


Abstract
4,4′-Bis-1-hydroxy-2-(4-methylpiperidin-1-yl)ethyl-biphenyl (A-4), a tertiary amine analog of hemicholinium-3 (HC-3), is an inhibitor of the sodium-dependent high-affinity choline uptake (HACU) system. We have evaluated 4-[1-hydroxy-2-(4-18F-fluoromethylpiperidin-1-yl)ethyl]-4′-[1-hydroxy-2-(4-methylpiperidin-1-yl)ethyl]biphenyl (18F-FA-4) and 4-[1-hydroxy-2-(4-11C-methylpiperazin-1-yl)ethyl]-4′-[1-hydroxy-2-(4-methylpiperidin-1-yl)ethyl]biphenyl (11C-pipzA-4), an 18F- and a 11C-labeled derivative of A-4 as potential in vivo tracers for the HACU system. Methods: The biodistribution of both compounds was determined in mice, and the intracerebral distribution was visualized by ex vivo and in vitro autoradiography. The in vitro affinity of the compounds was determined by a displacement study with 3H-HC-3 on mice brain slices. Results: In mice, both tracers show a high and persistent brain uptake. In vitro autoradiography shows binding to the striatum, whereas ex vivo autoradiography shows homogeneous binding throughout the brain. FA-4 and pipzA-4 inhibited the 3H-HC-3 binding with a 50% inhibitory concentration of 57 nmol/L and 320 nmol/L, respectively. Conclusion: The evaluated compounds have affinity for HACU and show high uptake in the brain. In vitro binding of the compounds to the striatum cannot be inhibited by the presence of HC-3, whereas binding of HC-3 was inhibited by the presence of both FA-4 and pipzA-4, suggesting allosteric binding.
Studies of choline transport have revealed the existence of both a low-affinity sodium-independent and a high-affinity sodium-dependent choline transport system (1). The sodium-independent low-affinity system is expressed in most cells, is thought to be of importance at high choline concentrations, and provides choline for phosphatidylcholine synthesis. The sodium-dependent high-affinity choline uptake (HACU) system is located specifically in presynaptic cholinergic nerve terminals and was found to be the regulating and rate-limiting step in the synthesis of acetylcholine (2–4). Thus, HACU is a marker of the functional status of the cholinergic presynaptic terminals (5). It has been found to be upregulated in brains of parkinsonian patients (6), whereas in brains of patients with Alzheimer’s disease there are reports of both downregulation (7) and upregulation (8). The latter conflicting results may be explained by the observation that activity related changes in the choline uptake undergo a rapid reversal in postmortem tissues (9). In rats, it was further found that HACU is upregulated after introduction of 6-hydroxydopamine lesions (10) and is inhibited by low concentrations of amyloid-β (11). The high-affinity choline transporters of Caenorhabditis elegans, rat (12), and man (13) have recently been characterized. The complementary DNA of the human high-affinity choline transporter encodes for a 580-amino acid protein and is distantly related to the Na+-coupled glucose transporter (SGLT) family.
The golden standard for studying the HACU system in vitro is hemicholinium-3 (HC-3), a reversible and competitive inhibitor of HACU. Because HC-3 is a quaternary ammonium salt, it is not able to cross the blood-brain barrier (BBB) and radiolabeled quaternary ammonium HC-3 derivatives cannot be used for in vivo visualization of the HACU transporter. Affinity studies of HC-3 analogs on rat brain slices revealed that 4,4′-bis-[1-hydroxy-2-(4-methylpiperidin-1-yl)ethyl]biphenyl ([A-4] Fig. 1), a 4-methylpiperidine analog of HC-3, inhibits the HACU system competitively and reversibly. Moreover, this compound was found to be more potent than HC-3 as a HACU inhibitor in neuroblastoma cells (14), whereas the reverse was found in rat brain synaptosomes (15) and neuromuscular junction preparations (16). Being a tertiary amine, A-4 should be able to cross the BBB and a radiolabeled derivative of A-4, therefore, potentially allows the in vivo investigation of the HACU system. For this reason, we have synthesized 2 derivatives of A-4 labeled with 18F and 11C, respectively—namely, 4-[1-hydroxy-2-(4-18F-fluoromethylpiperidin-1-yl)ethyl]-4′-[1-hydroxy-2-(4-methylpiperidin-1-yl)ethyl]biphenyl (18F-FA-4) and 4-[1-hydroxy-2-(4-11C-methylpiperazin-1-yl)ethyl]-4′-[1-hydroxy-2-(4-methylpiperidin-1-yl)ethyl]-biphenyl (11C-pipzA-4) (Fig. 1).
