


Abstract
Today, the standard approach for the kinetic analysis of dynamic PET studies is compartment models, in which the tracer and its metabolites are confined to a few well-mixed compartments. We examine whether the standard model is suitable for modern PET data or whether theories including more physiologic realism can advance the interpretation of dynamic PET data. A more detailed microvascular theory is developed for intravascular tracers in single-capillary and multiple-capillary systems. The microvascular models, which account for concentration gradients in capillaries, are validated and compared with the standard model in a pig liver study. Methods: Eight pigs underwent a 5-min dynamic PET study after 15O-carbon monoxide inhalation. Throughout each experiment, hepatic arterial blood and portal venous blood were sampled, and flow was measured with transit-time flow meters. The hepatic dual-inlet concentration was calculated as the flow-weighted inlet concentration. Dynamic PET data were analyzed with a traditional single-compartment model and 2 microvascular models. Results: Microvascular models provided a better fit of the tissue activity of an intravascular tracer than did the compartment model. In particular, the early dynamic phase after a tracer bolus injection was much improved. The regional hepatic blood flow estimates provided by the microvascular models (1.3 ± 0.3 mL min−1 mL−1 for the single-capillary model and 1.14 ± 0.14 min−1 mL−1 for the multiple-capillary model) (mean ± SEM mL of blood min−1 mL of liver tissue−1) were in agreement with the total blood flow measured by flow meters and normalized to liver weight (1.03 ± 0.12 mL min−1 mL−1). Conclusion: Compared with the standard compartment model, the 2 microvascular models provide a superior description of tissue activity after an intravascular tracer bolus injection. The microvascular models include only parameters with a clear-cut physiologic interpretation and are applicable to capillary beds in any organ. In this study, the microvascular models were validated for the liver and provided quantitative regional flow estimates in agreement with flow measurements.
Dynamic PET imaging involves sequential acquisition of data in several frames. Together, these reconstructed frames form a set of images from which physiologic parameters can be estimated by kinetic analysis. The choice of scanning protocol involves the selection of a set of acquisition times, for which the trade-off between good counting statistics and good temporal resolution must be considered. Continuous improvements in PET technology have led to scanners with increased sensitivity, which allows better temporal resolution but conserves good counting statistics. Thus, detailed information about early tracer distribution and metabolism can be obtained.
The standard method for kinetic analysis is a compartment model, with which the vast majority of dynamic PET studies are analyzed. In the standard method, the tracer and possible metabolites are considered together in compartments in which the substances are assumed to be in instantaneous spatial equilibrium. Mass is exchanged between compartments as a function of time, and the model parameters are rate constants that describe the exchange. The question is whether the standard compartment model used for kinetic analysis maximizes information gleaned from data obtained at a high temporal resolution, such as modern dynamic PET data. We show that this is not so.
At the microvascular level, the concentrations of a tracer and possible metabolites are distributed in both time and space. This fact is considered in distributed capillary models, in which the tracer is transported through capillaries by bulk flow and may be exchanged across capillary walls (1). Compared with the standard model, this scheme presents a more accurate description of tracer distribution and metabolic processes, which is necessary for a deeper physiologic understanding. We examine how spatially distributed capillary models can be unified with compartment theory. The traditional compartment model uses the organ inlet concentration as the concentration throughout the vascular compartment. In contrast, we consider models that make use of spatial averaging of the concentrations in the capillary volume, which is what a PET scanner actually registers. These models account for concentration gradients in capillaries during the dynamic phase of a bolus injection; we denote such models as microvascular models.
Microvascular models can be applied to any organ, and in this study, we validate their use for determining regional hepatic blood flow. Knowledge of hepatic blood flow is important for insight into liver diseases and understanding how substances are delivered to hepatocytes. Noninvasive quantitative methods for measuring hepatic blood flow are not available, and a PET method for measuring hepatic blood flow has not yet been satisfactorily validated (2). The problems are attributable in part to the liver having a dual blood supply, through the hepatic artery (HA) and the portal vein (PV); the inaccessibility of the PV makes direct noninvasive measurement of the dual-input function impossible. Because the hepatic dual-input function is essential for quantitative estimates of kinetic parameters (3), the development of a PET method for measuring total hepatic blood flow must start with animal models. This study was performed with pigs, which are suitable as an animal model because they have a hepatic vasculature similar to that of humans. Pigs were PET scanned with simultaneous dual-input blood sampling after 15O-carbon monoxide (CO) inhalation. We compare flow estimates from the PET microvascular models with ultrasound transit-time flow-meter measurements.
In summary, the study had 2 purposes: first, to compare the standard compartment model and 2 microvascular models for PET kinetic analysis by investigating the behavior of an intravascular tracer, and second, to examine whether microvascular models allow the determination of regional hepatic blood flow.
MATERIALS AND METHODS
Animals
The study procedures were approved by the Danish National Ethics Committee for Animal Research. Eight Danish Landrace pigs that were 3 mo old and had a body weight of 38–42 kg were used. The pigs were deprived of food 24 h before the experiments but had free access to water.
Animal Preparation
Each pig was anesthetized by injection of 20 mL of midazolam (Dormicum; Roche) (5 mg mL−1) and 12 mL of ketamine (Ketalar; Pfizer) (50 mg mL−1); this step was followed by intravenous infusion of a mixture of midazolam (10 mL h−1), ketamine (10 mL h−1), and isotonic saline (30 mL h−1). Every 4 h, the pig received an analgesic injection of 0.5 mg of fentanyl. The pig was ventilated with air containing 40% oxygen by use of a Servo 900 respirator (Siemens-Elema). The pig was placed on its back, and a catheter was placed in the caval vein via the right femoral vein for infusions. A catheter (0.7 mL) was placed into the aorta via the right femoral artery for arterial blood sampling corresponding to that of the HA and for blood pressure measurements. A catheter (0.7 mL) was placed through a 10-cm-long abdominal incision into the PV via the splenic vein for PV blood sampling. The position of the PV catheter was controlled by fluoroscopy before each experiment. Finally, 2 ultrasound transit-time flow-meter probes (In Vivo Aps; CardioMed) were placed around the HA and the PV through the abdominal incision. The pig was allowed to recover for 1 h before the first tracer administration. Altogether, the time interval between the initiation of narcosis and the start of PET was 3 h. Oxygen saturation and pH in arterial blood samples were measured every hour and were adjusted toward >98% and 7.45, respectively, by changing the amount of air delivered from the respirator. The pig was covered and placed on a thermostatically controlled heat blanket set to keep the body temperature at 38.5°C–39.5°C.
PET Examination and Blood Sampling
Each pig was scanned after inhalation of 500 MBq of 15O-CO. PET was performed by use of a Siemens ECAT EXACT HR-47 camera (CTI/Siemens Medical Systems, Inc.). The PET camera was calibrated by use of a phantom containing a 68Ge/68Ga solution with a known activity concentration. The pig was positioned with the liver inside the 15-cm field of view. A 15-min transmission scan with external rod sources was performed before the first emission scan and used for photon attenuation correction. Data acquisition was started at inhalation time and included 17 frames with a total scanning time of 310 s (eight 5-s, four 15-s, three 30-s, and two 60-s frames). The PET data were reconstructed with filtered backprojection by use of a Hanning filter with a cutoff at 0.3 Nyquist frequency. The resulting 3-dimensional images contained 128 × 128 × 47 voxels with a size of 2.4 × 2.4 × 3.1 mm3 and a central spatial resolution of 6.7 mm full width at half maximum. Arterial and PV blood samples of 1 mL each were collected manually for 5 min starting at inhalation time in eight 5-s, four 15-s, and six 30-s intervals. Blood radioactivity concentrations were measured by use of a well counter (Packard Instruments Co.), which was calibrated by use of a 68Ge/68Ga solution with a known activity concentration. Blood radioactivity concentrations were decay corrected to the start of tracer administration. Blood flow in the HA and the PV was measured continuously throughout the experiment with the ultrasound transit-time flow meters. After the measurements were completed, the liver was removed from the anesthetized animal and weighed.
Image Analysis
Regions of interest (ROIs) were defined by use of transaxial slices of the mean image consisting of the sum of all frames. This procedure made it easy to distinguish the liver from the surrounding tissue. ROIs were drawn to contain liver tissue without extrahepatic tissue and large vessels as described by Keiding et al. (4). The resulting liver ROI from which the liver tissue time–activity curve was generated had a volume of about 150 cm3.
Modeling
15O-CO is an intravascular tracer transported by red blood cells and cannot cross the endothelial cells lining the capillaries (hepatic sinusoids). We describe its kinetic behavior by using 3 models of increasing physiologic realism.
Standard Compartment Model.
