Imaging Reporting and Data System, have improved the sensitivity and specificity of mammography (2). Nonetheless, most women with suspicious mammograms undergo surgery only for the lesion to be found benign, and many other women undergo yearly mammograms interpreted as benign only to discover by self-examination of the breast a palpable lump that is found to be malignant.
Diagnostic efforts to identify women with precancerous changes or breast cancer are hindered by the fact that the evaluation of the breast traditionally has required a surgical biopsy. A nonoperative method to evaluate women for the presence of atypia or cancer of the breast would be very beneficial. Sestamibi has demonstrated utility in imaging lesions in a dense breast but is limited by the fact that other cell types with high mitochondrial activity avidly take up the tracer, leading to false-positive results in breasts with inflammation or infection (3), false-negative results attributable to sensitivity limits of the technology (in which tumors <8 mm in diameter are difficult to visualize) (4), and the potential for false-negative results with well-differentiated, slowly growing tumors because of their lower mitochondrial content.
Recent evidence from gene expression profiling suggests that alterations in messenger RNA (mRNA) conferring the potential for invasive breast cancer are already present in preinvasive disease, supporting the rationale for this approach (5). We hypothesized that noninvasive scintigraphic imaging of γ-particles emitted by decaying 99mTc chelated to an oligonucleotide hybridized to an oncogene mRNA overexpressed in breast cancer could identify sites of neoplastic transformation.
The insulin-like growth factor 1 (IGF1) receptor (IGF1R), HER2, CCND1, and MYC oncogenes, as well as mutant tumor suppressor p53, are early agents of malignant transformation and are frequently overexpressed in breast cancer cells. The overexpression of IGF1R and CCND1 was characteristic of estrogen and/or progesterone receptor-positive breast cancer cells (6), whereas the overexpression of HER2 and MYC is characteristic of estrogen receptor-negative breast cancer cells (7). However, transcription profiles obtained for breast cancer tissue by microarray analysis have not yet identified detectable overexpression of those markers (8), and that approach requires an invasive biopsy.
Nevertheless, oncogene-targeted oligonucleotide sequences have specifically decreased the expression of IGF1R (9), CCND1 (10), HER2 (11), MYC (12–14), and p53 (15), inhibiting cancer cell proliferation. Thus, we hypothesized that their mRNAs are significant markers of oncogenic transformation that can be used to distinguish precancerous and invasive cancerous changes from benign breast disease.
For noninvasive imaging of malignant lesions, we developed a method to label a vasoactive intestinal peptide with 99mTc by including an N4-chelating peptide, Glyd(Ala)GlyGly-4-aminobutyric acid (Aba) (TP3654) (16–18). In a series of 16 patients, we were able to image unequivocally in 2 separate patients 2 tumors that were not detectable by standard scintigraphic imaging agents. In the neck of a 20-y-old female, we detected a high-grade spindle-cell sarcoma that was not detectable by bone scanning or with 99mTc-sestamibi. In the left breast of a 42-y-old female who had undergone a mastectomy for cancer of the right breast 2 y previously, we detected atypical ductal epithelial hyperplasia that was not detectable with 99mTc-sestamibi. Both lesions were confirmed by histologic analysis and were imaged clearly and unambiguously (18), despite probe uptake in experimental tumors of only 0.3% injected dose per gram (%ID/g) (16).
Basu et al. examined SKBR3 cells overexpressing HER2 mRNA for the accumulation of a 99mTc-labeled HER2 phosphorothioate oligonucleotide and of a scrambled control but found the same amounts of label in all of the cellular preparations (S. Basu, PhD, E. Wickstrom, PhD, and M. L. Thakur, PhD, unpublished data, 1995). Similarly, there were no differences in the levels of uptake of a 99mTc-labeled 6-hydrazinonicotinate-conjugated MYC antisense phosphorothioate oligonucleotide in cell lines with high, normal, and low levels of MYC mRNA (19). Those negative results might have been attributable to a nonspecific affinity of phosphorothioate oligonucleotides for proteins, limiting the efflux of unbound label; to the destruction of the oncogene message being measured by ribonuclease (RNase) H attack on the RNA target because of hybridization of the labeled antisense DNA; or to the lack of a receptor-targeting ligand. On the other hand, the specific tumor uptake of 111In-MYC phosphorothioate (20) and 68Ga-KRAS phosphorothioate (21) has been described.
To address these drawbacks, we considered peptide nucleic acids (PNAs), which hybridize more strongly and more specifically to RNA, resist nuclease attack, and demonstrate antisense activity on microinjection into cellular nuclei (22). RNA hybridized to uncharged oligonucleotide derivatives, such as a PNA, is not recognized by RNase H; hence, the PNA does not catalyze the degradation of its analyte, the bound RNA (23), but inhibits mRNA translation solely through hybridization arrest and thus provides an opportunity for diagnostic application (22). The requirement for the microinjection of PNAs, however, stems from their poor cellular uptake, which has been reported to be 10 times less efficient than the uptake of phosphorothioates in a variety of mammalian cells (24).
Morpholino phosphorodiamidates also show poor uptake (25) and hybridize to RNA less strongly than do PNAs (26,27). However, radiolabeled morpholino oligonucleotides exhibit good pharmacokinetic and tissue distribution properties, particularly when cytosines in morpholino sequences are minimized to reduce renal accumulation (28). Conjugation of basic peptides elevates the cellular uptake of radiolabeled morpholino oligomers (29).
Cellular uptake of PNAs is also improved by the addition of a variety of ligands (30). Previously, Basu and Wickstrom observed that the synthesis of an IGF1R PNA dodecamer with an N-terminal d-peptide analog of IGF1, d(CysSerLysCys), provided cell type specificity and increased cellular uptake by cells overexpressing IGF1R 5- to 10-fold (31). A reverse sequence was synthesized with respect to the normal l-amino acid sequence to account for the reversal of chirality. Cellular uptake of the PNA-peptide chimera, a control PNA-peptide with 2 d-Ala residues in the peptide in place of d(SerLys), and a control PNA without a peptide adduct was studied in BALB/c3T3 cells transformed with human IGF1R (32) and in 2 cell lines with a low level of IGF1R expression. Transformed cells overexpressing IGF1R displayed 5- to 10-fold-higher uptake of the specific PNA-peptide chimera after 4 h of exposure at 1 μmol/L than did the control PNA or the control PNA-peptide (31). Only background levels of uptake were seen in the control cell lines. In prostate cancer cells, for comparison, the addition of dihydrotestosterone or a nuclear localization peptide to a MYC antisense PNA permitted some nuclear localization and MYC reduction in LNCaP cells expressing the androgen receptor after 24 h of exposure to PNA at 10 μmol/L (33). It would appear that a peptide analog specific for a cell surface receptor is far more effective than a steroid capable of binding to a cytoplasmic protein after unassisted uptake.
We pursued this cell-specific approach to enable the application of PNAs as gene expression diagnostic agents in vivo and to develop methods for the synthesis of peptide-PNA-peptide chimeras that exhibit the same melting temperatures with complementary RNA targets as do PNAs without ligands (34). Twelve bases are sufficient for statistical uniqueness among transcribed mRNAs, and the melting temperature results confirmed that 12 PNA residues hybridize strongly enough and specifically enough to serve as mRNA probes in vivo. Quantitative reverse transcription (QRT)-polymerase chain reaction (PCR) measurements of MYC mRNA in total RNA from MCF7 cells expressing IGF1R (MCF7:IGF1R cells) revealed 4-fold inhibition after preannealing with an N-Glyd(Ala)GlyGlyAba-MYC PNA-(Gly)4d(CysSerLysCys) probe at 0.1 μmol/L before addition of the PCR primers and 8-fold inhibition after preannealing with the probe at 1.0 μmol/L. The MYC mismatch control probe had no effect, consistent with the hypothesis that the complementary PNA probe would bind strongly to the mRNA target (23).
