


Abstract
Pharmacokinetics and metabolite analysis of radioiodinated 5-iodo-4′-thio-2′-deoxyuridine (ITdU), a potential tumor cell proliferation-imaging agent, were investigated in rodents. Methods: Blood and urine metabolites of radiolabeled ITdU were analyzed by thin-layer chromatography in rats. Tissue fractionation studies in mice were also performed to investigate the incorporation of 125I-ITdU into DNA. Results: The blood distribution study suggested the influx of 125I-ITdU into rat peripheral blood cells. The blood metabolism of 125I-ITdU in vitro was negligible. Plasma analysis at 60 min after injection showed that 50% of the radioactivity was intact 125I-ITdU. The plasma clearance of 125I-ITdU showed a biexponential function with half-lives of 0.2 and 29.5 min, representing 94.2% and 5.8% of plasma 125I-ITdU concentration, respectively. 125I-ITdU was excreted in urine 86% intact at 3.5 h after injection. Extraction of the tissues demonstrated that >70% of the activity in the duodenum, spleen, and thymus was incorporated into DNA at 8 h after injection. On the other hand, <8% of the activity in the liver was in the DNA fraction at that time. Conclusion: These data demonstrate that 125I-ITdU is less susceptible against catabolism and readily incorporated into DNA in proliferating tissues. Therefore, 125I-ITdU may be useful in measuring DNA synthesis with SPECT.
The noninvasive evaluation of tumor proliferation activity by imaging methods will provide information that is useful in patient management. For example, detection of the most rapidly growing regions of the tumor would be useful for planning the radiation fields of radiotherapy and for identifying a suitable portion for biopsy. Such methods would also permit an early and accurate determination of therapeutic effects, which are difficult to identify by CT- or MRI-based anatomic evaluation or FDG PET-based measurement of glucose metabolism. In particular, these methods will be useful for an early assessment of the therapeutic effects of anticancer agents that may have strong side effects. To realize these clinical applications, many attempts to develop proliferation imaging tracers have been made using radiolabeled thymidine and its C-5 radiohalogenated analogs for PET and SPECT imaging (1–3). However, a major disadvantage of these radiolabeled nucleoside analogs is that they are rapidly catabolized in vivo. This catabolism results in low tissue utilization of the labeled nucleosides and high background blood levels of the circulating radiolabeled metabolite.
To overcome this catabolism problem, we attempted the introduction of the “4′-thio strategy” as a drug design using a practical radioiodine-labeled nucleoside for proliferation imaging (4). The 5-125I-iodo-4′-thio-2′-deoxyuridine (125I-ITdU), which is a 4′-thio derivative of 5-125I-iodo-2′-deoxyuridine, exhibited a high resistance to the glycosidic bond cleavage reaction provoked by thymidine phosphorylase. Despite this 4′-thio substitution, 125I-ITdU sustained its affinity for nucleoside kinases. Compared with that of 5-125I-iodo-2′-deoxyuridine, the derivative showed higher in vivo stability against deiodination and more rapid DNA incorporation of 125I-ITdU and realized a selective and significant uptake of radioactivity in the proliferating organs. However, due to the rapid blood clearance of 125I-ITdU, the specific uptake in the proliferating organs needs to be generally improved. Therefore, to obtain information contributing to a clear understanding of 125I-ITdU and its disadvantages, we further examined the in vivo pharmacokinetics and the metabolism of 125I-ITdU in rodents. Herein, we also discuss the characteristics and improvement of the 125I-ITdU method used for proliferation imaging.
MATERIALS AND METHODS
Radiochemicals
Carrier-free sodium 125I-iodide was purchased from Amersham Bioscience. 125I-ITdU was prepared from 5-trimethylstannyl-4′-thio-2′-deoxyuridine by an electrophilic substitution reaction using iodine as an oxidant (4). Regrettably, this reaction dramatically lowered the specific activity of the final radiolabeled compound due to the resultant isotopic dilution. However, in this study, the specific activity sufficed for the evaluation. Briefly, sodium 125I-iodide and iodine (in the same molar concentration as that of the trimethylstannyl-precursor) were added to a reaction vial containing MeOH. The trimethylstannyl-precursor solution was added to the vial and allowed to stand for 2 h at room temperature. The labeling reaction was terminated by the addition of sodium metabisulfite, and the MeOH was evaporated. The labeled compound was purified by means of high-performance liquid chromatography (HPLC) on an ODS-80 column (4.5 × 250 mm; Toyo-soda, Co., Ltd.). A MeOH/H2O/trifluoroacetic acid solution (30:70:0.1) was used as the mobile phase, under a flow rate of 0.8 mL/min. The radioactive fraction, eluted with a retention time corresponding to that of the authentic sample, was collected. The HPLC solvent of the collected fraction was then evaporated by heating, and the purified radioiodinated compound was dissolved using sterile saline. For calculations of specific activity, standard solutions of unlabeled compounds were used to compare the area under ultraviolet absorption. The labeling yield of 125I-ITdU was approximately 20%, with a radiochemical purity of >99%. The resultant 125I-ITdU solution was preserved in a refrigerator. Under this condition, the rate of decomposition of 125I-ITdU was <3%/wk.