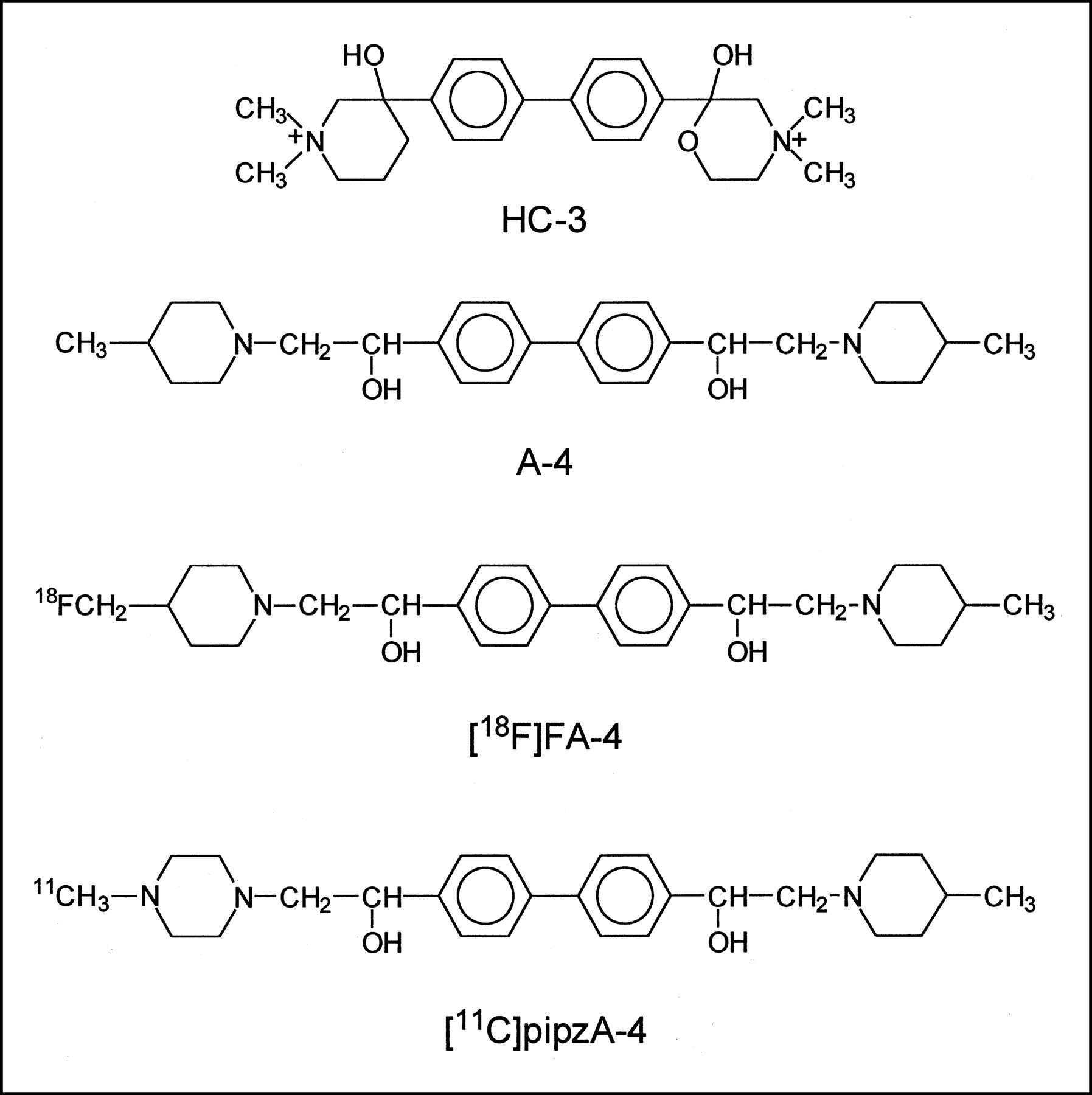
Structure of HC-3, A-4, 18F-FA-4, and 11C-pipzA-4.
Both agents have been obtained as a mixture of 4 stereoisomers that were not further separated, as a study of Scheff et al. (14) showed that the different isomers of A-4 do not show any difference in potency or reversibility for inhibition of choline uptake. The radiosynthesis of these tracers has been described elsewhere (17).
The cholinergic system is severely affected in Alzheimer’s disease and several specific radioligands have been developed for the in vivo visualization with PET of muscarinic cholinergic receptors, nicotinic acetylcholine receptors, vesicular acetylcholine transporters, and acetylcholinesterase (18). However, a suitable radiolabeled ligand for the visualization of HACU with PET would provide specific information with regard to the functional status of the presynaptic cholinergic nerve terminals in healthy individuals and in patients with Alzheimer’s disease.
Here we report the affinity of FA-4 and pipzA-4 for the HACU system measured by a displacement experiment with 3H-HC-3 on rat brain slices. Both tracers were evaluated in normal NMRI mice, with special attention to their ability to cross the BBB. Autoradiographic in vitro and ex vivo experiments were performed on coronal mouse brain sections through the striatal region to investigate the distribution pattern of the tracers throughout the brain.
MATERIALS AND METHODS
Materials
Fentanyl (50 μg/mL) was obtained from Janssen-Cilag (Beerse, Belgium), and ketamine (Ketalar, 50 mg/mL) was obtained from Parke-Davis (Orléans, France). 3H-HC-3 (4.44 TBq/mmol) was obtained from Dupont (Boston, MA), and HC-3 was obtained from RBI Sigma (Natick, MA). A-4, FA-4, pipzA-4, 18F-FA-4, and 11C-pipzA-4 were synthesized as described (17).
