


Abstract
Gastrin-releasing peptide (GRP) receptors have been shown to be expressed with high densities on several types of cancer cells including prostate, breast, small cell lung, and pancreas cancers. Bombesin (BBN) has been known to bind to GRP receptors with high affinity and specificity. The aim of these studies was to develop new 111In-labeled BBN analogs having high tumor uptake and optimal pharmacokinetics for specific targeting of human prostate cancers. Methods: A novel series of dodecanetetraacetic acid (DOTA)-X-BBN[7–14]NH2 (X = 0, β-Ala, 5-Ava, 8-Aoc, or 11-Aun) conjugates and their In(III)/111In complexes exhibiting high GRP-receptor-binding affinities were synthesized and characterized. Results: In vitro competitive binding assays, using PC-3 androgen-independent human prostate cancer cells, demonstrated values of <2.5 nmol/L for inhibitory concentration of 50% for analogs with β-Ala, 5-Ava, and 8-Aoc spacers. In vivo biodistribution studies of the 111In-DOTA-X-BBN[7–14]NH2 conjugates performed on CF-1 mice at 1 h after injection have revealed that the uptake of radioactivity in the pancreas, a GRP-receptor-expressing tissue, increased as a function of hydrocarbon spacer length (i.e., from 0.20 ± 0.04 percentage injected dose [%ID] per gram for the analog with no spacer to a maximum of 26.97 ± 3.97 %ID/g for the analog with 8-Aoc spacer). The radioactivity was cleared efficiently from the blood pool by excretion mainly through the renal/urinary pathway (e.g., 71.6 ± 1.8 %ID at 1 h after injection for 8-Aoc spacer analog). In vivo pharmacokinetic studies of the 111In-DOTA-8-Aoc-BBN[7–14]NH2 conjugate conducted on PC-3 human prostate cancer-derived xenografts in SCID mice showed a specific uptake of radioactivity in tumor, with 3.63 ± 1.11 %ID/g observed at 1 h after injection. High tumor-to-blood and tumor-to-muscle ratios of approximately 6:1 and 45:1, respectively, were achieved at 1 h after injection. Relative to the radioactivity observed in the tumor at 1 h after injection, 43%, 19%, and 9% of the radioactivity was retained at, respectively, 24, 48, and 72 h after injection. Conclusion: These studies showed that radiometallated DOTA-X-BBN[7–14]NH2 constructs with hydrocarbon spacers ranging from 5 to 8 carbon atoms are feasible candidates for further development as diagnostic and therapeutic radiopharmaceuticals for patients with GRP-positive cancers.
Radiolabeled small receptor-avid peptides have attracted considerable interest because of their wide applicability in the development of target-specific radiopharmaceuticals (1,2). Several malignant tumors overexpress certain types of receptors on their surface that make the tumors potential targets for imaging and therapy with radiolabeled receptor-avid peptides (1,2). Bombesin (BBN), a 14-amino-acid peptide, is an amphibian analog of human gastrin-releasing peptide (GRP) that binds with high affinity and specificity to the GRP-receptor subtype (BB2) of the BBN-receptor family (3,4). GRP receptors have been shown to be expressed with high densities on several types of cancer cells including prostate, breast, small cell lung, and pancreas cancers (5–7). Many synthetic BBN peptide antagonists that bind with high affinities to GRP receptors have been synthesized to evaluate their potential to reduce or minimize the rate of growth of GRP-receptor-expressing cancers (3–8). Insights from this research guide the design of radiolabeled BBN conjugates that maintain high in vitro and in vivo GRP-receptor-binding affinities.
Significant progress has been made over the past few years in developing effective strategies to produce radiolabeled BBN analogs that specifically target GRP-receptor-expressing cancer cells in vitro and in vivo (9–24). Our laboratory has focused on developing radiometallated BBN analogs in which radiometal chelates are linked to the truncated BBN[7–14]NH2 sequence that serves as the highly specific GRP-receptor-binding motif (2,12–18). Use of this truncated BBN[7–14]NH2 sequence was selected since the BBN derivatives containing this sequence have been shown to bind in an agonist manner to the GRP receptors (7,14,17). As with other G-protein-coupled, 7-transmembrane segment (7-TMS) receptors, specific binding of an agonist to cell-surface GRP receptors typically leads to internalization of the agonist-receptor complex (25–27), providing an effective method to produce residualization of the radioactive metal atom that will be specifically delivered to the cancer cell by the BBN[7–14]NH2 moiety targeting vector (28). Studies with BBN[7–14]NH2 conjugates radiolabeled with 105Rh, 99mTc, and 111In have demonstrated receptor-mediated trapping of these radiotracers by GRP-receptor-expressing cancer cells (14,22,24). These results demonstrate the feasibility of producing BBN[7–14]NH2 analogs labeled with a variety of radiometals that hold potential for effective in vivo targeting of GRP-receptor cancer cells.