In the standard compartment approach, the tissue activity M(t) is the product of the vascular volume V0 and the inlet activity concentration in the blood Ci(t):
 Eq. 1 For most tissues, Ci(t) is an arterial time–activity curve, and for liver kinetics, as in this study, it is the hepatic dual-input time–activity curve (3) (Equation 11). The compartment model is an inlet equilibration model that describes M(t) as a vascular compartment containing a uniform concentration seen at the inlet only with parameter V0.
Eq. 1 For most tissues, Ci(t) is an arterial time–activity curve, and for liver kinetics, as in this study, it is the hepatic dual-input time–activity curve (3) (Equation 11). The compartment model is an inlet equilibration model that describes M(t) as a vascular compartment containing a uniform concentration seen at the inlet only with parameter V0.
Single-Capillary Microvascular Model.
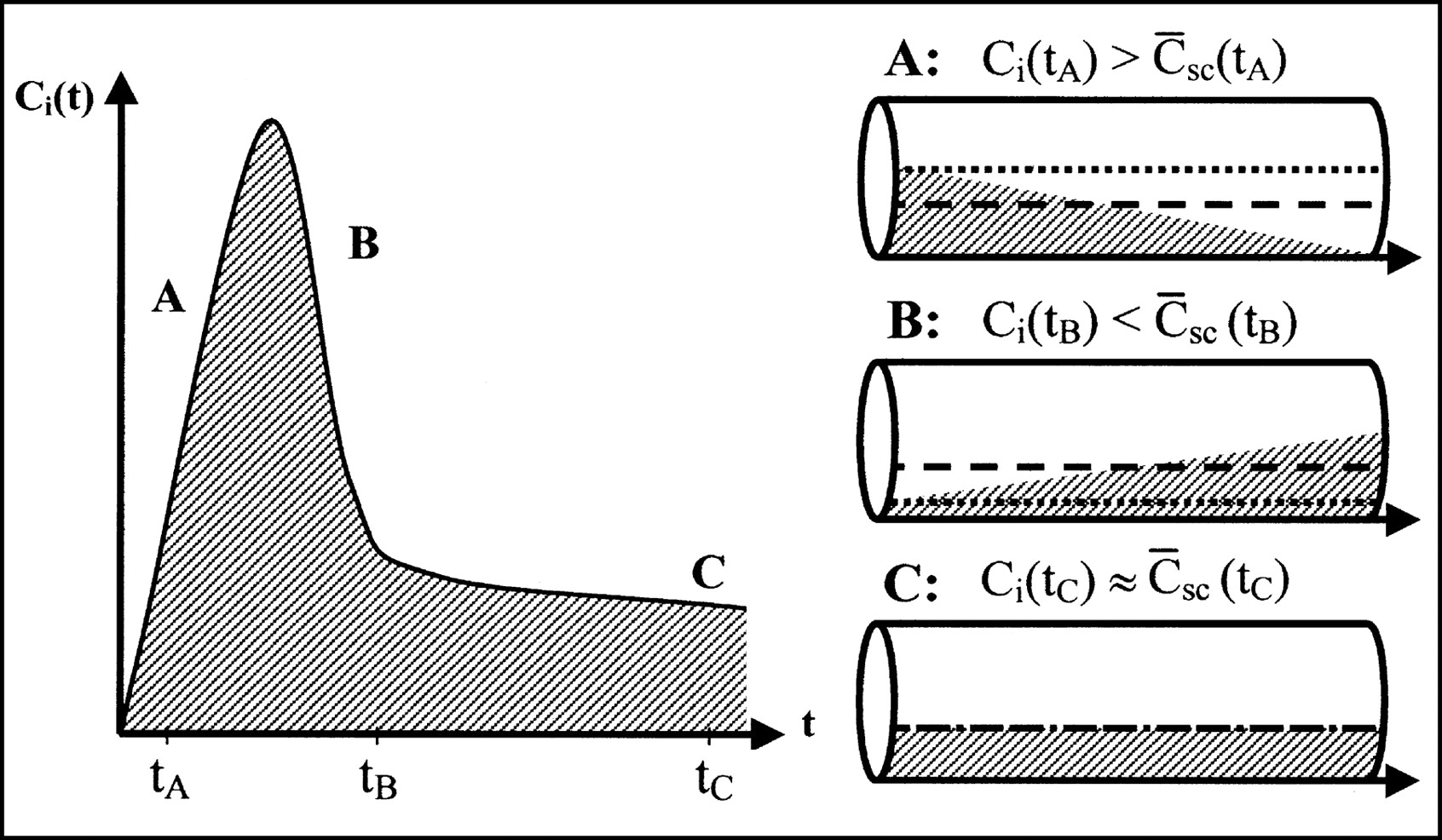
The vascular compartment should not always be treated as a perfect compartment containing the organ inlet concentration. In particular, for early times after a tracer bolus injection, it is important to consider the concentration gradients within capillaries. Instead of the use of the inlet concentration, a more physiologic estimate of the tracer content of the vasculature can be obtained by calculating the time-dependent spatially averaged concentration within capillaries (Fig. 1). Fick’s conservation law states that the change in the amount of tracer inside the tissue is equal to the difference between what enters the tissue and what leaves the tissue. When the ROI inlet concentration is denoted by Ci(t) and the ROI outlet concentration is denoted by Co(t),
 Eq. 2 where F is flow. Here and below a dot over a designation indicates differentiation with respect to time. Integration of a single capillary or a set of capillaries with the same transit time T, where T = Vb/F (where Vb is blood volume), yields
Eq. 2 where F is flow. Here and below a dot over a designation indicates differentiation with respect to time. Integration of a single capillary or a set of capillaries with the same transit time T, where T = Vb/F (where Vb is blood volume), yields
 because Co(t) = Ci(t − T) in the absence of dispersion. By substituting u = t′ − T and setting Ci(t) at 0 for t < 0,
because Co(t) = Ci(t − T) in the absence of dispersion. By substituting u = t′ − T and setting Ci(t) at 0 for t < 0,
 where t′ and u are dummy variables. Therefore, the operational equation for the single-capillary microvascular model is
where t′ and u are dummy variables. Therefore, the operational equation for the single-capillary microvascular model is
 Eq. 3 where the spatially averaged capillary concentration C̄sc(t) can be expressed as:
Eq. 3 where the spatially averaged capillary concentration C̄sc(t) can be expressed as:
 Eq. 4 The inlet concentration is averaged over transit time, T = Vb/F. The single-capillary microvascular model for an intravascular tracer, described by Equations 3 and 4, has flow F and blood volume Vb as its parameters.
Eq. 4 The inlet concentration is averaged over transit time, T = Vb/F. The single-capillary microvascular model for an intravascular tracer, described by Equations 3 and 4, has flow F and blood volume Vb as its parameters.

Differences in tracer concentrations in vascular volume determined with compartment model and single-capillary microvascular model. (Left) Measured inlet time–activity curve after tracer bolus. During dynamic phase, ROI inlet concentration Ci(t) rises (A), reaches its maximum, and falls (B) toward steady state (C). (Right) Concentration profiles in capillary at times tA, tB, and tC, corresponding to situations A, B, and C, respectively. Arrows show direction of flow. Dotted lines show inlet concentrations used in compartment model. Dashed lines show spatially averaged concentrations used in microvascular models. Use of inlet concentrations overestimates amount of tracer present in organ in rising part of bolus (A) and underestimates it in falling part of bolus (B). Error is most significant in early dynamic phase with large gradients. Later, when steady state is approached (C), error becomes smaller.
Multicapillary Microvascular Model.
In order to describe an organ as a multiple-capillary system, we take a step further toward physiologic realism and allow for a distribution of transit times f(T) through different pathways. We describe the distribution of transit times f(T) by a mean transit time T̄ and a variance of transit times σ2. According to Fick’s law (Eq. 2), the venous outlet concentration Co,mc(t) is now
 Eq. 5 where f(T)dT is the probability that the transit time is in the interval [T, T + dT]. Expansion of Ci(t − T) around the mean transit time T̄ gives
Eq. 5 where f(T)dT is the probability that the transit time is in the interval [T, T + dT]. Expansion of Ci(t − T) around the mean transit time T̄ gives

=Ci(t−T̄)−C̈i(t−T̄)(T−T̄)
 Eq. 6 where the double dot indicates double differentiation with respect to time.
Eq. 6 where the double dot indicates double differentiation with respect to time.