Biodistribution in Mice
Solutions of 18F-FA-4 and 11C-pipzA-4 obtained after high-performance liquid chromatography (HPLC) purification were diluted with normal saline to a concentration of 370 kBq/mL and 37 MBq/mL, respectively. The biodistribution of both tracers was determined in normal male NMRI mice (body mass, 30–40 g). The animal studies were performed according to the Belgian code of practice for the care and use of animals. The mice were sedated by intramuscular injection of 0.1 mL of a solution containing 34 μg/mL fentanyl and 16 mg/mL ketamine. A volume of 0.1 mL of the diluted tracer solution was injected in the mice via a tail vein. The mice were killed by decapitation at 2, 10, or 30 min after injection (n = 5 at each time point). Blood was collected in a tared tube and weighed. All organs and other body parts were dissected and weighed, and their activity was counted in a 3-in (7.62 cm) NaI(Tl) well crystal, coupled to a multichannel analyzer (Wallac, Turku, Finland). Corrections were made for background radiation and physical decay during counting. Results are expressed as percentage of injected dose (%ID), equal to the sum of the net counts in all organs and, where possible, as percentage of injected dose per mass of tissue (%ID/g). For calculation of total blood activity, blood mass was assumed to be 7% of the body mass.
Metabolism of 18F-FA-4 in Mice
After intravenous injection of 7.4 MBq 18F-FA-4 in normal mice, metabolites of the tracer were determined in urine and blood, which was collected 10 and 30 min after injection. Blood was centrifuged for 5 min at 1,500g and plasma was isolated. Both urine and plasma were filtered through a 0.22-μm membrane filter (Acrodisc; Gelman Sciences, Ann Arbor, MI) and the filtrates were analyzed by HPLC. The eluate was collected in 1-mL fractions and the activity in each fraction was counted in a 3-in NaI(Tl) well crystal, coupled to a multichannel analyzer (Wallac). The amount of metabolites was expressed as percentage of total 18F activity in the sample.
3H-Hemicholinium Binding Inhibition Curve
The 3H-hemicholinium binding inhibition curve was determined following the procedure described by Sandberg and Coyle (1). A normal FVB mouse was sacrificed by decapitation, and the brain was rapidly removed and frozen in 2-methylbutane (cooled to −25°C with liquid nitrogen). Serial 16-μm frozen sections were cut in the coronal plane and mounted onto poly(lysine)-coated slides (Poly-Prep; Sigma Diagnostics, St. Louis, MO). The slices were air dried and stored at −70°C until used. For the determination of the affinity for the HACU system, the slide-mounted tissue sections were equilibrated to room temperature and incubated for 30 min with 400 μL of assay medium containing 50 mmol/L glycylglycine buffer, pH 7.8, 200 mmol/L NaCl, 10 nmol/L 3H-HC-3, and either HC-3, A-4, FA-4, or pipzA-4 in various concentrations (1 nmol/L, 5 nmol/L, 10 nmol/L, 50 nmol/L, 100 nmol/L, 500 nmol/L, 1 μmol/L, 5 μmol/L, 10 μmol/L). After incubation, slides were washed for 2 min in ice-cold 50 mmol/L glycylglycine buffer, pH 7.8, containing 200 mmol/L NaCl, followed by a 15-s rinse in ice-cold distilled water. This washing procedure was repeated once. For autoradiography studies, the sections were air dried and autoradiograms were obtained by exposing the sections to a 3H-sensitive film (Hyperfilm-3H; Amersham, Buckinghamshire, U.K.) for 2 wk. Quantitative autoradiographic analysis of the sections was performed by densitometric scanning of the different brain areas and analysis with suitable software (Pharmacia Biotech, Uppsala, Sweden). 3H-HC-3 binding, in the presence of different concentrations of the derivatives, was measured bilaterally for each concentration. The 3H-HC-3 inhibition curve was obtained by plotting the amount of 3H-HC-3 binding in the presence of increasing concentrations of HC-3, A-4, FA-4, or pipzA-4. The 50% inhibitory concentration (IC50) values were determined by linear regression of the linear part of the 3H-HC-3 inhibition curve.
In Vitro Experiments
Brain coronal sections of normal FVB mice obtained as described above were incubated for 30 min at room temperature with 400 μL of assay medium containing 74 kBq 18F-FA-4 or 0.7 MBq 11C-pipzA-4 in 50 mmol/L glycylglycine buffer, pH 7.8, containing 200 mmol/L NaCl or LiCl, followed by the washing procedure as described for the determination of the 3H-HC-3 binding inhibition curve. Nonspecific binding of 18F-FA-4 or 11C-pipzA-4 was determined by incubation of the slices with the radiotracers in the presence of 10 μmol/L HC-3 or 10 μmol/L A-4 in the same conditions. The sections were dried and exposed to a high-performance storage phosphor screen (Packard, Meriden, CT) that was scanned in a Phosphor Imager Scanner (Cyclone; Packard). Quantification of different brain areas was performed with Optiquant software (Packard).
Ex Vivo Experiments
To determine the in vivo intracerebral distribution, 0.1 mL of a saline solution containing either 3.7 MBq 18F-FA-4 or a mixture of 3.7 MBq 18F-FA-4 and A-4 (20 μmol/kg) was injected intravenously in sedated NMRI mice. The animals were killed by decapitation at 10 min after injection, and the brain was rapidly removed and immediately frozen in 2-methylbutane (cooled to −25°C with liquid nitrogen). Serial 50-μm frozen sections were cut in the coronal plane and mounted on microscope slides (Knittel, Braunschweig, Germany). The sections were air dried at 50°C and exposed overnight to a high-performance storage phosphor screen (Packard) that was scanned in a Phosphor Imager Scanner (Cyclone).