A fundamental concern in the design of radiometallated peptide constructs is to ensure in vivo stability of the radiometal incorporated by the ligand framework. In vivo stability can be controlled by virtue of the high thermodynamic stability and the kinetic inertness of a specific metal chelation framework (29–33). Because the dodecanetetraacetic acid (DOTA) macrocyclic ligand system is well known to form kinetically inert and thermodynamically stable chelates with indium, yttrium, and lanthanides, it has received wide acceptance for in vivo applications with several trivalent radiometals (29–33). Our research group is currently evaluating the potential of DOTA-BBN analogs labeled with trivalent radioactive metals, which are linked either directly to the N-terminal amine group of BBN[7–14]NH2 moiety or through hydrocarbon spacer groups, to specifically target GRP-receptor-expressing cancer cells (Fig. 1) (18). The length and composition of the spacer group, or tether, as well as the physicochemical properties of the radiolabeled moiety will influence the GRP-receptor-binding affinity, residualization of radioactivity in cancer cells, and pharmacokinetics of the BBN conjugate. The purpose of this study was to determine the effects of varying the length of the hydrocarbon spacer on the in vitro binding affinity of 111In+3-DOTA conjugates of BBN[7–14]NH2 with GRP receptors expressed on PC-3 androgen-independent human prostate cancer cells and their in vivo pharmacokinetics in CF-1 mice. The ultimate goal was identification of one or more lead candidates with optimum in vivo pharmacokinetics and high GRP-receptor-mediated uptake in tumor cells.

DOTA-X-BBN[7–14]NH2 analogs.
MATERIALS AND METHODS
Reagents and Apparatus
All chemicals were obtained from either Aldrich Chemicals or Fisher Scientific, and all chemicals and solvents were reagent grade and used without further purification. The Rink amide 4-methylbenzhydrylamine (MBHA) resin and 9-fluorenylmethyl chloroformate (Fmoc)-protected amino acids were purchased from Calbiochem-Novabiochem Corp., and the other peptide reagents, from Applied Biosystems, Inc. The DOTA-tris(t-butyl ester) was purchased from Macrocyclics, and the Fmoc-protected ω-amino alkyl carboxylic acids, from Advanced ChemTech. 125I-Tyr4-BBN was obtained from NEN Life Sciences Products, Inc., 111InCl3 was obtained from Mallinckrodt Medical, Inc., as a 0.05N HCl solution. Electrospray mass spectral analyses were performed by Synpep Corp. PC-3 cells were obtained from American Type Culture Collection and were maintained and grown in the University of Missouri Cell and Immunobiology Core Facility. CF-1 mice were purchased from Charles River Laboratories and were maintained in an in-house animal facility.
High-Performance Liquid Chromatography (HPLC)
HPLC analyses were performed on a Waters 600E system equipped with a Varian 2550 variable absorption detector, a Packard Radiometric 150TR flow scintillation analyzer, a sodium iodide crystal radiometric detector, an Eppendorf TC-50 column temperature controller, and Hewlett-Packard HP3395 integrators. HPLC solvents consisted of H2O containing 0.1% trifluoroacetic acid (solvent A) and acetonitrile containing 0.1% trifluoroacetic acid (solvent B). A Phenomenex Jupiter C-18 (5 μm, 300 Å, 4.6 × 250 mm) column was used with a flow rate of 1.5 mL/min. This HPLC gradient system began with an initial solvent composition of 80% A and 20% B followed by a linear gradient to 70% A and 30% B in 30 min, after which the column was reequilibrated.
Solid-Phase Peptide Synthesis
DOTA-X-BBN[7–14]NH2 conjugates (Fig. 1) were synthesized on a Perkin Elmer-Applied Biosystems model 432 automated peptide synthesizer using traditional Fmoc chemistry, with 2-(1-H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate activation of carboxyl groups on the reactant, and with the N-terminal amino group on the growing peptide anchored through the C-terminus to the resin. Rink amide MBHA resin (25 μmol), Fmoc-protected amino acids with appropriate side-chain protections (75 μmol), Fmoc-protected ω-amino alkyl carboxylic acids (75 μmol), and DOTA-tris(t-butyl ester) (75 μmol) were used for the synthesis. The final products were cleaved by a standard procedure using a cocktail containing thioanisole, water, ethane dithiol, and trifluoroacetic acid in a ratio of 2:1:1:36 and were precipitated into methyl-t-butyl ether. Typical yields of the crude peptides were 80%–85%. Crude peptides were purified by HPLC, and the solvents were removed on a SpeedVac concentrator (Thermo Savant). The purified peptides were characterized by electrospray mass spectrometry (Table 1).