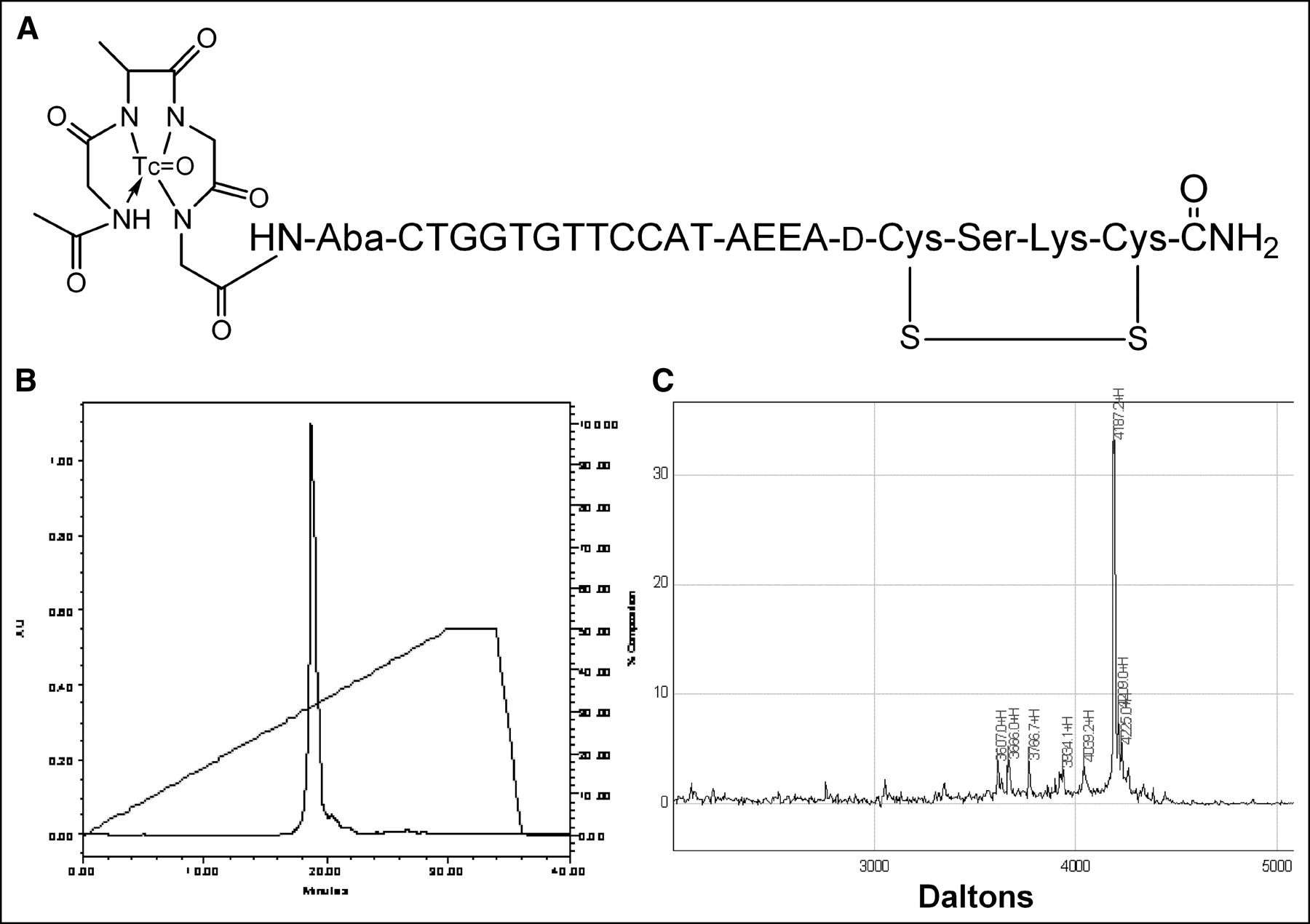
We report here the results of administration to nude mice bearing human estrogen receptor-positive MCF7 breast cancer xenografts 99mTc-peptide-PNA-peptide chimeras that bind to IGF1R, are internalized, and hybridize to CCND1 mRNA (Fig. 1A).
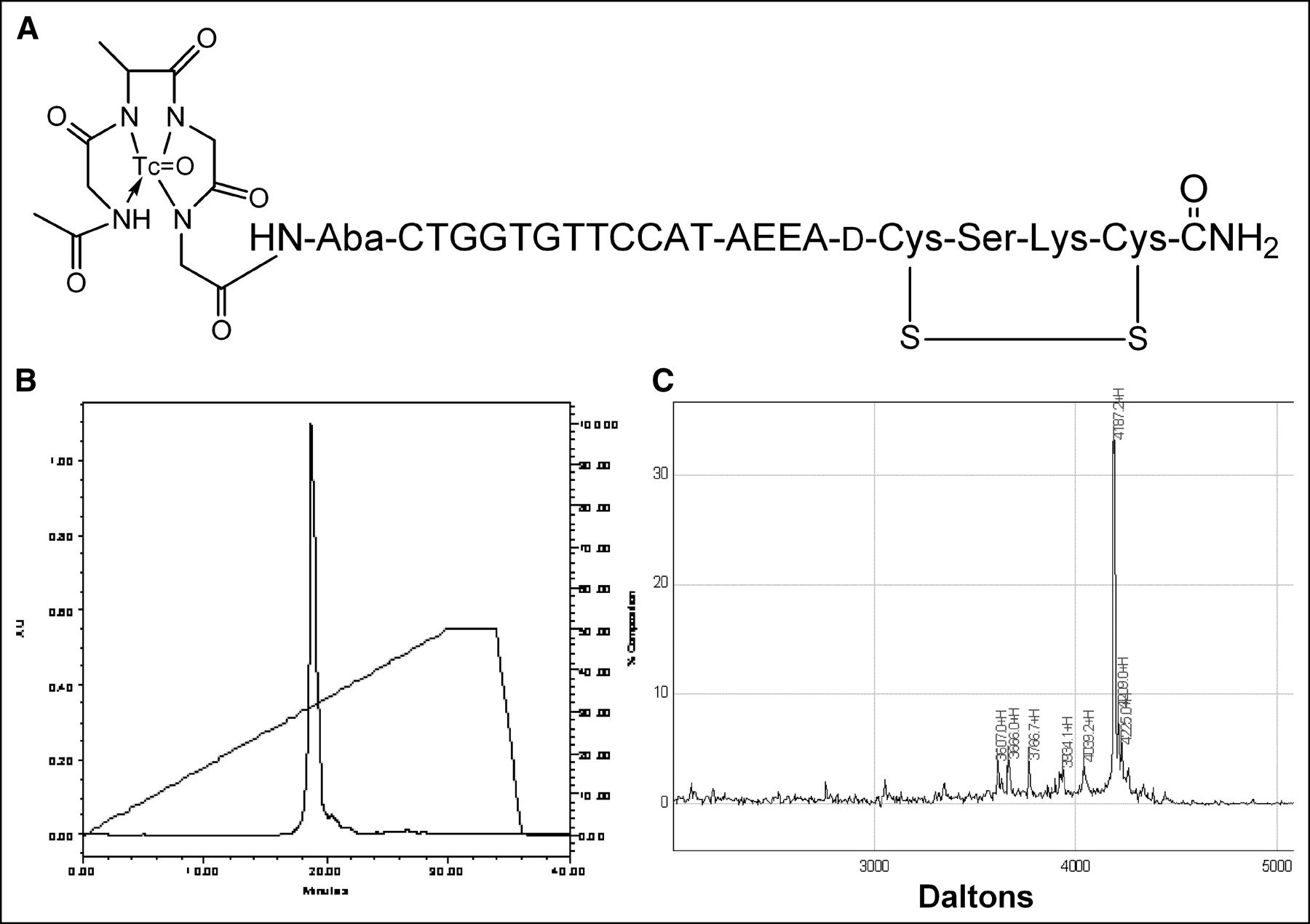
99mTc-chelator-PNA-peptide designed to bind to IGF1R, to be internalized, and to hybridize with CCND1 mRNA. Scintigraphic imaging of γ-rays emitted on decay of 99mTc will identify sites with high levels of CCND1 expression. (A) Schematic structure of 99mTc-AcGlyd(Ala)GlyGlyAba-CTGGTGTTCCAT-AEEA-d(CysSerLysCys), WT4185. (B) Preparative C18 HPLC of cyclized chimera AcGlyd(Ala)GlyGlyAba-CTGGTGTTCCAT-AEEA-d(CysSerLysCys), WT4185. (C) MALDI-TOF MS analysis of purified chimera AcGlyd(Ala)GlyGlyAba-CTGGTGTTCCAT-AEEA-d(CysSerLysCys), WT4185. Experimental mass was 4,187.2 Da; the calculated mass was 4,185.0 Da.
MATERIALS AND METHODS
Peptide-PNA-Peptide Synthesis
The peptide-PNA-peptide chimeras were assembled, purified, and characterized as described previously (34). Briefly, the IGF1 analog d(CysSerLysCys) or the mismatch control d(CysAlaAlaCys) was assembled by 9-fluorenylmethoxy carbonyl (Fmoc) coupling on NovaSyn TGR resin (0.2–0.3 mmol/g) (Novabiochem) by use of an Applied Biosystems 430A peptide synthesizer. Next, the linker Fmoc-aminoethoxyethoxyacetic acid (AEEA) and PNA monomers (Applied Biosystems) were sequentially coupled to the N terminus of the peptide-resin by use of an Applied Biosystems 8909 DNA synthesizer with the manufacturer’s 2-μmol PNA protocol. Finally, the chelator peptide Fmoc-Glyd(Ala)GlyGlyAba (Novabiochem) was coupled to the N terminus of the PNA-peptide by use of a long-coupling-cycle protocol (34) and then acetylated (Ac). The cysteine residues were cyclized on the solid phase with 10 equivalents of I2 in (CH3)2NCHO for 4 h at room temperature (34). For analysis of cellular uptake, fluorescent derivatives were prepared by solid-phase coupling of 6-(fluorescein-5-carboxamido)hexanoic acid, succinimidyl ester (SFX; Molecular Probes), to the AEEA-PNA-peptide after solid-phase cyclization of the disulfide bridge but before deprotection of lysine or PNA bases.