RESULTS
Biodistribution in Mice
The biologic behavior of 18F-FA-4 and 11C-pipzA-4 was assessed by studying their biodistribution in mice. In Table 1 the distribution of the radioactivity at 2, 10, and 30 min after injection, expressed as %ID and as %ID/g, is given for the different organs and urine. Clearance occurred mainly via the liver and, to a lower extent, via the kidneys. High uptake was found in lung, heart, kidney, and liver.
Biodistribution of 18F-FA-4 and 11C-pipzA-4 in Normal Mice at 2, 10, and 30 Minutes After Injection
Metabolism of 18F-FA-4 in Mice
HPLC analysis of the urine obtained 10 and 30 min after injection revealed that 18F-FA-4 was present in the intact form at 93% 10 min after injection and at 92% 30 min after injection. HPLC analysis of plasma showed that 18F-FA-4 was present in the intact form at 88% 10 min after injection and at 80% 30 min after injection.
Affinity for HACU System
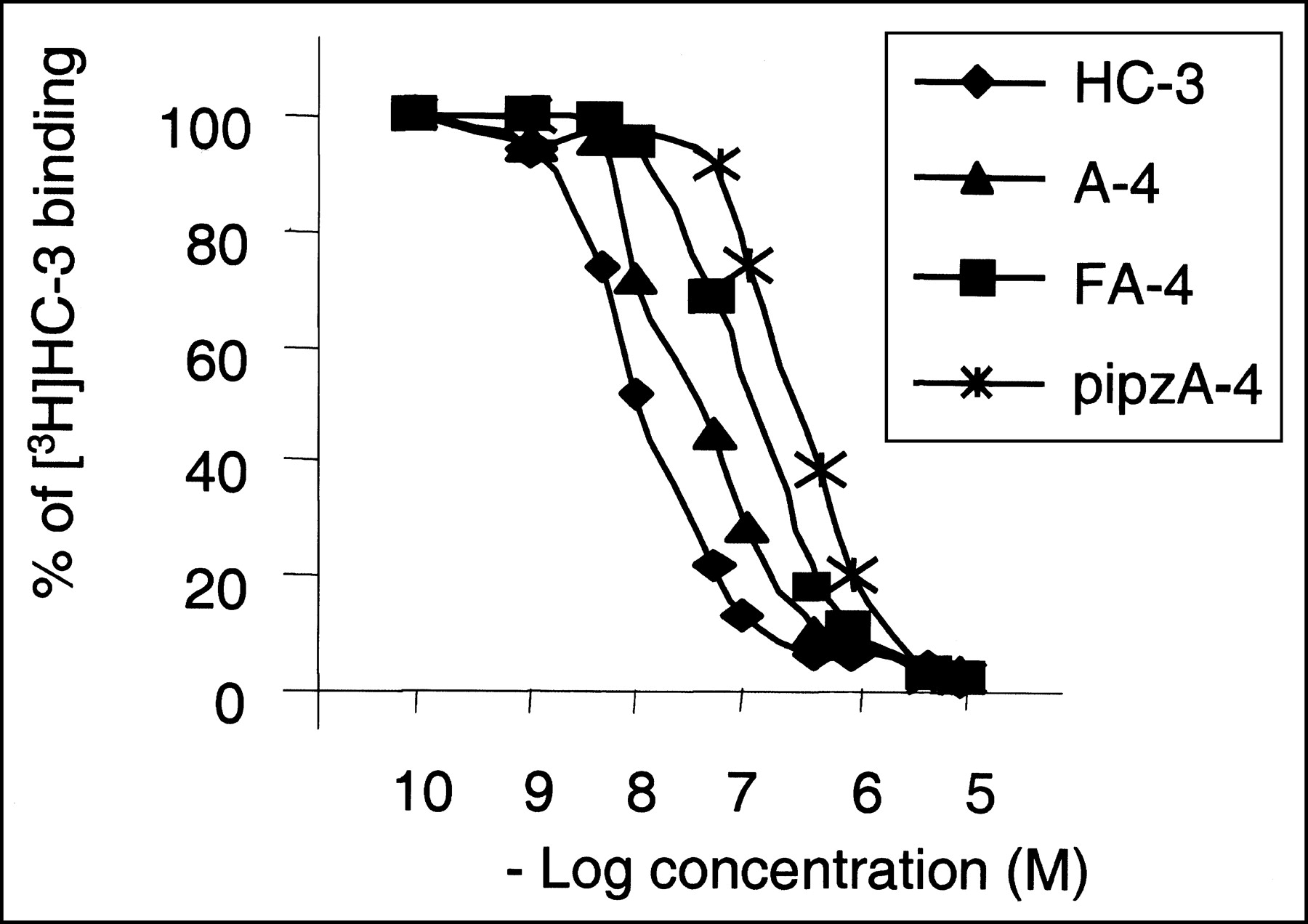
After quantitative autoradiographic analysis of the brain sections, the 3H-HC-3 inhibition curves in the presence of HC-3, A-4, FA-4, or pipzA-4 was obtained and the IC50 values were determined graphically by linear regression. Figure 2 shows the inhibition of 3H-HC-3 binding as a function of increasing concentrations of the different unlabeled compounds. The resulting IC50 values are as follows: HC-3, 10 nmol/L; A-4, 32 nmol/L; FA-4, 57 nmol/L; and pipzA-4, 320 nmol/L.