Electrospray Mass Spectrometry and HPLC Data
In+3 Metallation and 111In Labeling
A solution of each DOTA-X-BBN[7–14]NH2 conjugate (5.0 mg) in 0.2 mol/L tetramethylammonium acetate (0.5 mL) was added to indium chloride (10.0 mg). The pH of the reaction mixture was adjusted to 5.8. The reaction mixture was incubated for 1 h at 80°C (Fig. 2). The resultant In-DOTA-X-BBN[7–14]NH2 conjugate was purified by reversed-phase HPLC and was analyzed by electrospray mass spectrometry. The results of mass spectral analysis are shown in Table 1. The pure product was obtained as a white powder with a typical yield of 50%–60%.

Scheme for 111In labeling of DOTA-X-BBN[7–14]NH2 conjugates. EDTA = ethylenediaminetetraacetic acid.
For 111In labeling, an aliquot of 111InCl3 (37 MBq, 50 μL) was added to a solution of DOTA-X-BBN[7–14]NH2 (100 μg) in 0.2 mol/L tetramethylammonium acetate (500 μL). The pH of the reaction mixture was adjusted to 5.8. The reaction mixture was incubated for 1 h at 80°C. An aliquot of 0.002 mol/L ethylenediaminetetraacetic acid (50 μL) was added to the reaction mixture to complex the unreacted 111In+3 (Fig. 2). The resultant 111In-DOTA-X-BBN[7–14]NH2 conjugate was obtained as a single product and was purified by reversed-phase HPLC (Fig. 3). The purified 111In-DOTA-X-BBN[7–14]NH2 conjugate (27.8–29.6 MBq, 75%–80% recovery) was then concentrated by being passed through an Empore C18-HD high-performance extraction disk (7 mm/3 mL) cartridge and being eluted with 33% ethanol in 0.1 mol/L NaH2PO4 buffer (400 μL). The concentrated fraction was then diluted with 0.1 mol/L NaH2PO4 buffer (2.3 mL, pH 7) to make the final concentration of ethanol in the solution < 5%.

HPLC chromatograms of purified 111In-DOTA-8-Aoc-BBN[7–14]NH2 (radiometric detector) (A) and DOTA-8-Aoc-BBN[7–14]NH2 (ultraviolet detector at λ = 280 nm) (B).
In Vitro Competitive Cell-Binding, Internalization, and Efflux Studies
The inhibitory concentration of 50% (IC50) of In-DOTA-X-BBN[7–14]NH2 conjugates was determined by a competitive displacement cell-binding assay using 125I-Tyr4-BBN. Briefly, 3 × 104 PC-3 cells suspended in Roswell Park Memorial Institute (RPMI) medium 1640 at pH 7.4 and containing 4.8 mg/mL N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES), 0.1 μg/mL Bacitracin (Pharmacia and Upjohn Co.), and 2 mg/mL bovine serum albumin (BSA) were incubated at 37°C and in 5% CO2 for 40 min in the presence of 125I-Tyr4-BBN (20,000 cpm, 370 Bq) and increasing concentrations of the In-DOTA-X-BBN[7–14]NH2 conjugates. After the incubation, the reaction medium was aspirated and the cells were washed 3 times with medium. The radioactivity bound to the cells was counted in a Packard Riastar γ-counting system. The percentage of 125I-Tyr4-BBN bound to cells was plotted versus increasing concentrations of In-DOTA-X-BBN[7–14]NH2 conjugates to determine the respective IC50 values.
In vitro studies to determine the degree of internalization of 111In-DOTA-8-Aoc-BBN[7–14]NH2 conjugate were performed by a method similar to that described by Reile et al. (9). These studies were performed by incubating 3 × 104 PC-3 cells (suspended in RPMI medium 1640, at pH 7.4, and containing 4.8 mg/mL HEPES, 0.1 μg/mL Bacitracin, and 2 mg/mL BSA) for 40 min at 37°C and in 5% CO2 in the presence of 20,000 cpm 111In-DOTA-8-Aoc-BBN[7–14]NH2 conjugate. After 40 min of incubation, the binding was stopped through aspiration of the reaction medium and washing of the cells with culture medium. The radioactivity-bound cells were then incubated in culture medium at 37°C. After 1, 2, 3, and 4 h of incubation in culture medium, the percentage of radioactivity trapped in the cells was determined after removal of activity bound to the surface of the cells by washing with a pH 2.5 (0.2 mol/L acetic acid and 0.5 mol/L NaCl) buffer.
In Vivo Biodistribution Studies
The biodistribution and uptake of 111In-DOTA-X-BBN[7–14]NH2 conjugates in healthy CF-1 mice was studied. Each mouse (average weight, 25 g) was injected with an aliquot (50–100 μL) of the HPLC-purified radiolabeled peptide solution (55–75 kBq) through the tail vein. Tissues and organs were excised from the animals sacrificed at 1 h after injection. The activity was counted in a NaI counter, and the percentage injected dose (%ID) per organ and per gram was calculated. The %ID in the blood was estimated assuming a blood volume equal to 6.5% of the total body weight. Receptor-blocking studies were also performed when excess (100 μg) BBN was administered to animals along with the 111In-DOTA-8-Aoc-BBN[7–14]NH2 conjugate.