On the assumption of a narrow distribution of transit times f(T), only small deviations from the mean transit time T̄ contribute to the venous outlet concentration Co,mc(t). By terminating the series after the third term and substituting Equation 6 into Equation 5, we find
 Eq. 7 where the mean transit time T̄ and the variance of transit times σ2 are
Eq. 7 where the mean transit time T̄ and the variance of transit times σ2 are

By inserting the venous outlet concentration Co,mc(t) (Eq. 7) into Fick’s law (Eq. 2) and integrating, we obtain the operational equation for the multiple-capillary microvascular model:
 Eq. 8 where the spatially averaged capillary concentration C̄mc(t) is
Eq. 8 where the spatially averaged capillary concentration C̄mc(t) is
 Eq. 9 The first term on the right side of Equation 9 is the spatially averaged capillary concentration for a single capillary, and the second term corrects for the distribution of transit times, which changes sign at least once during the passage of a bolus. The multiple-capillary microvascular model has 3 parameters: flow F, blood volume Vb, and variance of transit times σ2.
Eq. 9 The first term on the right side of Equation 9 is the spatially averaged capillary concentration for a single capillary, and the second term corrects for the distribution of transit times, which changes sign at least once during the passage of a bolus. The multiple-capillary microvascular model has 3 parameters: flow F, blood volume Vb, and variance of transit times σ2.
In summary, the 3 models that we compare in this study are the standard compartment model (Eq. 1), the single-capillary microvascular model (Eqs. 3 and 4), and the multiple-capillary microvascular model (Eqs. 4, 8, and 9).
Synchronizing Blood Samples and PET Scanning
Measurements used to estimate model parameters are obtained by 2 different procedures. Tissue concentrations are obtained by scanning the organ, whereas blood concentrations in vessels supplying the organ are measured through sampling catheters. The time points pertaining to blood samples on the time axis of the PET scan depend on various parameters, such as the dimensions of the sampling catheter, the speed of blood withdrawal, the distance from the blood sampling site to the organ, and the blood flow. Before model parameters are estimated, it is necessary to synchronize the 2 data sets, those for the inlet concentration Ci(t) and tissue activity M(t), to a common time axis.
Sometimes the problem is recognized by including an adjustable parameter, a time shift, in the fitting procedure, but we approach the problem without introducing additional parameters by using model-independent considerations. We make use of mass conservation and take advantage of knowledge about flow from microvascular models. On a time scale shorter than transit time, no appreciable amount of tracer has left the tissue; consequently, the ROI outlet concentration Co(t) = 0. In this case, Fick’s conservation law (Eq. 2) leads to

During each frame, a PET scanner sums up the radioactive events, leaving no information about tissue activity M(t) at specific times. Because only the sums of events within frames are available, we integrate once more to obtain the operational equation
 Eq. 10 By use of a sufficiently dense frame structure with one or more frames within a transit time, Equation 10 can be applied to estimate the time shift for the inlet concentration Ci(t) by matching the left and right sides of the equation. This model-independent synchronization, based on mass conservation, is related to flow and made possible by the use of flow as a parameter in microvascular models.
Eq. 10 By use of a sufficiently dense frame structure with one or more frames within a transit time, Equation 10 can be applied to estimate the time shift for the inlet concentration Ci(t) by matching the left and right sides of the equation. This model-independent synchronization, based on mass conservation, is related to flow and made possible by the use of flow as a parameter in microvascular models.
Parameter Estimation and Statistical Criteria
Model parameters were estimated by the Levenberg-Marquardt method by minimizing the weighted residual sum of squares (WRSS); PET data were weighted proportional to the frame durations. As part of the iterations, when microvascular models were used, the input time–activity curve and the PET time–activity curve were synchronized according to Equation 10. Plots of the WRSS against time were examined for systematic errors. The ability of model parameters to be identified was examined with a sensitivity function:
 where Sp(t) is the sensitivity function of model parameter p, and M′(t) is the model solution. The mathematic independence of the model parameters is demonstrated by the fact that no sensitivity function is proportional to or reciprocal for another. When we display sensitivity functions, they are individually scaled to emphasize shapes and thereby evaluate possible correlations. Consequently, our sensitivity plots allow easy evaluation of the time scale within which the parameters are identified, but evaluation of absolute sensitivities is obscured.
where Sp(t) is the sensitivity function of model parameter p, and M′(t) is the model solution. The mathematic independence of the model parameters is demonstrated by the fact that no sensitivity function is proportional to or reciprocal for another. When we display sensitivity functions, they are individually scaled to emphasize shapes and thereby evaluate possible correlations. Consequently, our sensitivity plots allow easy evaluation of the time scale within which the parameters are identified, but evaluation of absolute sensitivities is obscured.
In comparisons of models, it is important to take into account that the best model fitted to the data is not necessarily the model producing the smallest WRSS. The addition of more parameters in general decreases the WRSS. We therefore identified the statistically favorable model on the basis of criteria that included penalty functions proportional to the number of parameters in the model (5). The Akaike information criterion (AIC) (6) was used to measure the quality of fit with the equation
 where P is the number of parameters in the model and N is the number of data points. It can be seen that small AIC scores should be preferred.
where P is the number of parameters in the model and N is the number of data points. It can be seen that small AIC scores should be preferred.
Results are reported as mean ± SEM.
RESULTS
time–activity curves
The dual-input concentrations were calculated as the flow-weighted input concentrations from HA and PV according to
 Eq. 11 where FHA and FPV are flow rates in the hepatic artery and portal vein, respectively, and CHA and CPV are concentrations in the corresponding vessels.
Eq. 11 where FHA and FPV are flow rates in the hepatic artery and portal vein, respectively, and CHA and CPV are concentrations in the corresponding vessels.
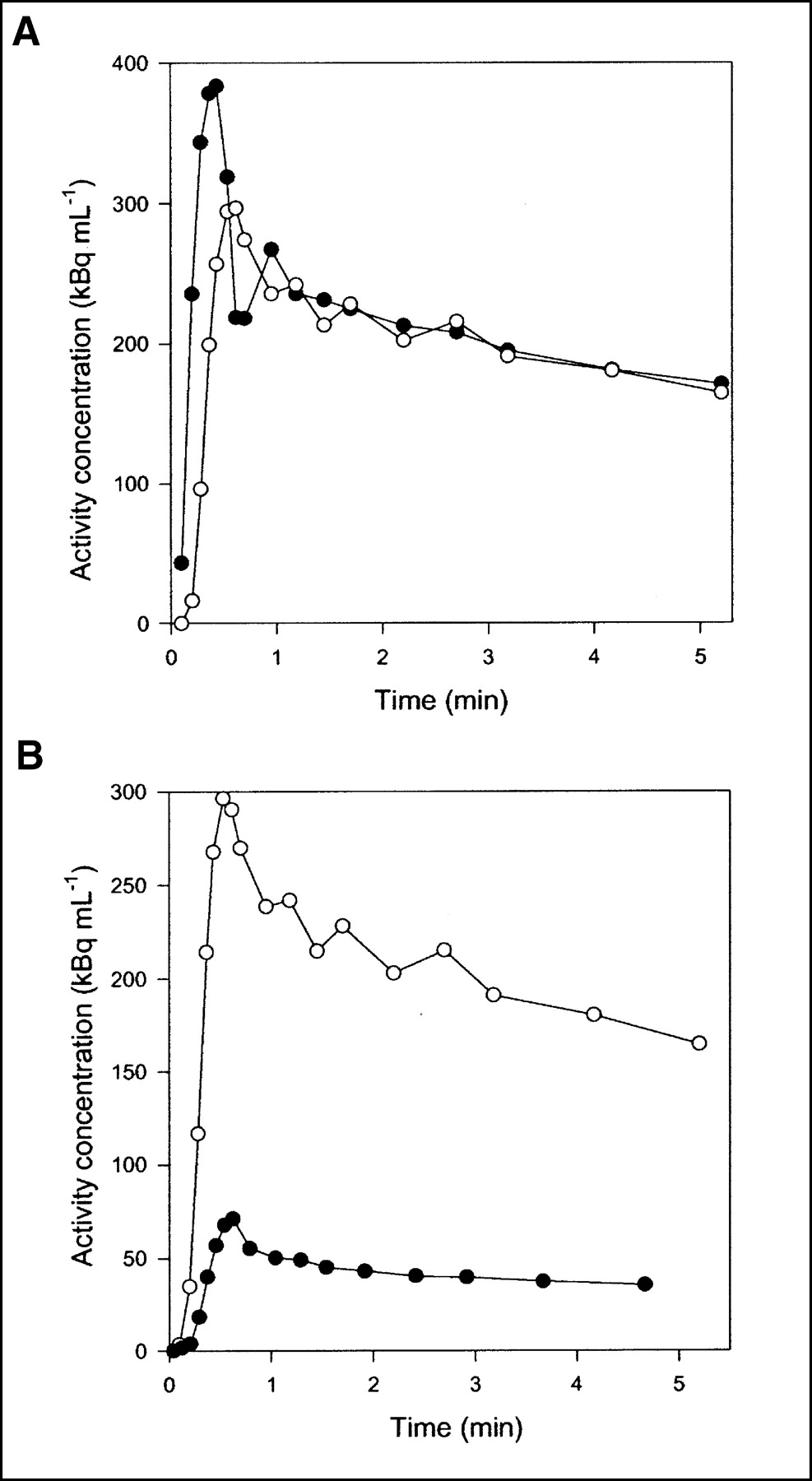
Flow was measured continuously during each experiment. Typical flow fractions were 20% from HA and 80% from PV. The mean hepatic blood flow was 1.03 ± 0.12 mL min−1 mL−1 (mean ± SEM mL of blood min−1 mL of liver tissue−1). It was measured with transit-time flow meters and normalized to liver weight. HA time–activity curves have a rapid phase with a narrow peak, whereas PV time–activity curves are delayed and dispersed during passage of the bolus through the intestines. The calculated dual-input time–activity curve is intermediate, because it is composed of the 2 input curves (3). Typical HA and PV time–activity curves are shown in Figure 2A, and a dual-input time–activity curve with a corresponding PET time–activity curve after CO inhalation is shown in Figure 2B. A possible mismatch between the time axes for blood samples and PET measurements was too short to be apparent on the time scale of Figure 2B, but the time axes nevertheless were synchronized when the microvascular models were fitted to the data.