Determination of IC50 values of HC-3, A-4, FA-4, and pipzA-4 in mouse brain. Inhibition curves of 10 nmol/L 3H-HC-3 binding to mouse brain slices in presence of unlabeled HC-3, A-4, FA-4, or pipzA-4 are shown.
In Vitro Experiments
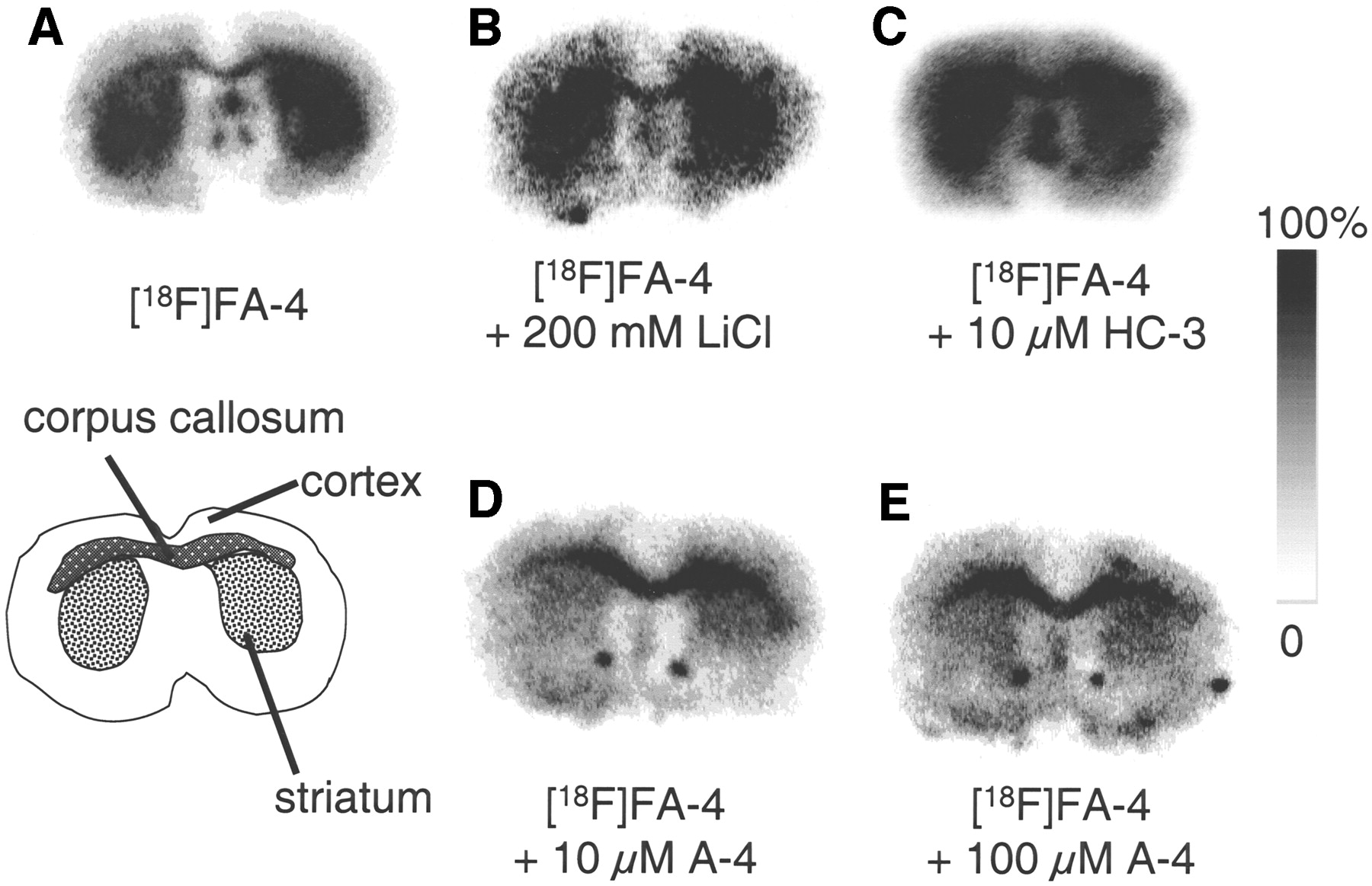
Incubation of normal mouse brain sections with 18F-FA-4 resulted in the autoradiograms shown in Figure 3. They clearly reveal a high binding of the tracer to the striatum. The autoradiograms also revealed binding of the tracer to the corpus callosum, a white matter structure.

Autoradiograms of FVB mouse coronal brain sections incubated with 18F-FA-4 in standard incubation buffer (A), 18F-FA-4 with incubation buffer containing LiCl instead of NaCl (B), and 18F-FA-4 in presence of 10 μmol/L HC-3 (C), 10 μmol/L A-4 (D), or 100 μmol/L A-4 (E). Images are scaled to maximum pixel intensity within each individual image.