Pharmacokinetic Studies of the 111In-DOTA-8-Aoc-BBN[7–14]NH2 Analog in PC-3 Tumor-Bearing SCID Mice
Four- to 5-wk-old female ICR SCID (severely compromised immunodeficient) outbred mice were obtained from Taconic. Five mice were housed per sterile Microisolator cage (Alternative Design) in a temperature- and humidity-controlled room with a daily schedule of 12 h of light followed by 12 h of darkness. The animals were fed autoclaved rodent chow (Ralston Purina Co.) and water ad libitum. They were housed for 1 wk before inoculation of tumor cells, and they were anesthetized for injections with isoflurane (Baxter Healthcare Corp.) at a rate of 2.5% with 0.4 L oxygen through a nonrebreathing anesthesia vaporizer.
PC-3 cells were injected bilaterally and subcutaneously in the flank, with ∼5 × 106 cells in a suspension of 100 μL normal sterile saline injected per site. The PC-3 cells were allowed to grow in vivo for 2–3 wk after inoculation, and tumors ranging from 0.2 to 1.0 g developed. The pharmacokinetic studies of the 111In-DOTA-8-Aoc-BBN[7–14]NH2 conjugate were determined in SCID mice bearing PC-3 tumors. Each mouse (average weight, 25 g) was injected with an aliquot (50–100 μL) of the HPLC-purified 111In-labeled peptide solution (55–75 kBq) through the tail vein. The mice were euthanized, and tissues and organs were excised at 15 min; 30 min; and 1, 4, 24, 48, and 72 h after injection. Subsequently, the tissues and organs were weighed and counted in a NaI well counter, and the %ID and %ID/g of each organ or tissue were calculated. The %ID in whole blood was estimated assuming a whole-blood volume of 6.5% of the total body weight. Receptor-blocking studies were also performed when excess (100 μg) BBN was administered to the animals along with the 111In-DOTA-8-Aoc-BBN[7–14]NH2 conjugate. All studies were conducted in accordance with protocols approved by the institutional animal care and use committee.
RESULTS
A series of DOTA-X-BBN[7–14]NH2 (X = 0, β-Ala, 5-Ava, 8-Aoc, or 11-Aun) analogs containing no spacer (0) and 3, 5, 8, and 11 carbon chain spacers was synthesized by solid-phase peptide synthesis using traditional Fmoc chemistry (Fig. 1). The metallation of the DOTA-X-BBN[7–14]NH2 analogs with both radioactive indium (111InCl3) and nonradioactive indium (InCl3) was performed using the conditions described in Figure 2. The 111In-DOTA-X-BBN[7–14]NH2 conjugates were obtained in 90%–95% radiochemical yields as single products. The 111In-DOTA-X-BBN[7–14]NH2 analogs were purified by reversed-phase HPLC before in vivo studies in order to remove corresponding excess nonmetallated DOTA-X-BBN[7–14]NH2 and obtain the high-specific-activity no-carrier-added 111In-DOTA-X-BBN[7–14]NH2 conjugates (Fig. 3). All the new DOTA-X-BBN[7–14]NH2 analogs and their nonradioactive indium complexes were purified by reversed-phase HPLC and characterized by electrospray mass spectrometry (Table 1).
In vitro competitive cell-binding assays performed on PC-3 cells for the In-DOTA-X-BBN[7–14]NH2 analogs against 125I-Tyr4-BBN demonstrated a high specificity and affinity (i.e., IC50 < 2.5 nmol/L; Table 2 and Fig. 4) for the analogs in which the spacer contains 3–8 carbon atoms.