The chimeras were cleaved and deprotected with 85% CF3CO2H, 5% CH2Cl2, 9.5% m-cresol, and 0.5% (C2H5)3SiH for 2 h at room temperature (34); the chimeras then were purified by reversed-phase liquid chromatography on an Alltima C18 column (10 × 250 mm; Alltech) eluted with a gradient from 5% to 70% CH3CN in aqueous 0.1% CF3CO2H at 1 mL/min over 25 min at 50°C, with monitoring at 260 nm. Surface-enhanced laser desorption ionization–time-of-flight mass spectrometry (MALDI-TOF MS; Ciphergen Corp.) was performed with an α-cyano-hydroxycinnamic acid matrix excited at 338 nm and calibrated with porcine neuropeptide Y.
Radiolabeling
Purified peptide-PNA-peptide chimeras were labeled with 99mTc essentially as described previously (18). Briefly, 600 μL of Na3PO4 (0.05 mol/L; pH 12) and 0.1% Tween 80 were added to 426 MBq (11.5 mCi) of freshly eluted 99mTc-O4− in 200 μL of NaCl (0.15 mol/L). The radionuclide mixture was added to 20 μg of chelator-PNA-peptide dissolved in 45 μg of SnCl2 · 2H2O in 15 μL of HCl (0.05 mol/L) and mixed. The mixture was incubated for 30 min at 22°C, and the pH was adjusted to ≈7 by the addition of 1 mL of NaH2PO4 (0.05 mol/L; pH 4.5). The reaction mixture was analyzed to determine free 99mTc and chelated 99mTc by reversed-phase liquid chromatography on a reversed-phase Microbond C18 high-pressure liquid chromatography (HPLC) column (4.5 × 250 mm) (Rainin) coupled to a UV detector, an NaI (Tl) radioactivity monitor, and a rate meter. The column was eluted with a gradient from 10% CH3CN in aqueous 0.1% CF3CO2H to 100% CH3CN in 0.1% CF3CO2H at 1 mL/min over 28 min at 25°C. Unchelated free 99mTc (Rf, 1.0) was also determined by instant thin-layer chromatography on silica gel (Gelman Sciences) developed with methylethyl ketone. Colloid formation (Rf, 0.0) was determined by instant thin-layer chromatography on silica gel developed with pyridine:acetic acid:H2O (3:5:1.5). These preparations were stable at 22°C for more than 4 h, as determined by HPLC, and were stable to challenges with 100-fold molar excesses of diethylenetriaminepentaacetic acid, human serum albumin, or cysteine. Radiolabeled chimeras and mock-treated unlabeled chimeras were analyzed by denaturing gel electrophoresis on polyacrylamide (10%–20%)–Tris–Tricine–sodium dodecyl sulfate (SDS) gels (Bio-Rad) (31). Duplicate gels were autoradiographed or stained with Coomassie blue.
Cell Lines and Xenografts
Human MCF7:IGF1R estrogen receptor-positive breast cancer cells, clone 17, transformed to express 106 IGF1Rs per cell constitutively from a cytomegalovirus promoter (35), were maintained in Dulbecco minimal essential medium (DMEM; Sigma) containing 5% calf serum, penicillin at 50 U/mL, streptomycin at 5 μg/mL, glutamine at 2 mmol/L, and 17-β-estradiol at 7.5 nmol/L at 37°C under 5% CO2. For tumor induction, 5 × 106–6 × 106 cells in 0.2 mL of culture medium were implanted intramuscularly through a sterile 27-gauge needle into the thighs of female BALB/c nude mice obtained from the National Institutes of Health. Tumors were allowed to grow to no more than 0.5 cm in diameter. Each injection included 10 mg of Matrigel (Becton-Dickinson). A pellet that releases 4.5 mg of 17-β-estradiol (Innovative Research of America) over 60 d was implanted subdermally in each mouse. All animal studies were conducted in accordance with federal and state guidelines governing laboratory animal use and under approved protocols reviewed by the Animal Care and Use Committee at Thomas Jefferson University. All animals were anesthetized by approved methods and, when required, the animals were restrained by use of methods and devices specifically designed to provide a minimum of discomfort to the animals. The animals were euthanized in a halothane chamber consistent with U.S. Department of Agriculture regulations and American Veterinary Medical Association recommendations.
Cellular Internalization of Fluorescent Probes
Internalization of peptide-PNA-peptide chimeras by MCF7:IGF1R cells was analyzed essentially as described for p6 cells (31). Briefly, MCF7:IGF1R cells were grown in DMEM:F12 medium (Sigma) (1:1) containing 5% calf serum, penicillin at 100 U/mL, streptomycin at 10 μg/mL, and glutamine at 2 mmol/L. Cells were detached from the flask with 0.02% ethyldiaminetetraacetic acid (EDTA) solution (Sigma) and plated on a LabTek 8-well chamber slide (Nalge Nunc International Corp.) at a concentration of 1.5 × 104 cells per well. Cells were allowed to attach, were grown to 50% confluence, and then were synchronized in phenol red-free and serum-free DMEM (Life Technologies, Inc.) containing 0.1% bovine serum albumin, holotransferrin (Sigma) at 50 μg/mL, penicillin at 100 U/mL, streptomycin at 10 μg/mL, and glutamine at 2 mmol/L (PRF-SFM) for 24 h. After synchronization, the cells were stimulated with 17-β-estradiol at 10 nmol/L for 24 h. Cells were washed once with PRF-SFM and then incubated for 4 or 8 h with fluoresceinyl-peptide-PNA-peptide at 1 or 5 μmol/L in PRF-SFM. At the end of the incubation, the cells were washed 3 times with phosphate-buffered saline (PBS). Cells then were fixed with 1% paraformaldehyde in PBS for 1 h at 37°C and washed 3 times with PBS. The chamber superstructure was removed, and 40 μL of ProLong Antifade reagent (Molecular Probes) was applied to cover the fixed cells in each well on the slide. A coverslip was applied and sealed to the slide. The slide was examined on an MRC-600 laser scanning confocal microscope (Bio-Rad) interfaced to an Axiovert 100 inverted microscope (Zeiss) with a PlanApo 63 × 1.40NA oil-immersion lens (Zeiss).
Cellular Oncogene mRNA Levels
MCF7:IGF1R cells were grown to 80% confluence, detached with trypsin-EDTA (Sigma), and washed with DMEM. For QRT–PCR analyses, cells were lysed with Trizol (Sigma) and RNA was extracted with phenol and chloroform. From each sample, 0.5 μg of RNA was amplified and analyzed as described previously (36). For the uptake of 99mTc-peptide-PNA-peptide probes, samples of 11–12 × 106 cells in 0.5 mL of culture medium were dispensed in test tubes. To each tube, 2.8 pmol (1.66 × 1012 molecules) of 99mTc-peptide-PNA-peptide were added, mixed, and incubated at 22°C for 30 min. Cells were centrifuged at 450g for 5 min, and the supernatant was saved for counting. Cells were washed twice with DMEM. The 3 supernatants were combined. Radioactivity associated with the supernatants and with the cell pellets then was determined. Knowing the percentage of 99mTc taken up by the cells (A; cell pellet counts divided by total counts) and assuming that 1 99mTc atom is bound per peptide-PNA-peptide molecule, the number of peptide-PNA-peptide molecules bound was calculated as follows: B = A × 1.66 × 1012. The number of peptide-PNA-peptides bound per cell (C = B/11.5 × 106 cells) yields the number of CCND1 mRNAs per cell, assuming that all molecules are uniformly distributed per cell, internalized, and hybridized.
Intratumoral Injection of Probes
For study of the effect of regional high delivery of an agent, groups of 5 mice each were injected intratumorally with 2 μg of each of 3 peptide-PNA-peptide chimeras. After 24 h, mice were euthanized, immediately after which the tumors were excised, frozen in liquid nitrogen, and then shipped on dry ice to the University of Missouri for the evaluation of CCND1 RNA and cyclin D1 protein expression. For Western blot analyses, tumor slices were placed in liquid nitrogen and pulverized with a mortar and pestle, an appropriate amount of Cellytic MT lysis buffer (Sigma) was added, and then protein was extracted. From each sample, 100 μg of protein were electrophoresed on 12% polyacrylamide–SDS gels, transferred to nitrocellulose membranes (Bio-Rad), and blocked with 5% powdered milk in Tris-buffered saline. Blocked membranes were incubated with cyclin D1 antibody (Ab-3; Oncogene Research) at a 1:200 dilution. After being washed, the membranes were treated with a secondary antibody conjugated with horseradish peroxidase (sc-2031; Santa Cruz Biotechnology) at a 1:20,000 dilution. Detection was done with a chemiluminescence kit (34095; Pierce Chemicals), and the intensities of the bands on the film were quantitated with a Kodak Image Station 2000R. For QRT–PCR analyses, tumors were placed in liquid nitrogen and pulverized with a mortar and pestle, RNA was extracted with phenol and chloroform, and then 0.5 μg of RNA from each tumor was amplified and analyzed as described previously (36).