When NaCl in the incubation medium was substituted by LiCl, 18F-FA-4 still binds to the striatum. Addition of HC-3 to the 18F-FA-4 solution (final concentration, 10 μmol/L) did not inhibit the binding of 18F-FA-4 to the striatum and the corpus callosum. When the same experiment was performed with A-4 as a displacing agent, binding of 18F-FA-4 to the striatum was inhibited, whereas binding to the corpus callosum was not inhibited, resulting in a decreased striatum-to-cortex ratio (Table 2).
Ratios of In Vitro Binding of 18F-FA-4 to Mouse Brain Structures in Presence of A-4
Incubation of brain sections of normal mouse with 11C-pipzA-4 resulted in the autoradiograms shown in Figure 4. The binding of 11C-pipzA-4 to the striatum was higher than the binding to the cortex. In analogy with our findings with 18F-FA-4, addition of increasing concentrations of HC-3 (10 μmol/L, 100 μmol/L, 1 mmol/L, and 10 mmol/L) to the incubation medium did not result in an inhibition of 11C-pipz-A-4 binding to the striatum.

Autoradiograms of FVB mouse brain sections incubated with 11C-pipzA-4 in standard incubation buffer (A) and in presence of 10 μmol/L HC-3 (B), 100 μmol/L HC-3 (C), 1 mmol/L HC-3 (D), or 10 mmol/L HC-3 (E). Images are scaled to maximum pixel intensity within each individual image.
Ex Vivo Experiments
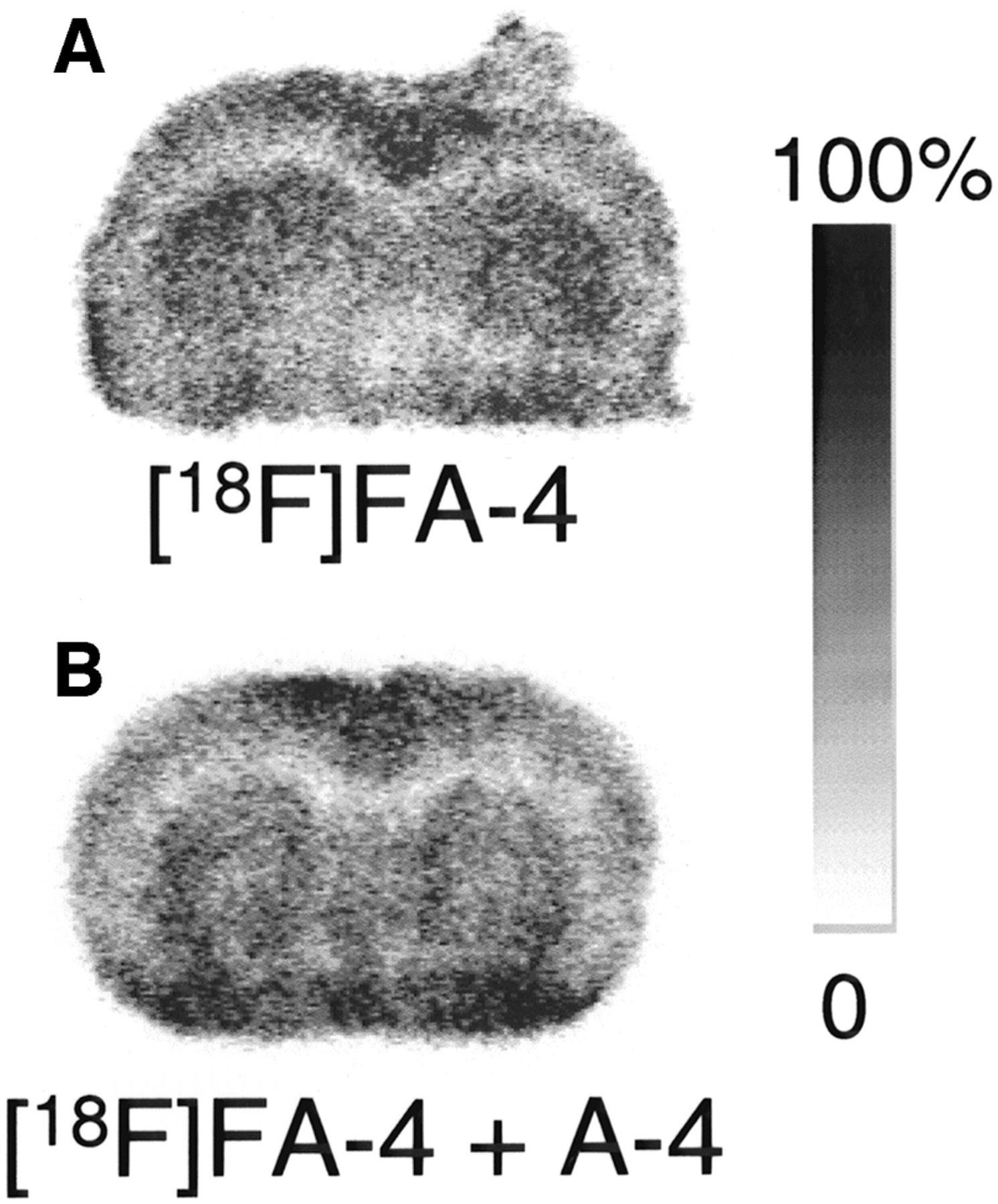
Autoradiograms of brain sections obtained 10 min after intravenous injection of 18F-FA-4 in a normal mouse are shown in Figure 5. 18F-FA-4 binds to the striatum but, to a lesser extent, also to the cortex, in contrast with our in vitro observations. No binding to the corpus callosum was found, indicating that in vitro binding of 18F-FA-4 to this structure was nonspecific. To determine whether in vivo binding of 18F-FA-4 in the brain could be inhibited by A-4, a solution containing both 18F-FA-4 and A-4 (20 μmol/kg) was injected in a normal mouse. Radioactivity measured in the brain of the mouse injected with the solution containing a mixture of 18F-FA-4 and A-4 (3.5 %ID) was higher than that in the brain of the mouse injected with only 18F-FA-4 (1.4 %ID).