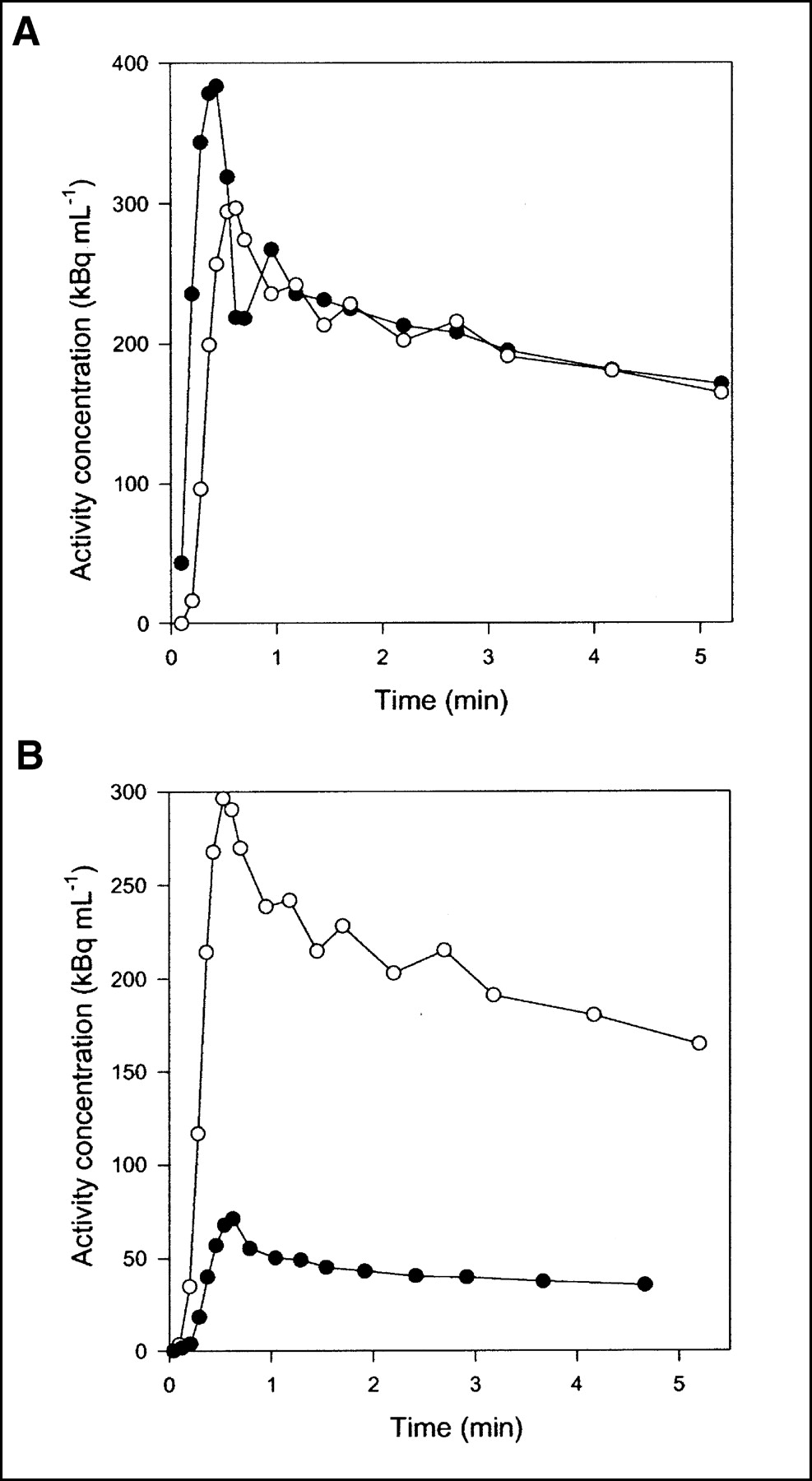
Typical time–activity curves after 15O-CO inhalation (pig 4). (A) Black circles = HA blood time–activity curve; white circles = PV blood time–activity curve. (B) Black circles = liver tissue PET time–activity curve; white circles = dual-input blood time–activity curve.
Compartment Model
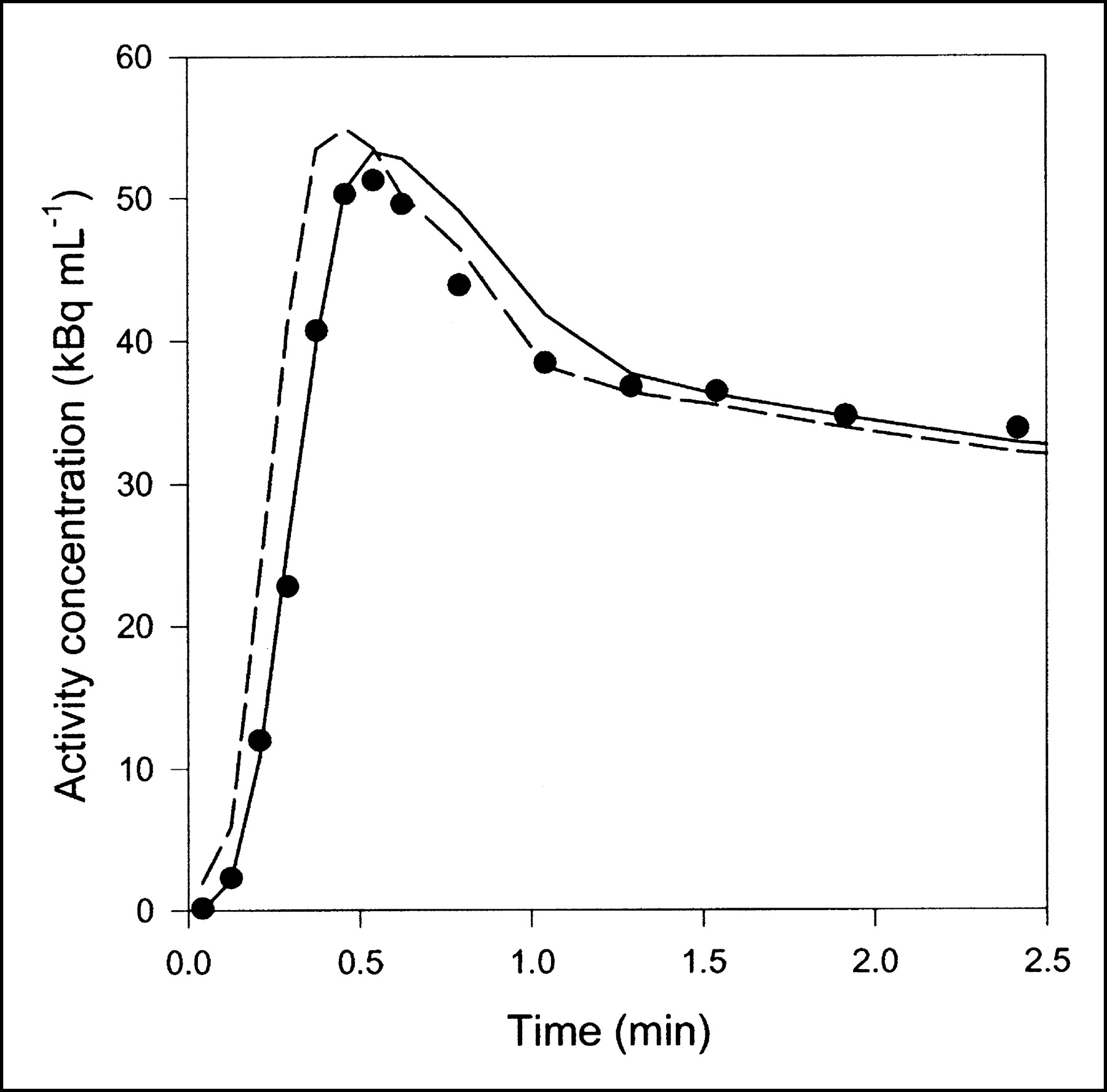
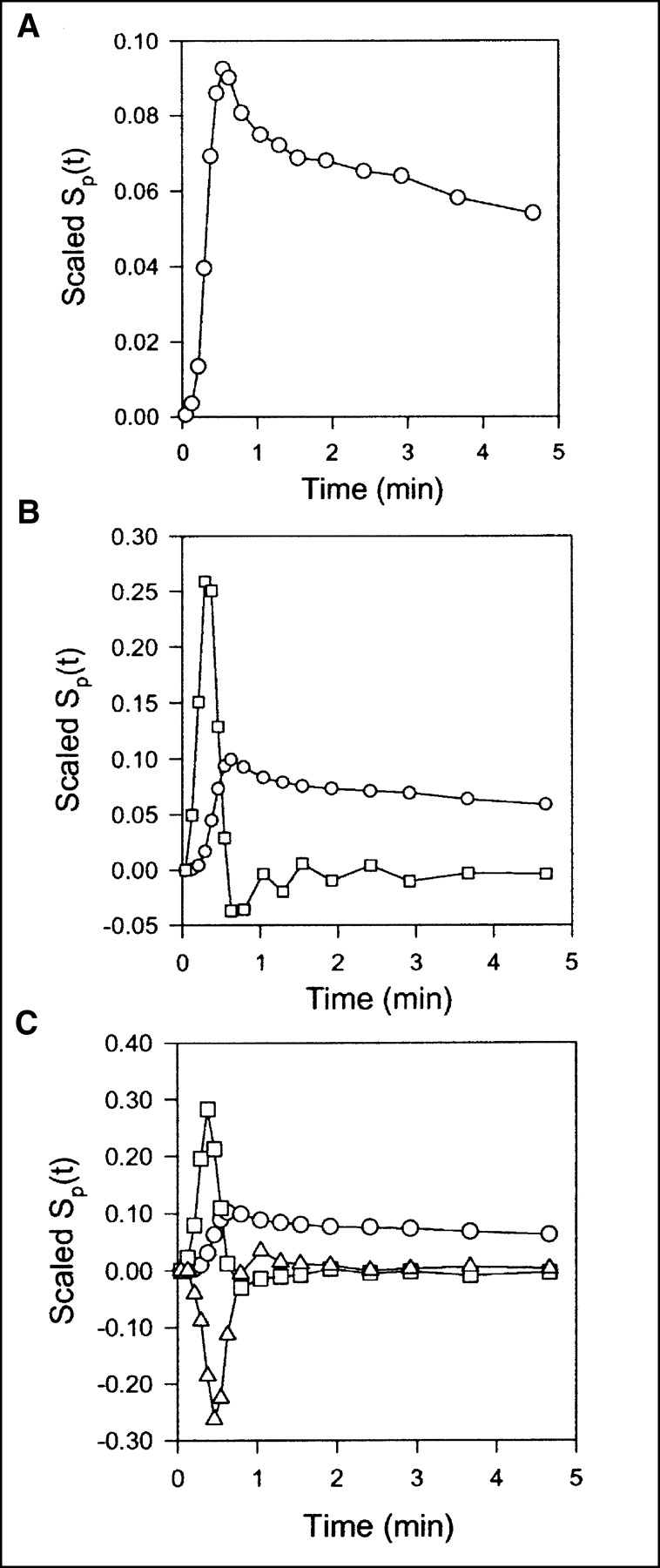
Data obtained after CO inhalation were analyzed by use of a vascular compartment containing the dual-input activity concentration. This model contains one parameter, vascular volume V0. The mean vascular volume estimate for all experiments was 0.18 ± 0.01 mL mL−1 (mL of blood mL of liver tissue−1). The volume estimates and statistical criteria are shown in Table 1, and a typical fit is shown in Figure 3. This simple model is unable to predict the early dynamic phase during which the tracer bolus is distributed in capillaries. After