Systemic Administration and Tissue Distribution of Labeled Probes
For assessment of chimera distribution and imaging, 7.4–11.1 MBq (0.2–0.3 mCi) of the CCND1 99mTc-peptide-PNA-peptide in 0.2 mL of sterile Na2HPO4 (0.1 mol/L; pH 7) were administered to groups of 5 mice each through a lateral tail vein by use of a sterile 27-gauge needle. For the 24-h distribution, 29.6–34.2 MBq (0.8–0.9 mCi) of the probe was administered. At 4, 12, and 24 h after injection, mice were lightly anesthetized and then imaged by use of a Starcam (General Electric Medical) γ-camera equipped with a parallel-hole collimator. For each image, 300,000 counts were collected. Digital scanning of region-of-interest intensities with an interfaced Entegra computer (General Electric Medical) across each scintigraphic image from the tumor-free left flank to the tumor-bearing right flank provided quantitation of tumor images.
Mice were euthanized, and tissues were dissected, washed free of blood, blotted dry, and weighed. Radioactivity associated with each tissue sample was counted in an automatic Series 5000 γ-counter (Packard), together with a standard radioactive solution of a known quantity prepared at the time of injection. Results were expressed as the percentage injected dose per gram (%ID/g) of tissue.
Statistical Methods
Statistical analysis of differences among groups was done by applying the Student t test, the Kruskal-Wallis one-way ANOVA on ranks, or the Dunn pairwise multiple-comparison procedure by use of SigmaStat 3.0 (SPSS).
RESULTS
Peptide-PNA-Peptides
We prepared an antisense probe specific for oncogene CCND1 mRNA and IGF1R. The probe is a cyclized peptide-PNA-peptide chimera, AcGlyd(Ala)GlyGlyAba-CTGGTGTTCCAT-AEEA-d(CysSerLysCys), designated WT4185 (Fig. 1A). Preparative reversed-phase HPLC of the crude product yielded a main peak containing 95% of the absorbance at 260 nm (Fig. 1B). The overall yield of the HPLC-purified chimera relative to the initial solid support was 30.6%. MALDI-TOF MS of the purified chimera confirmed the identity of the main product peak at 4,187.2 Da (Fig. 1C). Similarly, we prepared a CCND1 PNA control with 4 mismatches (PNA mismatch control), AcGlyd(Ala)GlyGlyAba-CTGGACAACCAT-AEEA-d (CysSerLysCys), designated WT4172, an IGF1 peptide alanine substitution control (peptide mismatch control), AcGlyd(Ala)GlyGlyAba-CTGGTGTTCCAT-AEEA-d(CysAlaAlaCys), designated WT4113, and a PNA-free control, AcGlyd(Ala) GlyGlyAba(Gly)4 d(CysSerLysCys), designated WT990. Table 1 shows the characteristics of the peptide-PNA-peptide chimeras.
Peptide-PNA-Peptide Chimera Characterization
99mTc-Peptide-PNA-Peptides
We radiolabeled the CCND1 probe with the scintigraphic nuclide 99mTc. Samples (20 μg) of the CCND1 peptide-PNA-peptide antisense probe, WT4185, were labeled well at 22°C (Fig. 2A). Free 99mTc was present at only 1.5%, and colloids were present at only 2.5%. The PNA mismatch control (WT4172), the peptide mismatch control (WT4113), and the PNA-free control (WT990) were labeled similarly (data not shown). Denaturing SDS gel electrophoresis (31) of the 99mTc-WT4185 9.3-min peak from Figure 2A showed a radioactive band that coelectrophoresed with the main radioactive band from the 99mTc-WT4185 labeling mixture; unchelated WT4185 or WT4185 mock chelated without 99mTc was slightly faster (Fig. 2B).
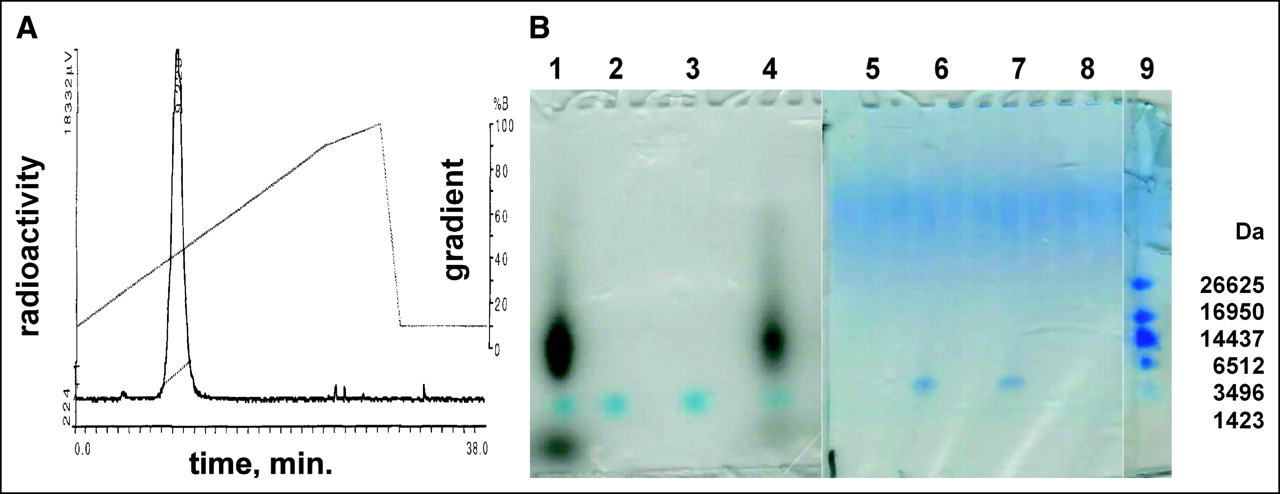
Analysis of 99mTc-AcGlyd(Ala)GlyGlyAba-CTGGTGTTCCAT-AEEA-d(CysSerLysCys), WT4185. (A) A sample from the labeling reaction mixture was analyzed by reversed-phase HPLC on a Microbond C18 column (10 × 250 mm) eluted with a gradient from 10% to 100% CH3CN in aqueous 0.1% CF3CO2H at 1 mL/min over 28 min at 25°C. %B (CH3CN) is shown on right axis, and NaI (Tl) radiometric γ-emission is shown on left axis. The single labeled peak eluted at 9.3 min. (B) Denaturing gel electrophoresis on polyacrylamide (10%–20%)–Tris–Tricine–SDS gels. Left panel is an autoradiogram; right panel is stained with Coomassie blue. Lanes 1 and 5: 99mTc labeling reaction; lanes 2 and 6: mock reaction without 99mTc; lanes 3 and 7: purified WT4185; lanes 4 and 8: 9.3-min 99mTc peak from A; lane 9: peptide mass standards.
Cellular Internalization
We examined the ability of the fluorescent CCND1 probe to enter breast cancer cells. At 8 h after exposure of MCF7:IGF1R cells to the fluorescent peptide mismatch chimera, WT4361, at 1 μmol/L, no cellular uptake was apparent (Figs. 3A–3C). On the other hand, significant uptake of the fluorescent PNA antisense chimera, WT4433, was observed throughout the cells (Figs. 3D–3F). These results agree with those reported previously for p6 cells (31). The same results were seen in cells treated with fluorescent probes at 5 μmol/L for 4 h (data not shown).