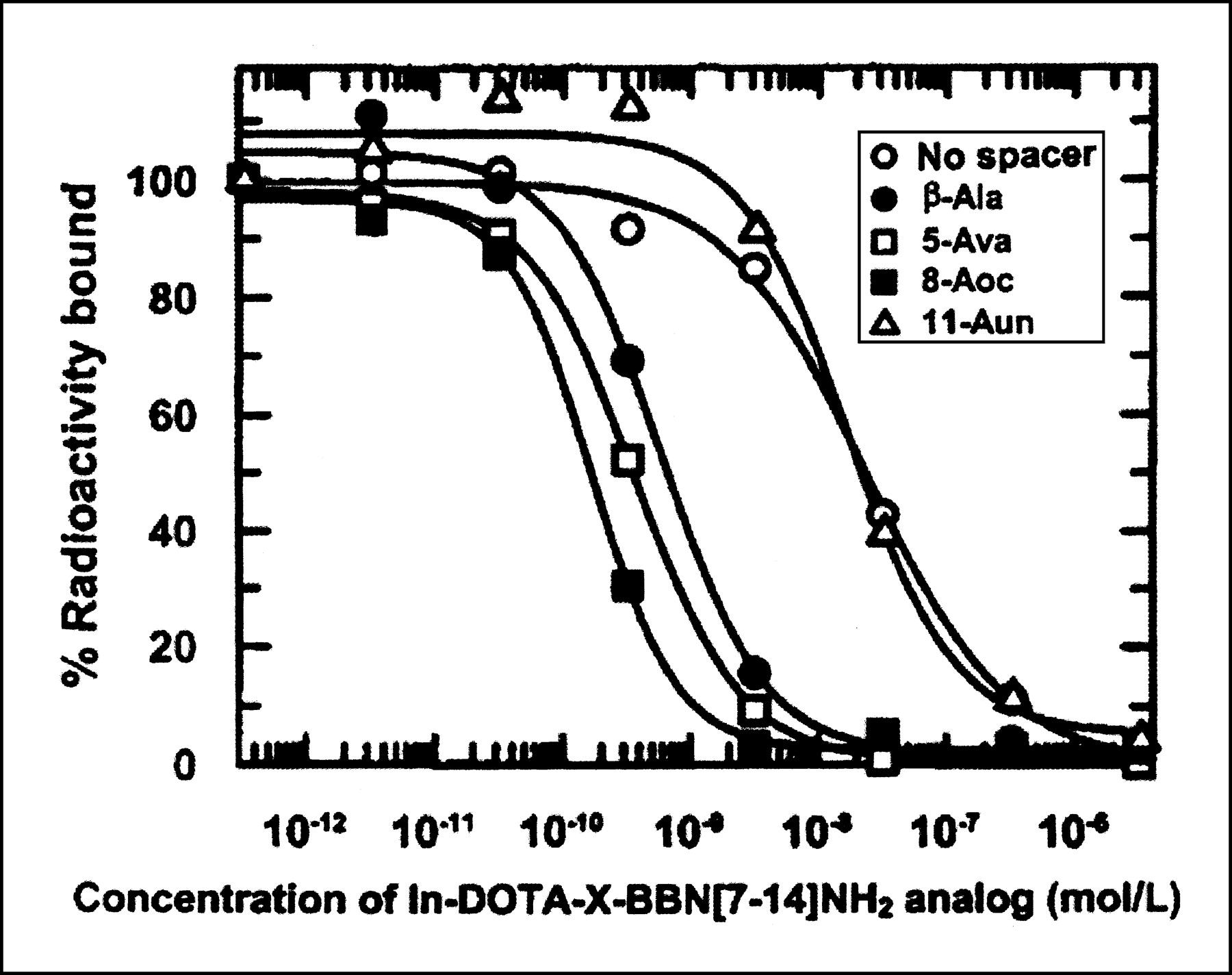
Competitive binding assay of In-DOTA-X-BBN[7–14]NH2 analogs vs. 125I-Tyr4-BBN in PC-3 cells.
IC50 Values of In-DOTA-X-BBN[7–14]NH2 Analogs vs. 125I-Tyr4-BBN in Human Prostate PC-3 Cells
Incubation of the 111In-DOTA-8-Aoc-BBN[7–14]NH2 analog with PC-3 cells demonstrated rapid internalization of the radioactivity, with measurable surface bound and internalized fractions (Fig. 5). The total residual radioactivity remained almost constant for more than 2 h (Fig. 6). Stability of the 111In-DOTA-8-Aoc-BBN[7–14]NH2 conjugate showed a human serum half-life of 17.3 h.

Internalization of 111In-DOTA-8-Aoc-BBN[7–14]NH2 in PC-3 cells (n = 5).

Efflux of 111In-DOTA-8-Aoc-BBN[7–14]NH2 in PC-3 cells (n = 5).
Results from in vivo biodistribution studies of the 111In-DOTA-X-BBN[7–14]NH2 analogs performed on healthy CF-1 mice at 1 h after injection are summarized in Table 3. All 111In-DOTA-X-BBN[7–14]NH2 conjugates cleared efficiently from the blood, with the primary route of radioactivity clearance being renal/urinary. Uptake of radioactivity in the pancreas increased as the lipophilicity of the conjugate increased; the value was 0.20 ± 0.04 %ID/g at 1 h after injection. Studies in which 100 μg of BBN were coadministered with the 111In-DOTA-8-Aoc-BBN[7–14]NH2 conjugate blocked nearly 98% of the pancreatic uptake.
111In-DOTA-X-BBN[7–14]NH2 Analog Biodistribution in CF1 Normal Mice at 1 Hour After Injection
The results from in vivo pharmacokinetic studies for the analog containing 8-Aoc spacer in PC-3 tumor-bearing SCID mice are summarized in Table 4. The radioactivity cleared efficiently from the bloodstream within 1 h after injection. Most of the activity was excreted through the renal/urinary pathway. A tumor uptake of 3.63 ± 1.11 %ID/g was observed at 1 h after injection, and retention over time was significant. Studies in which 100 μg of BBN were coadministered with the 111In-DOTA-8-Aoc-BBN[7–14]NH2 conjugate blocked approximately 64% of tumor uptake.