a few minutes, when the steady state is approached, the model prediction is improved. When only data from 2.5 min were used, the vascular volume estimate was slightly larger: 0.20 ± 0.01 mL mL−1. A sensitivity analysis of the model is shown in Figure 4A. V0 acts as a simple scale factor for the input concentration, and the shape of the dual-input time–activity curve is directly reflected by the shape of the sensitivity function.
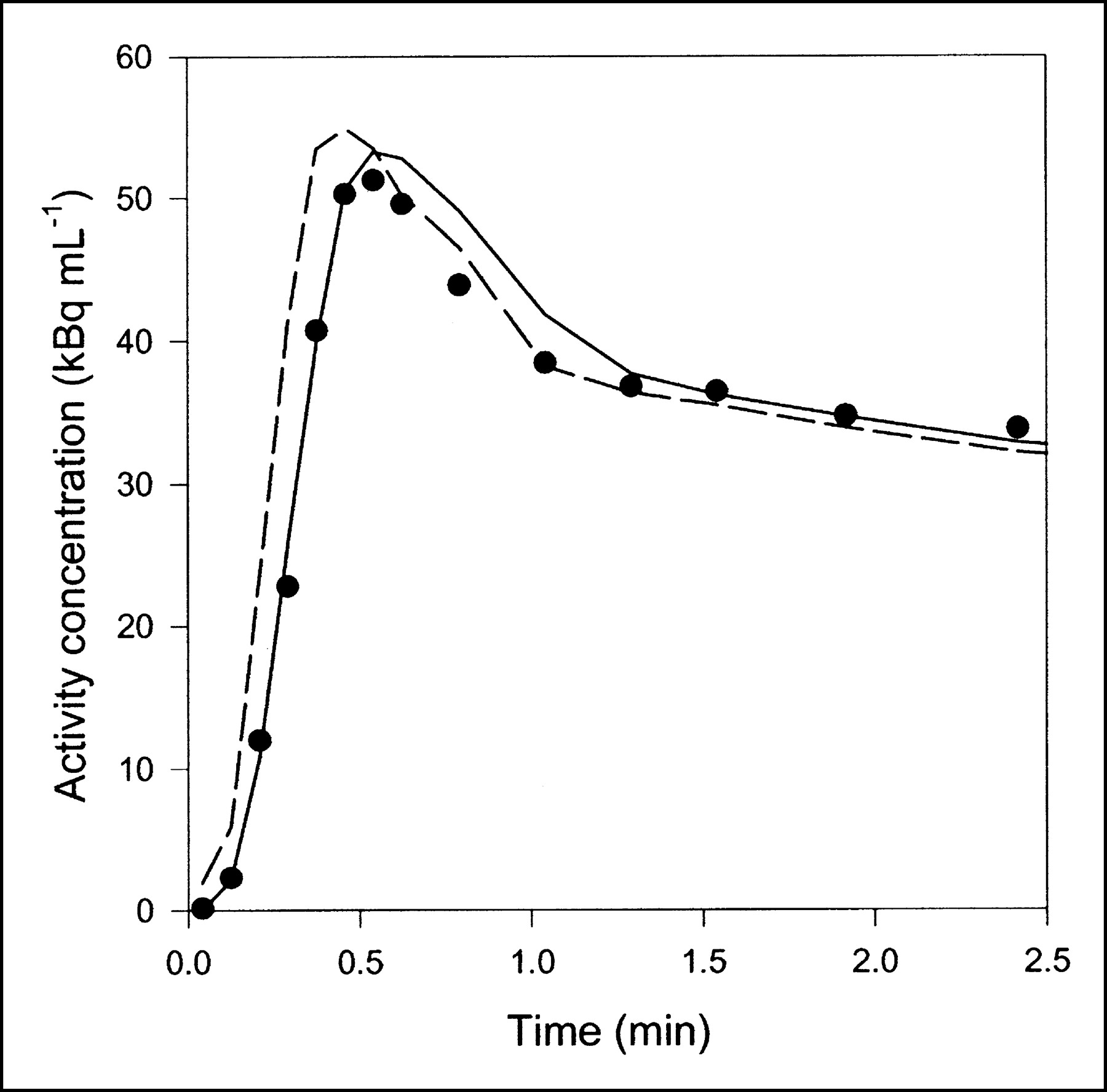
Typical liver tissue PET time–activity curve after 15O-CO inhalation (pig 2), fitted with 2 models. Only initial 2.5 min are shown, when differences between models were most pronounced. Black circles = liver PET data; dashed line = compartment model with dual input; solid line = multiple-capillary microvascular model. Fit obtained with single-capillary microvascular model has time course very similar to that of fit obtained with multiple-capillary microvascular model.