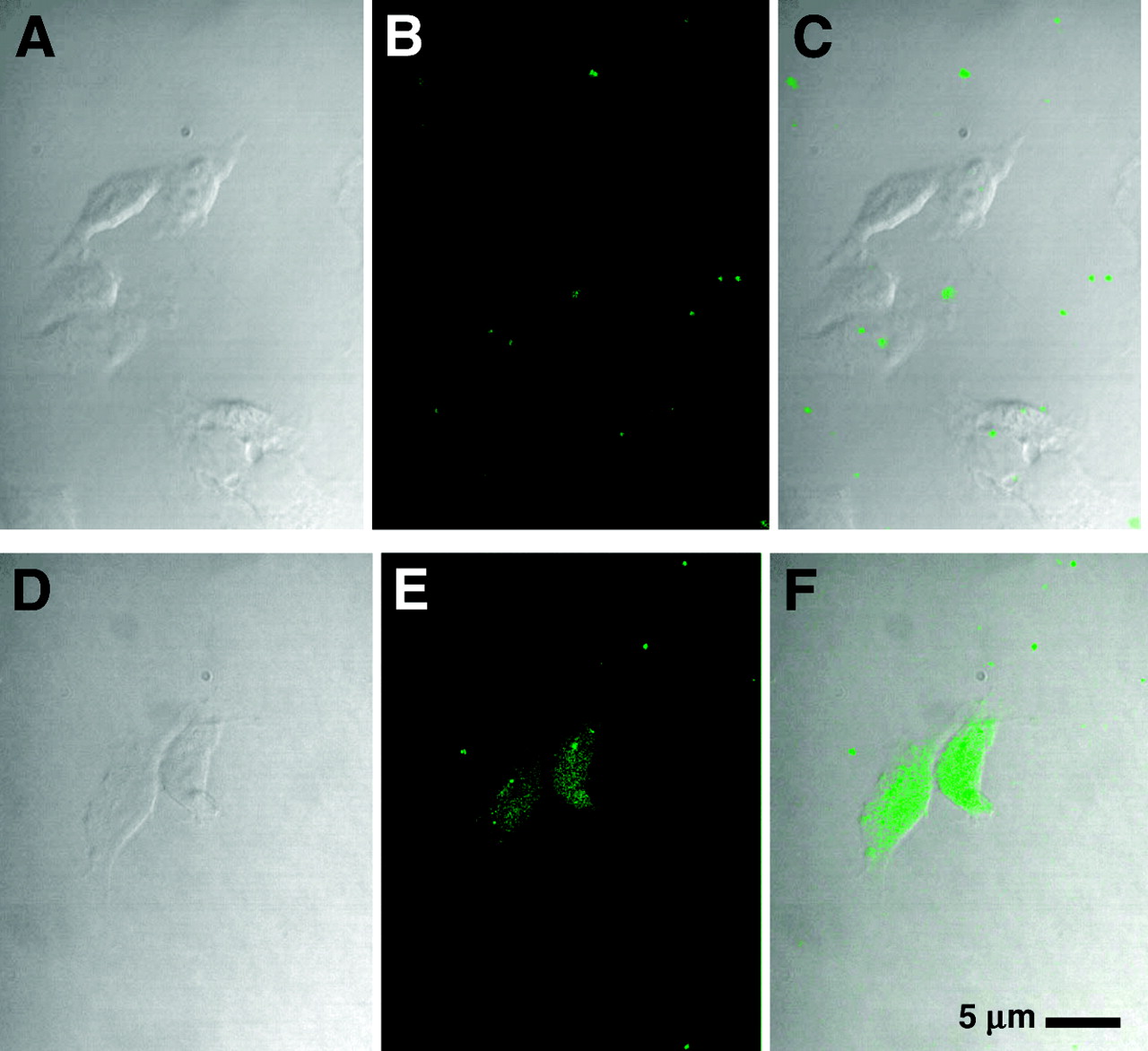
MCF7:IGF1R cell uptake of CCND1 fluoresceinyl-PNA-mismatch peptide probe, WT4361 (A–C), and CCND1 fluoresceinyl-PNA-IGF1 peptide probe, WT4433 (D–F). Cells were incubated with fluoresceinyl-PNA-peptide at 1 μmol/L for 8 h at 37°C in PRF-SFM, fixed, and examined by confocal microscopy. (Left) Phase contrast. (Middle) Fluorescence. (Right) Overlay.
Cellular Oncogene mRNA Copy Number Determined by QRT–PCR
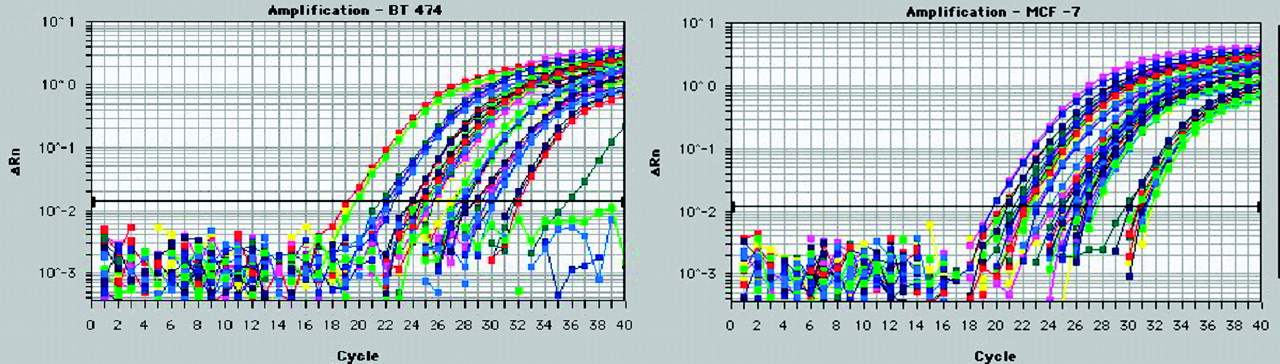
We measured oncogene expression levels in untreated cells by QRT–PCR. We compared BT474 human breast cancer cells, which overexpress HER2 and MYC oncogenes but not the estrogen receptor or IGF1R, with MCF7:IGF1R human breast cancer cells, which overexpress the estrogen receptor, IGF1R, and CCND1 and MYC oncogenes (Fig. 4). A comparison of threshold cycle numbers for CCND1 mRNA and those for TATA box–binding protein mRNA yielded ratios of 32 for BT474 cells and 8 for MCF7:IGF1R cells. On the basis of the previously reported number of TATA-box binding protein mRNA copies per breast cancer cell of 1,000–2,000 (37), we estimated that there were at least 32,000 CCND1 mRNA copies per BT474 cell and 8,000 CCND1 mRNA copies per MCF7:IGF1R cell.

BT474 (left) and MCF7:IGF1R (right) breast cancer cell mRNAs (10, 1, and 0.1 ng) were analyzed by QRT–PCR with a Prism 7700 (Applied Biosystems) to determine the levels of expression of HER2 (blue), CCND1 (green), IGF1R (red), MYC (pink), and TATA-box binding protein (yellow). ΔRn = relative difference in fluorescence at cycle n.
Cellular Oncogene mRNA Copy Number Determined by 99mTc Binding
We also determined oncogene expression levels in untreated cells by measuring the binding of radiolabeled oncogene probes to breast cancer cells. The uptake of 99mTc-peptide-PNA-peptide by MCF7:IGF1R cells was measured by counting radioactivity in cell pellets after 3 washes. Assuming that each 99mTc-peptide-PNA-peptide molecule was internalized and hybridized, we used the specific activity of 99mTc to calculate the number of CCND1 mRNAs per cell. For 12 cell suspensions measured on 3 different days, the CCND1 mRNA copy number was 6,089 ± 2,000 per cell for the antisense probe, 99mTc-WT4185; in contrast, for 8 cell suspensions, the copy number was only 2,388 ± 615 per cell for the PNA mismatch control probe, 99mTc-WT4172. The difference of 3,701 ± 2,615 CCND1 mRNA copies per cell was the same order of magnitude as that estimated by QRT–PCR above and by QRT–PCR of tumors below.
Intratumoral Injection
We measured the ability of our CCND1 probe to reduce the level of cyclin D1 protein in tumors. Cyclin D1 protein levels were determined by Western blotting of MCF7:IGF1R tumor xenografts 24 h after direct injection of the PNA mismatch chimera, WT4172, the peptide mismatch chimera, WT4113, and the PNA antisense chimera, WT4185 (Fig. 5). Kruskal-Wallis ANOVA determined that the 3 groups were different (P = 0.0308). When each group was compared against another, the Dunn multiple-comparison method determined that the PNA mismatch chimera, WT4172, and the PNA antisense chimera, WT4185, groups were different (P = 0.02). The peptide mismatch chimera, WT4113, and the PNA antisense chimera, WT4185, groups were different (P = 0.04). The PNA mismatch chimera, WT4172, and the peptide mismatch chimera, WT4113, groups, however, were not different (P = 0.39). The PNA antisense chimera, WT4185, significantly reduced cyclin D1 protein expression in MCF7:IGF1R xenografts—by approximately 50% (Table 2).

Western blots of 100 μg of protein extracted from MCF7:IGF1R estrogen receptor-positive breast tumor cell xenografts at 24 h after direct injection of the PNA mismatch chimera, WT4172, the peptide mismatch chimera, WT4113, and the PNA antisense chimera, WT4185. CD1 = cyclin D1; B-actin = β-actin.
Cyclin D1 Protein in Tumors Injected Intratumorally with Peptide-PNA-Peptide Chimeras
As expected for PNA-RNA hybridization, which does not provide a substrate for RNase H, real-time PCR results illustrated that the CCND1 mRNA expression levels were not significantly different in tumor xenografts treated with the PNA antisense chimera, WT4185, in those treated with the PNA mismatch chimera, WT4172, and in those treated with the peptide mismatch chimera, WT4113 (Table 3).