Pharmacokinetics of 111In-DOTA-8-Aoc-BBN[7–14]NH2 Analog in PC-3 Tumor–Bearing SCID Mice
DISCUSSION
There are 2 reasons for selecting carbon chain spacers in the DOTA-X-BBN[7–14]NH2 conjugates. First, these spacers will position the DOTA chelate sufficiently far from the GRP-receptor-binding region of BBN to prevent impedance of the binding affinity of the BBN[8–14]NH2 motif with GRP receptors, and second, these spacers will optimize pharmacokinetic properties by fine-tuning the lipophilicity of the DOTA-X-BBN[7–14]NH2 conjugate.
The biologic activity of the DOTA-X-BBN[7–14]NH2 analogs was determined through in vitro competitive cell-binding assays on PC-3 cells against 125I-Tyr4-BBN. Results from these cell-binding studies demonstrated that the In-DOTA-X-BBN[7–14]NH2 analogs containing β-Ala, 5-Ava, and 8-Aoc spacers bind to the GRP receptors on PC-3 cells with high specificity and affinity (i.e., IC50 < 2.5 nmol/L; Table 2 and Fig. 4). The In-DOTA-X-BBN[7–14]NH2 analogs containing no spacer and 11-Aun spacer exhibited high GRP-receptor specificity, but the affinity was more than 100-fold less than that of the 8-carbon-spacer analog (Table 2 and Fig. 4). These results were consistent with those obtained in our previous studies with 105Rh-S4-X-BBN[7–14]NH2 and 99mTc-N3S-X-BBN[7–14]NH2 conjugates—studies showing that increasing the hydrophobicity of the linker group to excessive levels will reduce the receptor-binding affinity (14,16,17). These cell-binding studies indicated that analogs of this design with β-Ala, 5-Ava, and 8-Aoc spacers have a dissociation constant suitable for further in vivo targeting studies of cells expressing GRP receptors.
For optimizing the diagnostic and therapeutic efficacy of a radioactive drug, it is important that the radioactivity be residualized in the tumor cell after drug localization at the tumor site (21,22,25,28,34). Internalization and efflux studies in PC-3 cells were performed with the 111In-DOTA-8-Aoc-BBN[7–14]NH2 analog, the analog that exhibited the lowest IC50 values with PC-3 cell–expressed GRP receptors (Table 2). GRP receptors are 7-TMS-G-protein-coupled receptors that can internalize agonist-receptor complexes (26,27,35). Since the BBN[7–14]NH2 peptide sequence confers agonistic GRP-receptor-binding capability, the GRP-receptor-mediated endocytosis of the 111In-DOTA-X-BBN[7–14]NH2 conjugates used in this study was not unexpected. Previous studies with 111In-cycloheximide-B-diethylenetriaminepentaacetic acid-8-Aoc-BBN[7–14]NH2, 105Rh-S4-X-BBN[7–14]NH2, and 99mTc-N3S-X-BBN[7–14]NH2 conjugates also demonstrated GRP-receptor-mediated trapping of radioactivity (9,14,16,17). In all these derivatives, the BBN[7–14]NH2 binding motif was present in the conjugate constructs. The specific intracellular trapping mechanism of the 111In activity in these PC-3 cells is not understood. It is likely that after internalization of 111In-DOTA-8-Aoc-BBN[7–14]NH2, lysosomal proteases degrade the 111In-DOTA-8-Aoc-BBN[7–14]NH2 conjugate to peptide fragments (36,37). The identity of 111In-containing fragments retained by the cell is not known, and further work is needed to identify the structure of the 111In-fragments to help elucidate the trapping mechanisms (36,38).