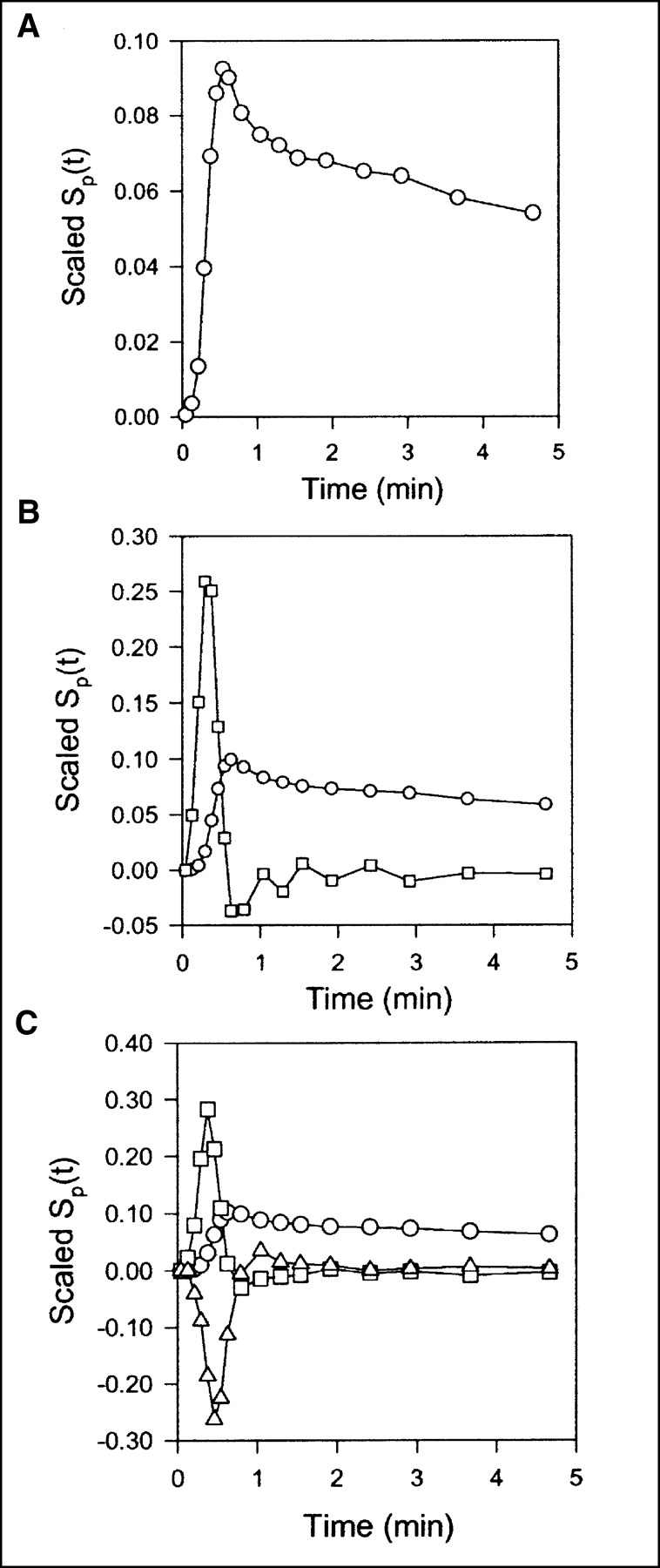
Scaled sensitivity function Sp(t) obtained with data from pig 2. Scaled Sp(t) is calculated at each point, and lines are linear interpolations. All sensitivity functions are scaled to emphasize shapes. (A) Standard compartment model. Circles = V0. (B) Single-capillary microvascular model. Circles = Vb; squares = F. (C) Multicapillary microvascular model. Circles = Vb; squares = F; triangles = σ.
Parameter Estimates and Statistical Criteria for Three Models
Single-Capillary Microvascular Model
With the single-capillary microvascular model, blood volume Vb and blood flow F are estimated. Fit obtained with this model is similar to fit obtained with the multiple-capillary microvascular model (Fig. 3); parameter estimates and statistical criteria are shown in Table 1. Figure 5 shows regional flow estimates plotted against transit-time flow-meter measurements of total flow. Flow was estimated both with and without synchronization. The mean synchronization time shift was 2.0 ± 0.8 s (n = 8), and the reproducibility of the synchronization method was verified by the stable estimates. A relatively small time shift was expected, because the blood sampling sites were close to the liver. In all instances, synchronization induced lower flow estimates. As shown on the sensitivity plot, F is most sensitive to the early dynamic phase, when the tracer is distributed in the organ. Consequently, determination of F requires data with a high temporal resolution and good counting statistics as well as accurate measurements of the input function. After a time scale of the order of mean transit time, the steady state is approached and the blood volume becomes the dominant parameter. Vb acts as a scale factor for the spatially averaged input concentration. Therefore, the early part of the sensitivity function of Vb is different from that of vascular volume V0. The different shapes of the sensitivity functions (Fig. 4B) show that both F and Vb can be identified.

Regional blood flow estimated by single-capillary microvascular model plotted against normalized total flow measured by flow meters (n = 8). Triangles = flow estimated without synchronization; squares = flow estimated with synchronization; solid line = identity line. In all instances, synchronization brings data sets together and induces lower flow estimates in closer agreement to normalized organ blood flow.
Multicapillary Microvascular Model
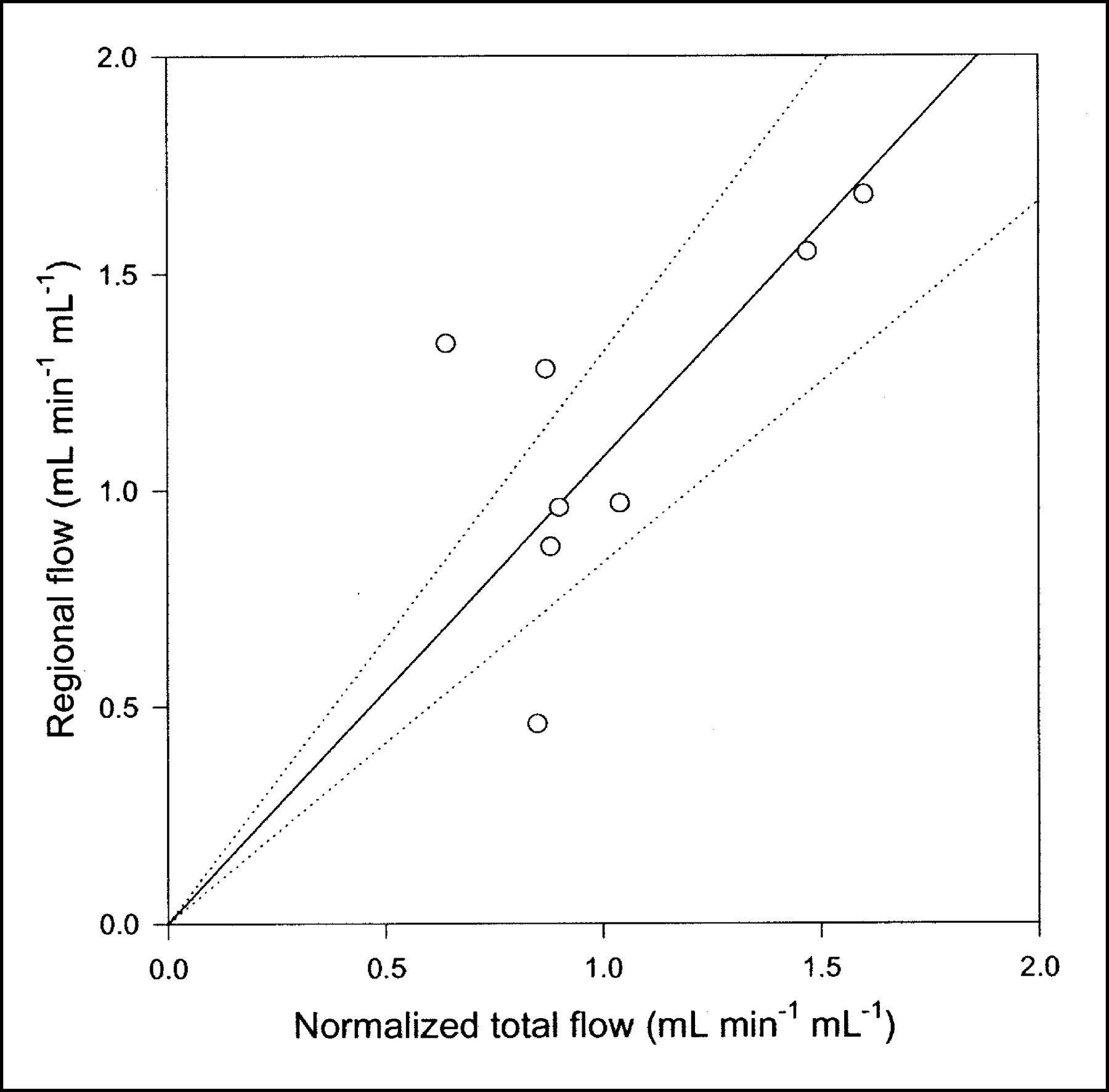
Compared with the single-capillary microvascular model, the multiple-capillary microvascular model has an extra parameter, σ, which describes the distribution of transit times. An example of the fit obtained with this model is shown in Figure 3, and parameter estimates and statistical criteria are shown in Table 1. Like flow F, σ is particularly sensitive to the early dynamic phase. Figure 6 shows regional flow estimates plotted against transit-time flow-meter measurements of total flow. Regression of a line through the origin yielded a slope of 1.07. Whereas increased flow tends to increase the model solution M′(t), a higher σ tends to decrease M′(t) because it causes more dispersion and thereby tends to flatten the peak of the model solution. A normalized sensitivity plot (Fig. 4C) shows a tendency for an inverse correlation between F and σ. The nonnormalized sensitivity functions of F and σ reveal that F is a far more sensitive parameter than is σ, so that a shift in σ causes only a minor change in M′(t) compared with a shift in F.