CCND1 mRNA in Tumors Injected Intratumorally with Peptide-PNA-Peptide Chimeras
Systemic Administration
We measured the ability of our radiolabeled CCND1 probe to visualize an active CCND1 oncogene in tumors from outside the body. To test our hypothesis that a complementary 99mTc-chelator-PNA-peptide probe specific for CCND1 mRNA and IGF1R could visualize a CCND1-expressing tumor, we administered approximately 18.5 MBq (0.5 mCi) of the 99mTc-PNA-peptide CCND1 probe to cohorts of 5 nude mice bearing approximately 0.5-cm MCF7:IGF1R xenografts to determine the specificity and sensitivity of scintigraphic imaging at 4, 12, and 24 h after administration (Fig. 6). To test whether the probe would bind to tumor cells expressing high levels of IGF1R, we administered the PNA-free chelator plus an IGF1 analog control, WT990, independent of internalization and mRNA binding. We did not observe tumor signals at 4, 12, or 24 h after injection.

Scintigraphic images of γ-rays emitted by decaying 99mTc in nude mice carrying human MCF7:IGF1R estrogen receptor-positive breast tumor cell xenografts at 4, 12, and 24 h after injection of the PNA-free control probe, WT990, the PNA mismatch control probe, WT4172, the peptide mismatch control probe, WT4113, and the PNA antisense probe, WT4185.
Similarly, to test whether a probe containing 4 central PNA mismatches would bind to tumor cells expressing high levels of IGF1R and CCND1 mRNA, we administered the PNA mismatch control, WT4172. We did not observe tumor signals at 4, 12, or 24 h after injection. Next, to test whether the complementary probe with 2 central peptide mismatches would bind to tumor cells expressing high levels of IGF1R and CCND1 mRNA, we administered the peptide mismatch control, WT4113. We did not observe tumor signals at 4, 12, or 24 h after injection.
Finally, to test whether the complementary probe with the correct IGF1 analog would bind to tumor cells expressing high levels of IGF1R and CCND1 mRNA, we administered the PNA antisense chimera, WT4185. In this experiment, we observed faint tumor signals at 4 h, strong tumor signals at 12 h, and intermediate tumor signals at 24 h after injection.
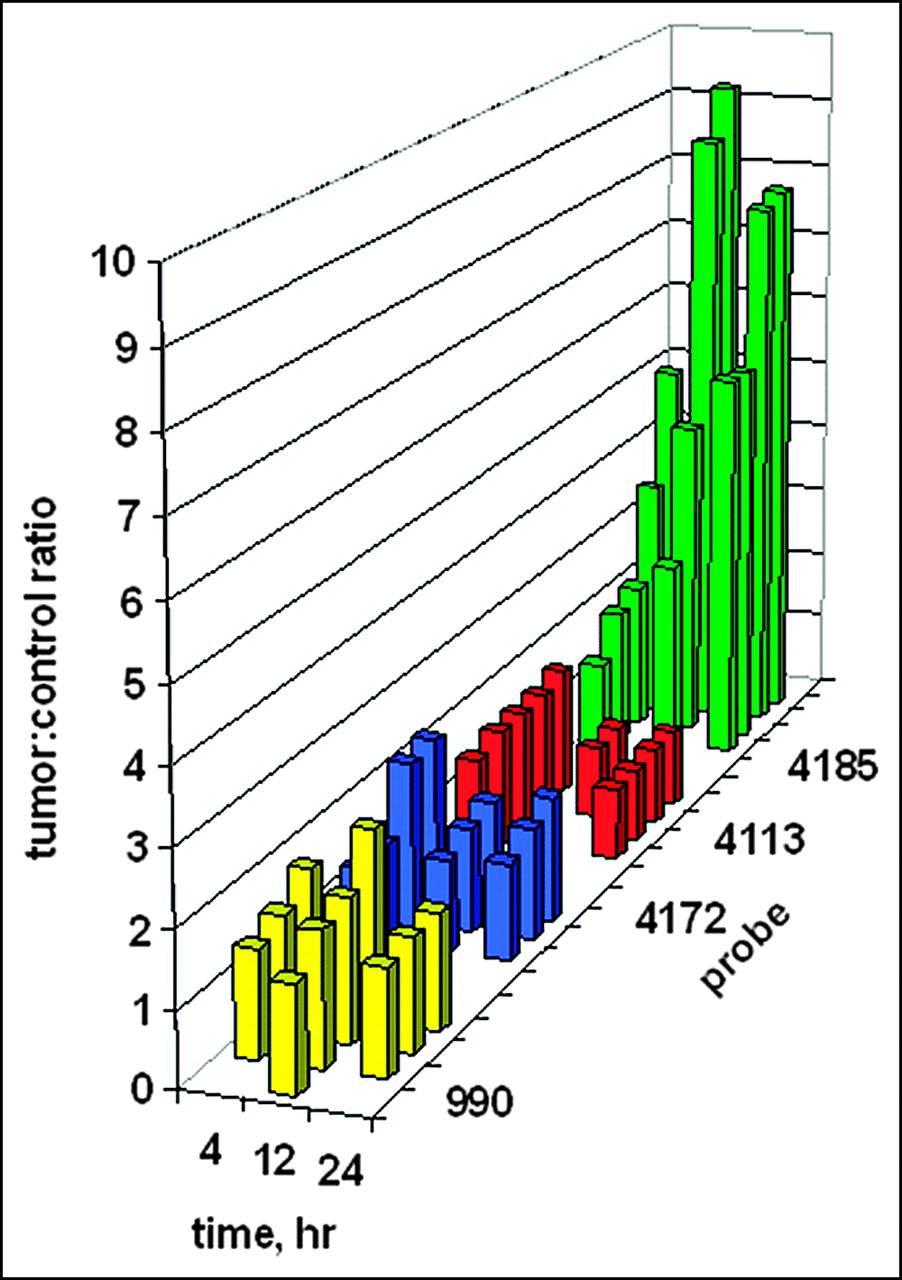
Digital scanning of the tumor site region of interest on the right flank versus the mirror-image tumor-free region of interest on the left flank enabled the quantitation of tumor images. The ratios of tumor site intensities to control site intensities are plotted in Figure 7 as a function of time. It is presumed that any ratio greater than 1 implies some preferential probe concentration. Although no tumor image was obvious in Figure 6 after administration of the PNA-free control, WT990, the measured ratios of about 1.5 imply some weak binding. Similar ratios of about 1.5 were found for the PNA mismatch control, WT4172, including the IGF1 peptide analog. For the peptide mismatch control, WT4113, some binding was apparent at 4 h after administration but not at 12 or 24 h. Only a faint tumor image was obvious in Figure 6 at 4 h after administration of the antisense probe, WT4185; this result correlated with the average scanning ratio of 2.75 ± 1.45. At 12 h after injection, the average scanning ratio increased to 6.32 ± 3.20, and a similar value of 6.65 ± 1.31 was seen at 24 h.
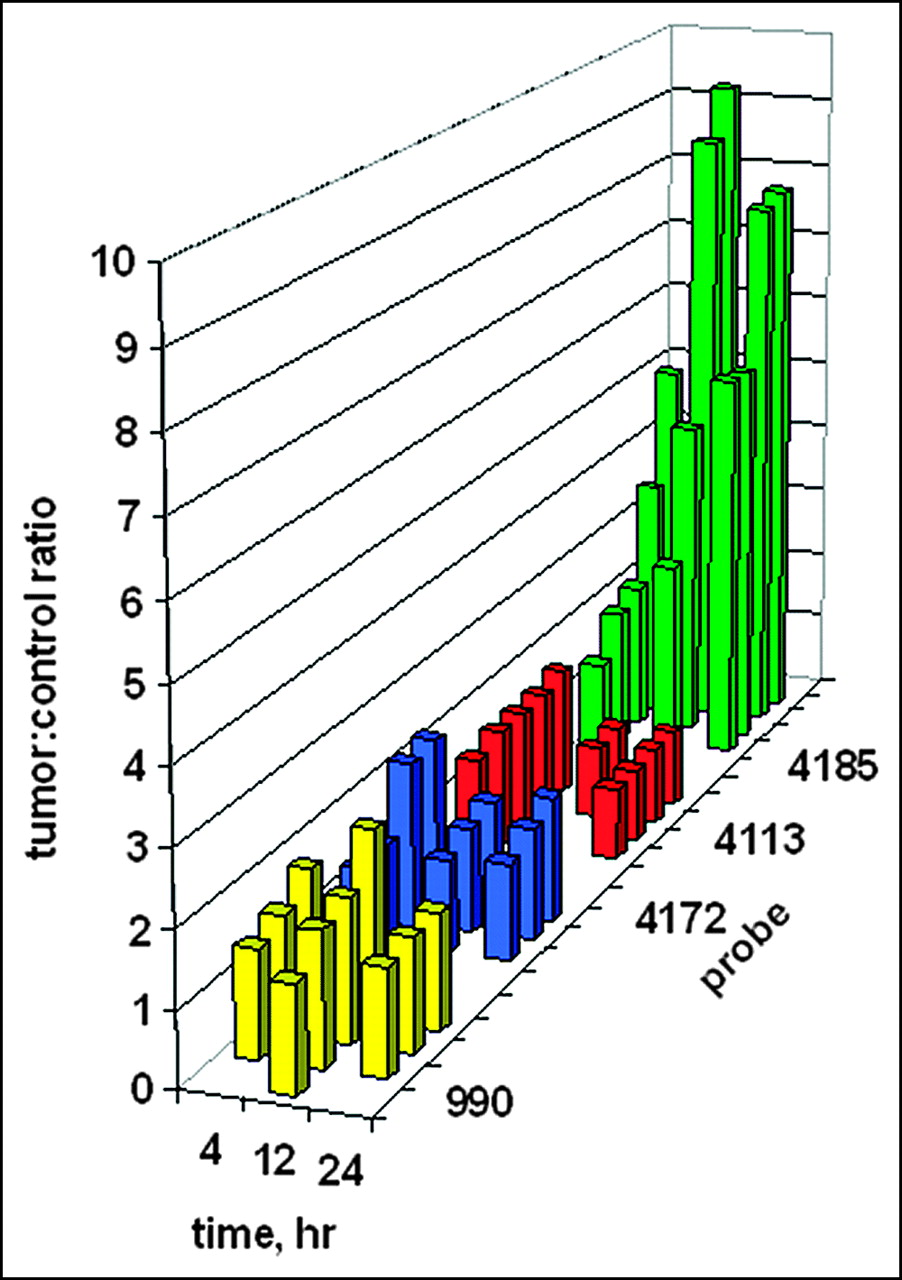
Ratios of tumor site γ-intensity to control site γ-intensity after systemic administration of 99mTc-peptide-PNA-peptide chimeras for all subjects in Figure 6. Bars indicate data for the PNA-free control probe, WT990 (yellow), the PNA mismatch control probe, WT4172 (blue), the peptide mismatch control probe, WT4113 (red), and the PNA antisense probe, WT4185 (green).