In vivo biodistribution studies of the 111In-DOTA-X-BBN[7–14]NH2 analogs were performed on healthy CF-1 mice. Table 3 summarizes the results of the biodistribution for the 111In-DOTA-X-BBN[7–14]NH2 analog series on CF-1 mice at 1 h after injection. Because the pancreas is the primary normal tissue in these animals that expresses a high density of bloodstream-accessible GRP receptors, the accumulation of 111In in the pancreas is a direct reflection of the efficacy of radiolabeled BBN analogs for in vivo targeting of cell-surface-expressed GRP receptors (10). In the present study, all 111In-DOTA-X-BBN[7–14]NH2 conjugates cleared efficiently from the blood, with <0.35 %ID/g remaining in the blood at 1 h after injection (Table 3). The primary route of radioactivity clearance from the body is renal/urinary, with more than 70 %ID observed in the urine at 1 h after injection for analogs with no spacer or with the β-Ala, 5-Ava, and 8-Aoc spacers (Table 3). 111In-DOTA-11-Aun-BBN[7–14]NH2 is cleared to a significantly greater extent through the hepatobiliary route (averaging 26 %ID at 1 h after injection) than are the other derivatives (Table 3); this finding is assumed to be related to the higher lipophilicity of this analog. Retention of radioactivity in the kidneys after administration of all these 111In-DOTA-X-BBN[7–14]NH2 analogs, at <2.5 %ID/g at 1 h after injection (Table 3), was lower than that of most other small peptide-based radioactive drugs (32,33,39). Uptake of radioactivity in the pancreas increased as the lipophilicity of the conjugate increased; uptake ranged from 0.20 ± 0.04 %ID/g at 1 h after injection for the analog with no spacer (the least lipophilic analog in the series) to 26.97 ± 3.97 %ID/g at 1 h after injection for the analog using the 8-Aoc spacer (second most lipophilic in the series) (Table 3). Biodistribution studies in which an excess (100 μg) of nonradioactive BBN was coinjected with the 111In-DOTA-8-Aoc-BBN[7–14]NH2 in CF-1 mice demonstrated that uptake in the pancreas is specific and receptor mediated. These studies showed that uptake of this analog was reduced to 0.43 ± 0.10 %ID/g in the pancreas at 1 h after injection (Table 3)—nearly 98% lower than pancreatic uptake of 111In-DOTA-8-Aoc-BBN[7–14]NH2 when no blocking agent was coinjected (Table 3). The pharmacokinetics and pancreatic accumulation of this short series of 111In-DOTA-X-BBN[7–14]NH2 analogs indicates that constructs in which the spacer group (X) ranges from 5 to 8 carbon atoms appear to be the most promising for further development as GRP-receptor-targeting radiopharmaceuticals.
On the basis of these observations, we selected the analog containing 8-Aoc spacer for in vivo pharmacokinetic studies on PC-3 tumor-bearing SCID mice. Table 4 summarizes the results of the pharmacokinetic studies on PC-3 tumor-bearing SCID mice. The 111In conjugate cleared efficiently from the bloodstream within 1 h after injection. For example, 0.60 ± 0.39 %ID/g remained in the blood at 1 h after injection. Most of the activity was excreted through the renal/urinary pathway (i.e., 68.6 ± 7.0 %ID at 1 h after injection and 87.2 ± 4.3 %ID at 24 h after injection), with the remainder of the radioactivity being excreted through the hepatobiliary pathway. Receptor-mediated tumor targeting of the PC-3-xenografted SCID mice resulted in tumor uptake and retention values of 3.63 ± 1.11, 1.56 ± 0.45, 0.68 ± 0.24, and 0.34 ± 0.10 %ID/g at, respectively, 1, 24, 48, and 72 h residualization (i.e., 43%, 19%, and 9% of the radioactivity in the tumor at 1 h after injection was retained at, respectively, 24, 48, and 72 h after injection). At 1 h after injection, tumor-to-blood and tumor-to-muscle ratios of approximately 6:1 and 45:1 were achieved. Although 111In in the tumor was not optimal, the fact that the half-life of retention was approximately 24 h indicates that the 111In- DOTA-8-Aoc-BBN[7–14]NH2 conjugate was internalized agonistically. The observation that coadministration of 100 μg of BBN along with the 111In-DOTA-8-Aoc-BBN[7–14]NH2 conjugate blocked about 98% of the receptor-mediated radioactivity uptake in pancreas but only 64% in tumor may be related to the fact that these human tumors are supported by murine vasculature architecture (Table 4). Student t tests of blocked-versus-unblocked in vivo tumor uptake showed a t value of 4.67 and a P value of 0.002, indicating a statistical difference among datasets. Accumulation of activity in the kidneys was significant (i.e., 5.66 ± 1.33 %ID/g at 1 h); however, the level of activity decreased with time (i.e., to 1.18 ± 0.14 %ID/g at 24 h and 0.48 ± 0.97 %ID/g at 48 h). It may be possible to reduce the initial accumulation of 111In conjugate in the kidney or accelerate its washout by administration of excess lysine or other positively charged molecules (38). Accretion of 111In-DOTA-8-Aoc-BBN[7–14]NH2 in other tissues or organs was minimal.
The DOTA chelator framework can form complexes with a variety of trivalent and divalent radiometals to produce radiolabeled bioconjugates with high in vitro and in vivo stability (29,30). Radiometallation of the DOTA-X-BBN[7–14]NH2 analogs with particle-emitting radionuclides available in high specific activities (including 90Y+3, 64Cu+2, 177Lu+3, 149Pm+3, and 166Ho+3) should produce GRP-receptor-avid conjugates with potential for therapeutic applications (31,40).