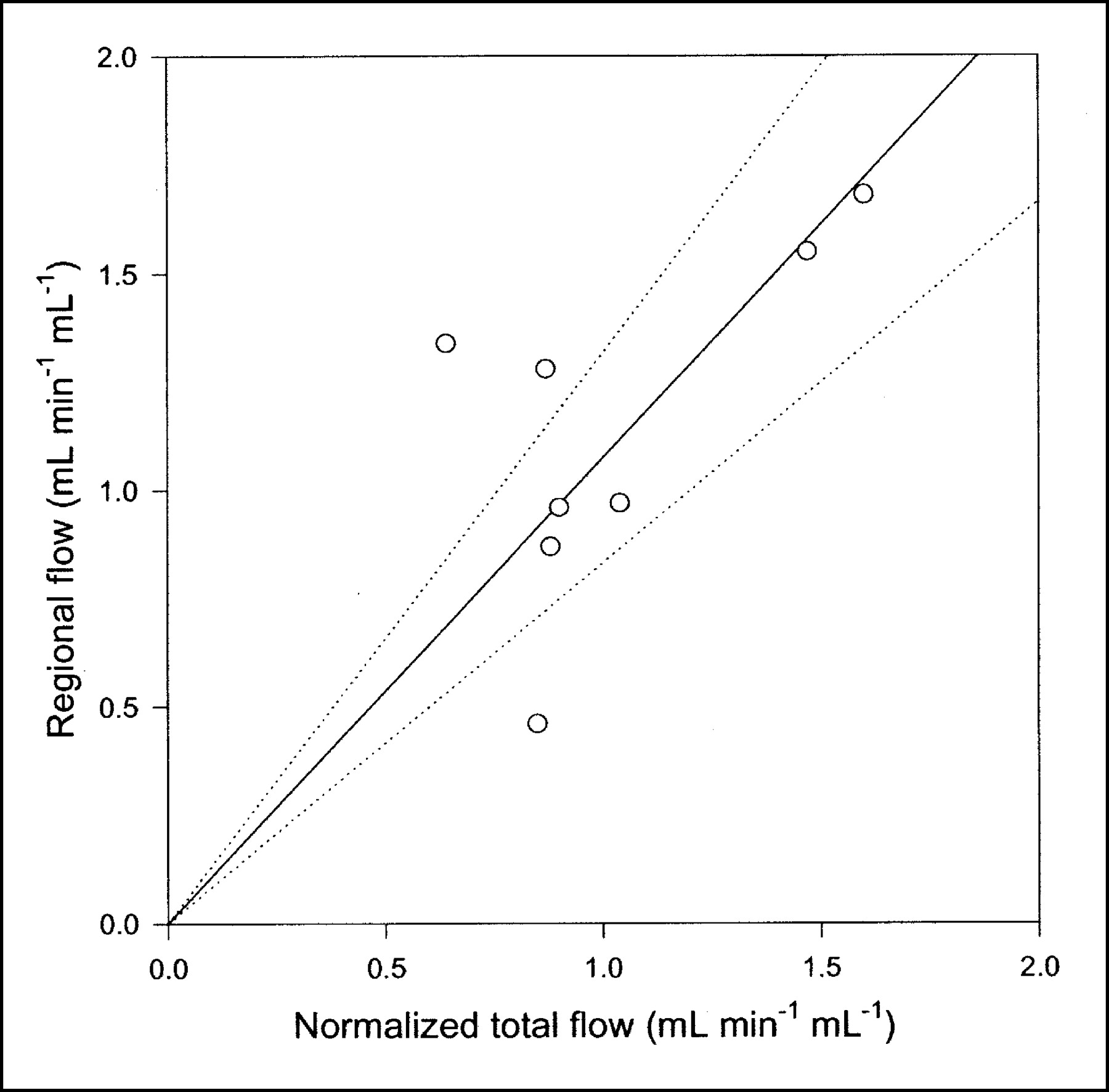
Regional blood flow estimated by multiple-capillary microvascular model with synchronization plotted against normalized total flow measured by flow meters (n = 8). Solid line = linear regression through origin; dotted lines = 95% confidence intervals.
The multiple-capillary microvascular model provides estimates of the mean transit time T̄ and the variance of transit times σ2, which are obtained by relating the inlet time–activity curve to the ROI time–activity curve. When these estimates are compared with independent measurements obtained by other methods, it is necessary to consider the interpretation of transit time. When inlet–outlet measurements are used, the transit time through the whole vascular structure is detected: artery, arterioles, capillaries, venules, and vein. When inlet–ROI measurements are used, it is not clear which transit time is estimated. First, we excluded large visible vessels from our liver ROIs. Second, the PET signal contains a mixture from the entire included vasculature. Consequently, estimates based on PET data and kinetic modeling do not describe the distribution of transit times in the entire organ, so that inlet–ROI estimates can be expected to be lower than inlet–outlet estimates.
DISCUSSION
Within the PET modeling community, much recent work has been devoted to the development of new techniques for calculating compartment parameters. A general trend toward parametric imaging has generated interest in new, advanced algorithms that are fast and insensitive to noise. However, the main portion of all of this work relies on the compartment design, and no matter how innovative the methods for optimizing goodness of fit might be, they are useful only when the underlying theory is sound. Compartment theory is a way of reducing the enormous complexity of real-life physiologic systems to simpler models that can be used for analysis of observed data. Like other models, this theory is based on assumptions that must be fulfilled approximately for the models to be effective. One of the assumptions is that compartments are instantaneously mixed with uniform concentrations. With this assumption, the time scales needed to achieve well-mixed compartments must be much shorter than the time scales of the exchange processes. Whether the time scale of mixing is sufficiently short depends on the physiologic system and the accuracy needed. These factors are among the reasons that successful forms of compartment models include linearizations (7–9) and reference tissue methods (10) that focus on estimating slow processes or steady-state volumes rather than describing early dynamics.
This study clearly demonstrates the deficiencies of a traditional compartment scheme for describing intravascular tracers in the liver. The early dynamic phase is poorly modeled, and only later, when a steady state is approached, does the model accurately predict measured tissue concentrations. Furthermore, interpretation of the vascular volume V0 is not trivial. In the early dynamic phase of a bolus injection, it is time dependent (11); later, close to the steady state, it approaches the physical blood volume Vb (Fig. 1).
Consequently, a physiologically richer theory was pursued. The distributed and compartment schemes were unified by spatial averaging of capillary concentrations. This process amended the compartment theory at its weakest point, which is the assumption of instantaneously well-mixed compartments (Fig. 1). The 2 microvascular models (Eqs. 3 and 4 and Eqs. 4, 8, and 9) include a physiologic description of the distribution of a tracer after the early dynamic phase of a bolus injection, which is needed when fast processes are estimated from early data. Analyses with the 2 microvascular models clearly show that it is indeed possible to obtain more information from dynamic PET data by using models that include more physiologic information than do traditional compartment models.