Kruskal-Wallis ANOVA determined that the 12 groups were significantly different from each other (P < 0.001). When each group was compared against another at 4 h after injection, Kruskal-Wallis ANOVA determined that there was not a statistically significant difference (P = 0.182) among them. However, at 12 h after injection, Kruskal-Wallis ANOVA determined that there was a statistically significant difference (P = 0.013) among the 4 groups. The Dunn multiple-comparison procedure determined that the PNA-free control, WT990, and the PNA mismatch, WT4172, groups were different (P < 0.05). The PNA mismatch, WT4172, and the PNA antisense, WT4185, groups were different (P < 0.05). However, the peptide mismatch, WT4113, and the PNA antisense, WT4185, groups were not different (P > 0.05). Similarly, at 24 h after injection, Kruskal-Wallis ANOVA determined that there was a statistically significant difference (P = 0.009) among the 4 groups. The Dunn multiple-comparison procedure again determined that the PNA-free control, WT990, and the PNA mismatch, WT4172, groups were different (P < 0.05). The PNA mismatch, WT4172, and the PNA antisense, WT4185, groups were different (P < 0.05). However, the peptide mismatch, WT4113, and the PNA antisense, WT4185, groups were not different (P > 0.05).
Tissue Distribution
We measured the distribution of our radiolabeled CCND1 probe in various tissues of the mice, including the tumors. The tissue distribution of each probe was measured in all subjects at 4, 12, and 24 h after administration. Table 4 shows the data for the PNA-free control probe, WT990; Table 5 shows the data for the PNA mismatch probe, WT4172; Table 6 shows the data for the peptide mismatch probe, WT4113; and Table 7 shows the data for the PNA antisense probe, WT4185. As the time after injection elapsed, the radioactivity in all tissues decreased, including that in the tumors. However, the ratio of radioactivity in tumors to radioactivity in blood and the ratio of radioactivity in tumors to radioactivity in muscle increased for WT4185 as time went on. This finding is consistent with the scintigraphic images in Figure 6 and the ratios of tumor intensities to muscle intensities shown in Figure 7, which were higher than the ratios of distribution in tumors to distribution in muscle determined by scintillation counting for WT4185 and shown in Table 7. Although the tumors were readily detectable by scintigraphy with WT4185 but not with WT990, WT4172, or WT4113, the tumor distribution counts for WT990, WT4172, and WT4185 were not significantly different. This apparent contradiction is considered below.
Tissue Distribution of PNA-Free Probe, WT990, After Systemic Administration (n = 5)
Tissue Distribution of PNA Mismatch Probe, WT4172, After Systemic Administration (n = 5)
Tissue Distribution of Peptide Mismatch Probe, WT4113, After Systemic Administration (n = 5)
Tissue Distribution of PNA Antisense Probe, WT4185, After Systemic Administration (n = 5)
The tissue distribution data indicated that renal excretion was the primary route of elimination and that renal uptake was the highest among all of the tissues, but the extent differed from agent to agent. In a previous experiment, urine was collected from 5 mice 4 h after injection with MYC antisense probe 99mTc-Glyd(Ala)GlyGlyAba-GCATCGTCGCGG (WT3613) for the assessment of probe stability in vivo (38). Analytic HPLC of radioactivity in the combined, deproteinized, and lyophilized urine revealed a voided-volume peak of free 99mTc with 17% of the radioactivity and an intact probe peak eluting at 10.7 min with 83% of the radioactivity. This result indicated the stability of the agent in vivo at 37°C. Breakdown fragments were not detected, consistent with the model that the 99mTc-peptide-PNA conjugate is resistant to proteases and nucleases in serum at 37°C (22).
DISCUSSION
In this study, we tested the feasibility of using a radiolabeled CCND1 oncogene probe to visualize a breast tumor from outside the body. We prepared a synthetic receptor-targeting vector with a PNA hybridization probe to provide access to breast cancer cells overexpressing IGF1R, a receptor that is overexpressed in a high percentage of breast cancer cells. We found by fluorescence microscopy that the peptide-PNA-peptide probes were taken up inside MCF7 breast cancer cells overexpressing IGF1R if and only if the IGF1 peptide analog sequence was correct.
Once the probes were inside the breast cancer cells in the murine xenografts, we found that the CCND1 antisense peptide-PNA-peptide binding was sequence specific, as the downregulation of cyclin D1 protein in tumors was seen only on intratumoral injection of the correct antisense sequence and not with either the peptide mismatch or the PNA mismatch sequence. The reduction in the level of cyclin D1 protein seen in Western blots of protein from tumors injected with the unlabeled antisense probe, WT4185, but not with any of the control probes is consistent with the hypothesis that the specific probe can enter MCF7:IGF1R cells and hybridize specifically with CCND1 mRNA. The unperturbed level of CCND1 mRNA is consistent with the knowledge that PNA bound to mRNA does not form a substrate for RNase H.
The measurements of fluorescent probe uptake, intratumoral downregulation of cyclin D1 protein, imaging of CCND1 mRNA in tumors, and intactness of the probe recovered from urine (38) demonstrated that the antisense peptide-PNA-peptide chimera was not obviously vulnerable in vivo for at least 24 h after administration. The downregulation of cyclin D1 protein by the antisense probe also established the possibility of therapeutic applications.
We performed a variety of tests of the hypothesis that the overexpression of CCND1 mRNA in MCF7:IGF1R breast cancer xenografts could be detected specifically and noninvasively by scintigraphic imaging of 99mTc-peptide-PNA-peptide probes. The number of copies of CCND1 mRNA in MCF7:IGF1R breast cancer cells was estimated to be 4,000–8,000 per cell, permitting a theoretic estimate of maximum possible labeling. If there are 6,000 mRNAs per cell and 109 cells per gram of tumor, then there would be an estimated 6 × 1012 mRNA copies available for binding in a 1-g tumor. Our data suggest that, on average, 0.2% of the injected 99mTc-WT4185 (specific activity, 74 GBq/μmol [2 Ci/μmol]) was associated with the tumors and that approximately 6 × 1011 molecules of 99mTc-WT4185 were bound to the mRNA in a 1-g tumor, or about 10% of the CCND1 mRNA, despite the nonuniform distribution of radioactivity in the tumor. In a high-specific-activity or carrier-free preparation, this value would be equivalent to 18.5 MBq (500 μCi) of 99mTc, suggesting the possibility of a sensitivity up to 500 times higher than that observed in this initial study.
To estimate the fraction of internalized PNA probes that could hybridize to available CCND1 mRNAs in live tumor cells, one must begin with the observed melting temperature of approximately 80°C for peptide-PNA-peptide 12-mers at 2.5 μmol/L and RNA at 2.5 μmol/L (34). A copy number of approximately 6,000 CCND1 mRNAs per picoliter of cells translates to 10 nmol/L, whereas the value for 99mTc-WT4185 appeared to be about 10% that value, 1 nmol/L. The melting temperature, the probe concentration, and the 10-fold excess mRNA concentration translated to a calculation (26) of virtually complete hybridization of the available intracellular probe.
Interestingly, concentration of the label in tumors was very faint at 4 h after injection of the CCND1 antisense PNA sequence with the correct IGF1 analog, WT4185, although the digital scan revealed significant concentration of the label. We suspect that uptake, intracellular trafficking, hybridization of label, and efflux of unbound label were all slow processes that did not reveal a strong tumor signal at 4 h but that concentration of labeled antisense PNA in tumors was prominent at 12 h, after the unbound probe had undergone efflux and been excreted, and was still detectable at 24 h. With respect to the hybridized label, the bound PNA probe dissociates negligibly from complementary RNA by 12–24 h (26). Therefore, we found that MCF7:IGF1R xenografts overexpressing CCND1 mRNA could be readily and optimally imaged at 12 h after injection.