The results of studies with these 111In-DOTA-X-BBN[7–14]NH2 constructs can be used as a basis to design potential therapeutic analogs labeled with β-particle-emitting radionuclides. In some cases, substitution of another metal for 111In+3 may not significantly modify the in vitro and in vivo properties of DOTA bioconjugates (29,33). However, the physicochemical properties of the appended radiometallated DOTA moieties may be metal specific and can measurably alter the receptor-binding and pharmacokinetic properties of the bioconjugate (2,33). Clearly, it will be important to formulate DOTA-X-BBN[7–14]NH2 analogs complexed with therapeutically useful radiometals and to individually evaluate their utility for specific in vivo targeting of GRP-receptor-expressing cancers.
CONCLUSION
The in vitro studies with PC-3 androgen-independent human prostate cancer cells showed that the 111In-DOTA-X-BBN[7–14]NH2 analogs in which the spacer group (X) is β-Ala, 5-Ava, and 8-Aoc exhibit high binding affinities (i.e., IC50 values ranging from 0.6 to 2.4 nmol/L) for GRP receptors. In vivo studies demonstrated that analogs in which X = 5-Ava and 8-Aoc exhibit high specific localization in the pancreas, normal GRP-receptor-expressing tissue, and efficient clearance from the blood primarily through the renal/urinary pathway. The 111In-DOTA-8-Aoc-BBN[7–14]NH2 analog binds to GRP receptors acting agonistically, resulting in receptor-mediated endocytosis. These results suggest that the 111In-DOTA-X-BBN[7–14]NH2 construct, in which X = a tether ranging between a 5-carbon and 8-carbon spacer, may form the basis for development of radiometallated diagnostic or therapeutic radiopharmaceuticals for selective in vivo targeting of GRP-receptor-expressing cancers.
Acknowledgments
This article is the result of work supported with resources and the use of facilities at the Harry S. Truman Memorial VA Hospital and the Departments of Radiology and Internal Medicine of the University of Missouri-Columbia School of Medicine. This work was also funded in part by grant RPG-99-331-01-CDD from the American Cancer Society and grants DHHS-1P20-CA72942 and DHHS-RO1-CA72942 from the National Cancer Institute. The University of Missouri holds a patent on the BBN agonists cited in this article. T.J. Hoffman, G.L. Sieckman, and W.A. Volkert are coinventors in this patent.
Footnotes
Received Jun. 17, 2002; revision accepted Nov. 27, 2002.
For correspondence or reprints contact: Wynn A. Volkert, PhD, Radiopharmaceutical Sciences Institute, 143 Major Hall, DC200.0 HSC, University of Missouri-Columbia, Columbia, MO 65212.
E-mail: VolkertW{at}health.missouri.edu
REFERENCES
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Introduction of an 8-Aminooctanoic Acid Linker Enhances Uptake of 99mTc-Labeled Lactam Bridge-Cyclized {alpha}-MSH Peptide in Melanoma
- Synthesis and In Vitro and In Vivo Evaluation of Hypoxia-Enhanced 111In-Bombesin Conjugates for Prostate Cancer Imaging
- PET of Tumors Expressing Gastrin-Releasing Peptide Receptor with an 18F-Labeled Bombesin Analog
- Effects of the Amino Acid Linkers on the Melanoma-Targeting and Pharmacokinetic Properties of 111In-Labeled Lactam Bridge-Cyclized {alpha}-MSH Peptides
- 18F-Labeled Bombesin Analog for Specific and Effective Targeting of Prostate Tumors Expressing Gastrin-Releasing Peptide Receptors
- Small-Animal PET of Tumors with 64Cu-Labeled RGD-Bombesin Heterodimer
- Bombesin Receptor Antagonists May Be Preferable to Agonists for Tumor Targeting
- In Vivo Evaluation and Small-Animal PET/CT of a Prostate Cancer Mouse Model Using 64Cu Bombesin Analogs: Side-by-Side Comparison of the CB-TE2A and DOTA Chelation Systems
- [64Cu-NOTA-8-Aoc-BBN(7-14)NH2] targeting vector for positron-emission tomography imaging of gastrin-releasing peptide receptor-expressing tissues
- Androgen-Dependent Expression of the Gastrin-Releasing Peptide Receptor in Human Prostate Tumor Xenografts
- 18F-Labeled Bombesin Analogs for Targeting GRP Receptor-Expressing Prostate Cancer
- GRP Receptor-Targeted PET of a Rat Pancreas Carcinoma Xenograft in Nude Mice with a 68Ga-Labeled Bombesin(6-14) Analog
- Candidates for Peptide Receptor Radiotherapy Today and in the Future
- A New Prostate Carcinoma Binding Peptide (DUP-1) for Tumor Imaging and Therapy
- microPET and Autoradiographic Imaging of GRP Receptor Expression with 64Cu-DOTA-[Lys3]Bombesin in Human Prostate Adenocarcinoma Xenografts
- PET Imaging of GRP Receptor Expression in Prostate Cancer