Autoradiograms of 50-μm NMRI mouse coronal brain sections taken 10 min after intravenous injection of 18F-FA-4 (A) or a solution containing a mixture of 18F-FA-4 and A-4 (20 μmol/kg) (B). Images are scaled to maximum pixel intensity within each individual image.
DISCUSSION
The biodistribution of 18F-FA-4 and 11C-pipzA-4 in mice reveals that both compounds behave similarly. They are cleared from the blood at comparable rates. The uptake in the heart is higher (2.5 %ID, 10 min after injection) in comparison with the uptake of a tracer that is retained in the heart as a function of myocardial blood flow (e.g., 99mTc-methoxyisobutylisonitrile, 1.2 %ID). The excretion in the urine is minimal for both radiotracers, but higher amounts of activity are found in the kidneys, which may indicate binding to the choline transporter in renal tubules. Choline undergoes a net reabsorption from the glomerular filtrate in mammalian renal tubules, and this uptake is specific, sodium independent, but chloride dependent, and located at the luminal cell membrane as shown earlier (19,20). HC-3 appears to be a potent inhibitor of choline uptake in the kidney (19), as revealed by Grunewald et al. (20), and the accumulation of the investigated tracers in the kidney may be due to binding of 18F-FA-4 and 11C-pipzA-4 to the choline transporter. A similar limited excretion in the urine (2.4 %ID, 10 min after injection) and higher amounts of activity retained in the kidneys (16%, 10 min after injection) were also found for 11C-labeled choline (G. Bormans, unpublished data, 1999).
Both 18F-FA-4 and 11C-pipzA-4 show a high brain uptake that increases between 2 and 10 min after injection and remains at the same level at 30 min. The brain uptake of 18F-FA-4 and 11C-pipzA-4 is comparable with that of a conventional cerebral perfusion tracer such as 99mTc-hexamethylpropyleneamine oxime (1.2 %ID).
Both tracers show a high uptake in the lungs that decreases as a function of time after injection. This uptake does not parallel the blood concentration, which indicates that it is not associated with the blood pool. The lung retention is unexpected, as only low levels of expression of mRNA of the high-affinity choline transporter were found in the rat lung (12). Because both 18F-FA-4 and 11C-pipzA-4 are lipophilic amines, it may not be excluded that the observed lung retention is caused by binding on endothelial cell membranes during the first pass through the pulmonary circulation, in analogy with other lipophilic amines such as isopropyl iodoamphetamine (21). Analyses of the urine and plasma indicated that 18F-FA-4 is not extensively metabolized. A degradation product, formed in small amounts, appeared to be more polar than 18F-FA-4, but its structure has not been elucidated.
The IC50 values of A-4 determined with 3H-HC-3 binding inhibition studies on mouse brain slices are of the same order of magnitude as that of HC-3 (32 nmol/L and 10 nmol/L, respectively). These values are comparable with the IC50 values obtained after determination of inhibited 14C-choline uptake in rat brain synaptosomes as shown by Chatterjee et al. (15) (IC50 values for HC-3 and A-4 were 18 nmol/L and 36 nmol/L, respectively). The introduction of the fluoro substituent (FA-4) is well tolerated, as indicated by the small increase in the IC50 value of the FA-4 derivative (57 nmol/L) in comparison with A-4 (32 nmol/L). However, replacement of a piperidine ring by a piperazine ring in pipzA-4 has a pronounced effect and reduces the affinity by a factor of 10 (IC50 = 320 nmol/L). All 3 ligands revealed a lower affinity than that of HC-3. Nevertheless, the affinity of FA-4 and pipzA-4 may be high enough to allow in vivo visualization of the HACU system.
To determine whether 18F-FA-4 and 11C-pipzA-4 bind specifically to the HACU receptor, coronal mouse brain sections of the striatal region were incubated with the tracer, and tracer binding was evaluated with autoradiography. The resulting autoradiograms show binding of 18F-FA-4 predominantly to the striatum (Fig. 3). Unexpectedly, a comparable binding was obtained when NaCl was substituted by LiCl in the incubation medium. Binding of 18F-FA-4 to the striatum in the absence of NaCl suggests a sodium-independent binding of 18F-FA-4, whereas the binding of HC-3 to synaptosomal preparations was found to be highly sodium dependent.