The tissue distribution results are comparable to previous observations obtained with the MYC PNA antisense probe 99mTc-Glyd(Ala)GlyGlyAba-GCATCGTCGCGG (23) and to the observations of Stalteri and Mather (19) as well as the tissue distribution of vasoactive intestinal peptide 99mTc-Glyd(Ala)GlyGlyAba (16–18). Nevertheless, the MCF7:IGF1R xenografts were imaged clearly with WT4185 but not with WT990, WT4172, or WT4113. That is because the tissue distribution data reflect radioactivity in the entire excised masses, without dissection of the actively proliferating cells on the periphery of the tumors from the necrotic cores, which exhibit poor uptake of macromolecules (39). Including the active and inactive portions of the tumors, as well as some surrounding normal tissues, overstated the masses of actively proliferating tumors and thereby decreased the apparent percent uptake of 99mTc per gram of tumor and the ratios of tumor intensity to muscle intensity, reducing them to the ranges seen with the control agents. This same difference between clear scintigraphic images and modest percent uptake of 99mTc per gram of tumor was also observed earlier for vasoactive intestinal peptide 99mTc-Glyd(Ala)GlyGlyAba, which revealed occult tumors in patients despite apparent tumor uptake of only 0.3% ID/g in xenografts in immunocompromised mice (16–18). However, digital scanning of scintigraphic intensities determined the radioactivity per pixel in the tumor region of interest, revealing CCND1 mRNA expression in the actively proliferating cells on the periphery of the tumors. In this situation, the necrotic centers presumably did not contribute to the measurement, eliminating the possibility of label dilution. Thus, the ratios of tumor site scintigraphic intensity to contralateral site scintigraphic intensity shown in Figure 7 probably provided more accurate results than the scintillation counting tissue distribution ratios shown in Tables 4–7.
For WT4113, with 2 d-Ala replacements in the peptide loop, the percent tumor distribution values were higher than those for WT4185, and the kidney distribution values were lower and statistically significantly different (P < 0.05). However, tumors with WT4113 were not detectable scintigraphically. Radioactivity uptake of WT4113 for all time points, not only for the tumor but also for blood and muscle, was 3–4 times higher (P < 0.05) than that for WT4185. This higher uptake in blood and muscle may have contributed to the apparently higher uptake in the excised tumor because of the contribution of blood activity remaining in the tumor vasculature and muscle, which could have contaminated the tumor during excision. The higher background radioactivity in the surrounding muscle rendered the tumor indistinguishable by scintigraphic scanning, even though the tissue counts showed higher WT4113 radioactivity than WT4185 radioactivity in muscle and tumors. Digital region-of-interest analyses (Fig. 7) also showed lower intensity ratios. The 2 d-Ala replacements in the peptide loop of WT4113, in lieu of d-Ser and d-Lys, precluded its binding to IGF1R and reduced the net charge on WT4113. Both of those factors could have slowed its blood clearance. This scenario was evidenced by renal uptake that was significantly lower than that seen with WT990, WT4172, or WT4185, each with the reversed, inverted IGF1 loop sequence, at each time point.
For most agents, renal clearance is predominantly determined by at least 2 parameters, the charge and the molecular size. In light of the neutrality of PNAs, the net charges on the PNA-free probe, WT990, the PNA mismatch probe, WT4172, and the PNA antisense probe, WT4185, at pH 7.4 are probably the same, +1 from the N-terminal amine and +1 from the d-Lys side chain. However, the peptide mismatch probe should only exhibit a charge of +1 from the N-terminal amine. The molecular masses of the 3 chelator-PNA-peptide agents are not very different from one another, whereas the mass of the PNA-free peptide agent is much smaller. Hence, one would predict that the smaller PNA-free probe would clear most quickly, followed by the PNA mismatch probe, the PNA antisense probe, and then the peptide mismatch probe. CCND1 expression has been reported to be high in renal cell carcinoma cells but low in normal renal tissue (40). Therefore, concentration of the PNA antisense probe, WT4185, in the kidneys was probably not attributable to labeling of CCND1 mRNA in kidney cells.
Our observation of scintigraphic imaging of CCND1 mRNA expression in xenografts could be applicable to patients at risk for new or recurrent breast cancer, including those with a strong family history of breast cancer, or when standard detection techniques provide ambiguous results. Imaging of mRNA expression with chelator-PNA-peptide chimeras labeled with 99mTc, 64Cu, or other radionuclides also might be applicable to other oncogenes or to other cell surface receptors.
The early detection of breast cancer currently relies on physical examination and mammography. Other imaging studies may be performed if there is a suspicious finding with either of these detection techniques. Incremental improvements in mammography have improved its sensitivity and specificity. The limitations of current approaches, however, include the following. It is difficult to discover by palpation masses smaller than 1 cm in the breast. Benign lesions are not distinguished from malignant lesions. Mammography does not identify isodense lesions, which are especially common in young women, whose cancers are often more aggressive, in women with dense breasts, and in women who have undergone prior surgery, in whom the scar may obscure a developing lesion.
Other studies for imaging the breast, usually ultrasound or MRI, rely on morphologic changes between normal and abnormal breast tissues that may or may not be present. Thus, not all malignant lesions are detected, and many invasive surgical procedures are performed to remove benign lesions. The latter problem results from the lack of specificity of current imaging techniques. The nature of masses detected by imaging studies must be confirmed through histologic analyses. More sensitive and specific techniques are needed to detect disease as early as possible, to minimize diagnostic procedures, and to lead directly to therapeutic intervention.
Targeting altered expression of oncogenes provides the promise of specificity, which may ultimately lead to the elimination of diagnostic surgical biopsies. Cyclin D1 overexpression is a common event in and is highly associated with the presence of breast cancer. Although not sufficiently sensitive to be used alone, noninvasive measurement of mRNA expression by one or more oncogenes to confirm the presence or absence of cancer by γ-emission or by PET when an imaging study is abnormal provides a mechanism for the more accurate diagnosis of breast cancer.
CONCLUSION
The results of this study establish the proof of principle for identifying oncogene activity in breast cancer xenografts from outside the body with a radiolabeled hybridization probe. Estrogen receptor-positive MCF7 human breast cancer xenografts were imaged scintigraphically as intracellular uptake by IGF1R and hybridization to CCND1 mRNA of a specific 99mTc-peptide-PNA-peptide. At 24 h after administration, tumor site intensity was 7 times higher than contralateral site intensity, and the level of cyclin D1 protein in the tumors was reduced by 56%. The data demonstrate noninvasive imaging of oncogene mRNA expression in solid tumors with receptor-targeted chelator-PNA-peptide chimeras.
Acknowledgments
We thank Eva Surmacz for advice and the gift of MCF7 cells constitutively overexpressing IGF1R, Zuping Qu for maintaining the MCF7 cells under a variety of conditions, Zhi-Xian Lu for the assembly of linear peptide-resin supports, Richard Wassell for assistance in measuring mass spectra of peptide-PNA-peptides, and Lois Wickstrom for assistance in rephrasing the final version of the manuscript. This work was supported by grants ER63055 from the Department of Energy and HL59769 from the National Institutes of Health.
Footnotes
Received Mar. 11, 2004; revision accepted Jul. 21, 2004.
For correspondence or reprints contact: Eric Wickstrom, PhD, Department of Biochemistry and Molecular Pharmacology and Kimmel Cancer Center, Thomas Jefferson University, Philadelphia, PA 19107.
E-mail: eric{at}tesla.jci.tju.edu
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Molecular Imaging of MDM2 Messenger RNA with 99mTc-Labeled Antisense Oligonucleotides in Experimental Human Breast Cancer Xenografts
- Molecular Imaging of bcl-2 Expression in Small Lymphocytic Lymphoma Using 111In-Labeled PNA-Peptide Conjugates
- Noninvasive Imaging of Human Telomerase Reverse Transcriptase (hTERT) Messenger RNA with 99mTc-Radiolabeled Antisense Probes in Malignant Tumors
- PET Imaging of CCND1 mRNA in Human MCF7 Estrogen Receptor Positive Breast Cancer Xenografts with Oncogene-Specific [64Cu]Chelator-Peptide Nucleic Acid-IGF1 Analog Radiohybridization Probes
- Receptor-mediated internalization of chelator-PNA-peptide hybridization probes for radioimaging or magnetic resonance imaging of oncogene mRNAs in tumours
- Cyclin D1 Is Necessary for Tamoxifen-Induced Cell Cycle Progression in Human Breast Cancer Cells
- Initial Mechanistic Studies of Antisense Targeting in Cells
- Meeting Report: The International Conference on Tumor Progression and Therapeutic Resistance