A common problem in all kinetic analyses based on data obtained from different measurement procedures is synchronization of the data sets. This problem is particularly important in the estimation of fast processes, such as blood flow, by use of measurements with a high temporal resolution. As shown in Figure 5, synchronizing has a dramatic effect on flow estimates even when the time shift is only a few seconds. We did not use synchronized data for fitting Eq. 1 because flow is not part of this model. However, the lack of synchronization is only slightly unfavorable for the compartment model because the vascular volume V0 is determined primarily at the steady state and therefore is not affected by minor time shifts. Even if the time shift of 2 s estimated with microvascular models were applied to the data sets before use of the compartment model, it could not account for the insufficient prediction of tissue concentrations at early times (Fig. 3). One way of synchronizing the data sets with the compartment model would be to include a time shift as an adjustable parameter. However, a time-shift parameter would correct not only for a mismatch between the time axes of the data sets but also, to some extent, for the inadequate physiologic description of the compartment model. We do not find the addition of an extra parameter with no clear interpretation desirable.
The microvascular models have more parameters than the compartment model. Despite appropriate penalties in the statistical criteria, these were at least as good as or better than those of the standard compartment model. Table 1 shows the results obtained by use of all 3 models with 5-min PET data. When judged only by AIC scores, the 3 models could seem equally good. This finding demonstrates that the choice of a model should never be based purely on goodness-of-fit criteria. The physiologic improvement obtained with the microvascular models compared with the standard compartment model has to do primarily with early times as the bolus is distributed in the tissues. Therefore, with a shorter PET scan, the statistical score would favor more clearly the physiologically richer microvascular models (Fig. 7). Still more remarkable are the findings that all parameters of the microvascular models have a clear-cut interpretation and provide actual physiologic information that is in agreement with independent measurements. Blood flow is estimated, and the correlation between regional blood flow in the liver ROI and normalized total blood flow measured by flow meters is evident from Figure 6. Some scatter is seen because of heterogeneous regional flow, possible vascular shunts, and uncertainty of the liver weight measurement. However, on average, a linear relationship is apparent.
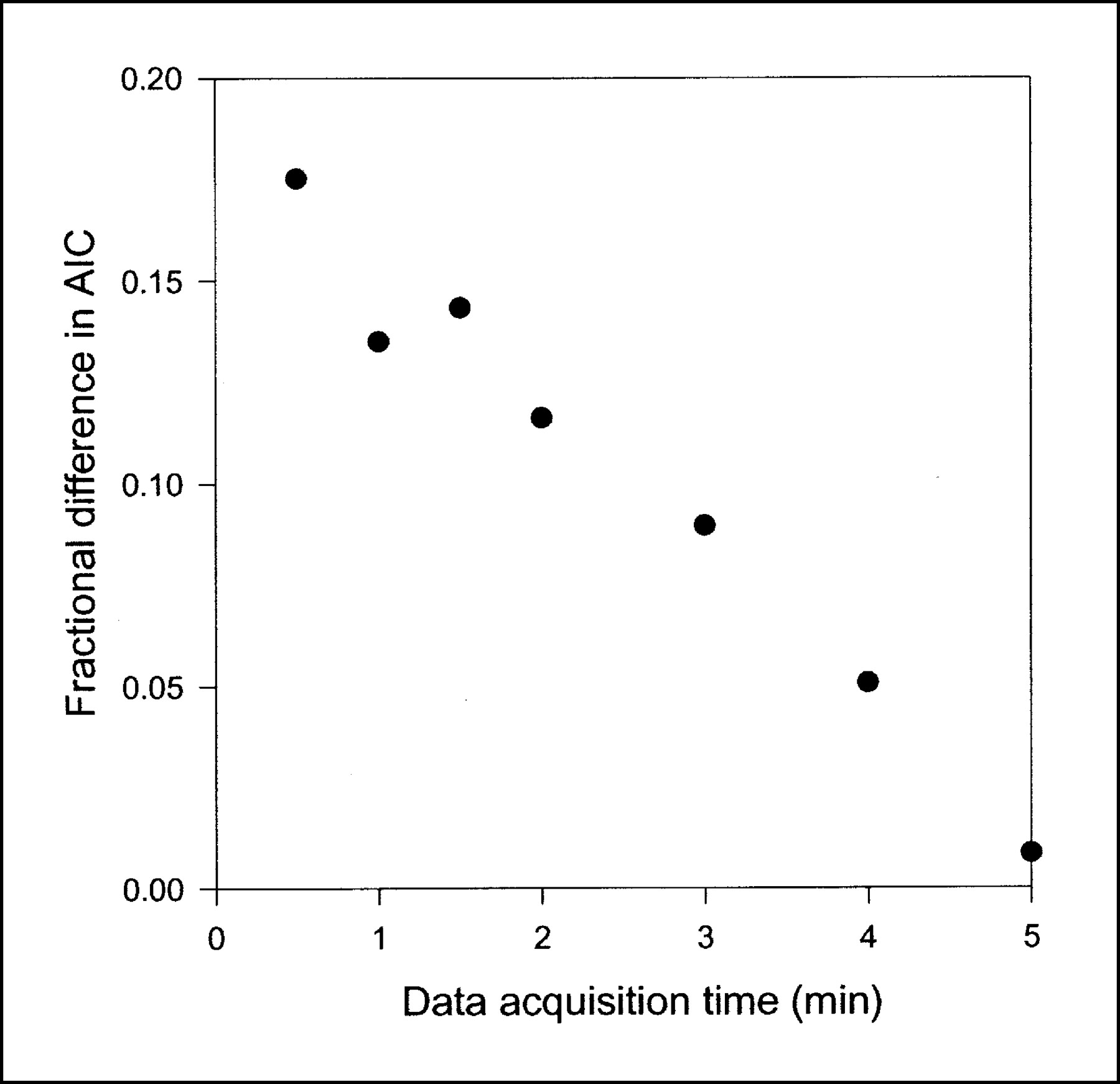
Fractional difference in AIC scores between compartment model and single-capillary microvascular model with synchronization as function of data acquisition time. Points represent mean fractional difference in AICs calculated as (AIC for the compartment model − AIC for the microvascular model)/AIC for the microvascular model (n = 8). Shorter data acquisition times were simulated by skipping late PET data. When all data up to 5 min were used, AICs were almost the same for 2 models, but as data acquisition time became shorter, the effect on AICs of using physiologically richer microvascular model became more apparent.
Previous attempts to measure hepatic blood flow used the traditional 15O-water injection method, which is widely used to estimate cerebral blood flow. Ziegler et al. (2) used PET scanning and dual-input samplings in dogs, but their results were not in accordance with independent microsphere measurements in the same animals. Taniguchi et al. (12) used a similar approach based on only arterial samplings in humans. Their model included portal systemic shunting and, because portal samplings were unavailable, the dual-input time–activity curve was modeled. Their flow estimates were not compared with other measurements, a process that could have validated the method.
The 2 microvascular models describe the distribution of a tracer by treating the liver as a set of vascular pathways acting in parallel, with a common transit time or a distribution of transit times. Furthermore, a mixed dual input was assumed, which requires portal venous sampling or modeling (13). Sensitivity analysis showed that the σ term of the multiple-capillary microvascular model was much less sensitive and consequently was predicted with less accuracy than were flow and blood volume. Therefore, the models presented included as much physiologic information as we thought the available data could support. The present scheme does not attempt to describe detailed physiologic features of the hepatic vasculature, such as separate arterial and portal systems with arterioloportal vascular shunting (14). In general, models for PET are formulated as ordinary or partial differential equations, which can be either expressed analytically or solved numerically (15). Analytic expressions have the advantage of transparently showing physiologic features in mathematic form, thereby allowing perception of model behavior and often leading to deeper insights. We prefer this approach to the degree that analytic expressions can be derived and fully understood. In some instances, the mathematic derivations can be too complicated or the analytic expression can be too difficult to interpret; in such instances, we consider a numeric approach to be preferable.
CONCLUSION
This study shows that work toward more physiologic kinetic models for the analysis of PET data is fruitful. We describe 2 microvascular models that, compared with the standard compartment model, provide a superior description of the tissue concentrations of an intravascular tracer. The microvascular models include only parameters with a clear-cut physiologic interpretation and provide quantitative regional flow estimates in agreement with flow measurements. Although the method of determining regional blood flow is validated here only for the liver, we expect it to be applicable to all capillary beds. The present approach for estimating hepatic blood flow uses dual-input blood sampling and require some invasive surgery. Therefore, it is not directly applicable for humans but provides information on hepatic blood flow that can be used in the further development of a noninvasive PET method for the liver.
Acknowledgments
This study was supported by grants from the Novo Nordisk Foundation, Aarhus University Hospital Research Initiative, and the Danish Medical Research Council (grant 22-02-0337). The authors thank the staff at the PET Center, Aarhus University Hospital, for excellent assistance.
Footnotes
Received Dec. 30, 2002; revision accepted Jul. 10, 2003.
For correspondence or reprints contact: Ole Lajord Munk, MSc, PET Center, Aarhus University Hospital, Nørrebrogade 44, DK-8000 Aarhus, Denmark.
E-mail: olmunk{at}pet.auh.dk




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}