Addition of HC-3 to the incubation medium did not prevent 18F-FA-4 binding to the striatum but, reversely, FA-4 was able to inhibit 3H-HC-3 binding to the striatum in a dose-dependent manner. Addition of 10 μmol/L A-4 to the 18F-FA-4 incubation solution inhibited binding of 18F-FA-4 to the striatum, but binding to the corpus callosum was not inhibited (Table 2). Inhibition of 18F-FA-4 binding to the striatum by A-4 thus indicates saturable binding to this structure. Inhibition of 18F-FA-4 binding to the striatum by A-4 but not by HC-3 suggests that FA-4 and HC-3 bind allosterically to the HACU protein.
Some pharmacologic studies have indicated that different subtypes of transporter may exist, at least with respect to pharmacologic sensitivity and regional specificity. For example, earlier studies with choline mustard, an irreversible inhibitor of the HACU transporter, revealed that only about half of the HC-3 binding sites were sensitive to alkylation by choline mustard (22,23). This suggests the presence of 2 distinct pools of HC-3 binding sites. It is also possible that FA-4 binding causes a conformational change in the transporter, which prevents HC-3 binding to the HACU receptor, but that HC-3 binding does not alter the FA-4 binding. The existence of both a slow and a fast dissociating conformational state of the HACU hemicholinium binding site has been suggested by Chatterjee et al. (15).
In vitro autoradiography experiments with 11C-pipzA-4 show predominant binding to the striatum, which, in analogy with 18F-FA-4, cannot be inhibited by the presence of 10 mmol/L HC-3 in the incubation medium (Fig. 4).
To investigate whether 18F-FA-4 binds to the HACU receptor in vivo, the radiotracer was injected in a normal mouse and its distribution in the brain was visualized with autoradiography. This experiment was performed only with 18F-FA-4 because the short half-life of 11C (20.4 min) precludes such study with 11C-pipzA-4. Autoradiograms of brain sections after intravenous injection of 18F-FA-4 showed equal binding of the tracer to the striatum and the cortex, whereas no binding to the white matter and the corpus callosum was found (Fig. 5).
The binding of 18F-FA-4 to the striatum and the cortex was not inhibited by intravenous coadministration of 20 μmol/kg of A-4 with the tracer. Quantitative analysis of the autoradiograms of brain sections after injection of a solution containing both 18F-FA-4 and A-4 revealed a relatively higher uptake of 18F-FA-4 into the brain. This finding is in contrast to the inhibition of 18F-FA-4 binding that was expected on the basis of our in vitro experiments. This observation suggests that the binding of 18F-FA-4 in peripheral binding sites or to plasma proteins is prevented, resulting in an increased availability for uptake in the brain. Even then it would be expected that the coadministered A-4 would also saturate the HACU binding sites in the brain.
Further elucidation of the in vivo binding specificity of FA-4 will require the use of animal models with local specific inhibition of cholinergic function by stereotactic injection of hemicholinium mustard (24) and a displacement study in a monkey visualized with an animal PET camera. Potential human PET application will also require the development of an alternative radiosynthesis method to provide higher yields of 18F-FA4 or the synthesis of a derivative of 18F-FA4 that can be prepared in a 1-step radiosynthesis method.
CONCLUSION
Our results may be summarized as follows: (a) Radiolabeled derivatives of A-4 may constitute potential tracers for the in vivo evaluation of the HACU system. (b) 3H-HC-3 binding inhibition studies on brain slices with HC-3, A-4, FA-4, and pipzA-4 showed that the affinity of FA-4 is in the same range as that of HC-3 and A-4, whereas the affinity of pipzA-4 is 10-fold lower. (c) Biodistribution experiments in mice revealed high and persistent brain uptake of both tracers, and in vitro autoradiographic studies of mice brain sections confirmed binding to the striatum. (d) However, the binding of 18F-FA-4 or 11C-pipzA-4 to the striatum was found to be sodium independent and was not inhibited by the presence of HC-3. 18F-FA-4 binding to the striatum was inhibited by the presence of A-4, indicating a saturable binding in vitro. Inhibition of 18F-FA-4 binding by A-4 but not by HC-3 may imply allosteric binding of FA-4 and HC-3 to the HACU transporter. (e) Ex vivo autoradiography after intravenous injection of 18F-FA-4 showed uptake in both striatum and cortex. Coinjection of A-4 with 18F-FA-4 resulted in an unexpected increase of brain uptake.
From these preliminary results we may conclude that 18F-FA-4 and 11C-pipzA-4 are potential probes for the visualization of the HACU system. However, additional experiments, including a monkey PET study and a study after introduction of hemicholinium mustard lesions in mice brain, are necessary to reveal the exact binding site of 18F-FA-4.
Footnotes
Received May 2, 2002; revision accepted Sep. 17, 2002.
For correspondence contact: Guy M. Bormans, PhD, Laboratory for Radiopharmaceutical Chemistry, Faculteit Farmaceutische Wetenschappen, Katholieke Universiteit Leuven, Herestraat 49, B-3000 Leuven, Belgium.
E-mail: guy.bormans{at}uz.kuleuven.ac.be
REFERENCES
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.