Metabolism of 125I-ITdU in Blood In Vitro
Seven-week-old male Sprague-Dawley rats (n = 5) were purchased from Japan SLC, Inc., and held for 1 wk before the study. All procedures were performed in accordance with the institutional guidelines (Guidelines for Animal Experiments, Fukui Medical University). Blood was obtained from the jugular vein of each rat using a heparinized 2.5-mL disposable syringe (Terumo Co. Ltd.) under thiopental sodium anesthesia ([RAVONAL] Tanabe; 30 mg/kg via peritoneal cavity injection). 125I-ITdU (55.5 kBq; radiochemical purity, 97%; specific activity, 27.4 MBq/μmol) was mixed into 1.5 mL of fresh, heparinized rat blood (1.3 nmol/mL). Blood samples were incubated at 37°C, and 0.2-mL aliquots were removed and transferred to precooled Eppendorf tubes at 1, 5, 10, 30, and 60 min of incubation. Continuously, whole blood was removed up to a volume of 20 μL and its radioactivity was counted by an auto-well gamma counter (ARC-300; Aloka). Residual blood was centrifuged at 12,000 rpm for 5 min at 4°C, and plasma was removed. Twenty microliters of plasma were collected and its radioactivity was counted by an auto-well gamma counter (ARC-300). Then, 30 μL of the plasma sample were treated with a 5-fold volume (w/v) of ice-cold 10% trichloroacetic acid (TCA), after which it was centrifuged at 12,000 rpm for 5 min. Three microliters of the supernatant were applied to a silica gel plate (Sil60 F254; Merck) with the standards for the nucleoside. The resultant metabolites were then separated from the parent compound, 125I-ITdU, by a thin-layer chromatography (TLC) method using a solvent system consisting of chloroform/isopropyl alcohol (3:1). TLC plates were analyzed with a bioimaging analyzer (BAS-2500; Fuji Photo Film Co.) and compared with standards of ITdU, 5-iodouracil (IU), and the sodium 125I-iodide.
Blood cell radioactivity distribution ratio was calculated as follows:
 where Cw and Cp are the radioactivity concentration in whole blood and in plasma, respectively. Cw and Cp were measured as described above. Sample hematocrit, H, was measured as follows: In brief, a small amount of whole blood was collected by a heparinized hematocrit-capillary tube (Terumo Co. Ltd.), one end of which was sealed by a Terumo seal. The capillary tube was then centrifuged at 12,000 rpm for 5 min (model-3100; Kubota). Afterward, hematocrit was calculated by a hematocrit reader (Kubota).
where Cw and Cp are the radioactivity concentration in whole blood and in plasma, respectively. Cw and Cp were measured as described above. Sample hematocrit, H, was measured as follows: In brief, a small amount of whole blood was collected by a heparinized hematocrit-capillary tube (Terumo Co. Ltd.), one end of which was sealed by a Terumo seal. The capillary tube was then centrifuged at 12,000 rpm for 5 min (model-3100; Kubota). Afterward, hematocrit was calculated by a hematocrit reader (Kubota).
In Vivo Kinetics of 125I-ITdU in Rats
In vivo metabolism of 125I-ITdU was studied in 7-wk-old Sprague-Dawley rats (n = 5). The rats were anesthetized by intraperitoneal cavity administration of thiopental sodium (30 mg/kg via peritoneal cavity injection), and the jugular vein was exposed by surgery for blood sampling. 125I-ITdU (668 kBq; radiochemical purity, 98%; specific activity, 98.9 MBq/μmol) was injected via the tail vain (6.94 nmol per head). Then, 150 μL of blood samples were collected periodically (at 1, 5, 10, 15, 30, 60, 120, and 180 min) via the jugular vein using a heparinized 1.0-mL disposable tuberculin syringe (Terumo). Twenty microliters of each blood sample were removed and its radioactivity was counted with an auto-well gamma counter (ARC-300). To evaluate the blood cell radioactivity distribution ratio, the hematocrit was also measured as described above. Residual withdrawn blood was centrifuged at 12,000 rpm for 5 min at 4°C and plasma was removed. Twenty microliters of plasma were collected and its radioactivity was counted with an auto-well gamma counter (ARC-300). Thirty microliters of plasma sample were then treated with a 5-fold volume (w/v) of ice-cold 10% TCA and centrifuged at 12,000 rpm for 5 min. Three microliters of the supernatant were applied to a silica gel plate (Sil60 F254; Merck) with standards for the nucleoside, and analysis was conducted as described above.
Urine samples were collected 3.5 h after injection by puncture of the urinary bladder using a 1.0-mL disposable syringe, and the radioactivity in the sample was counted with an auto-well gamma counter (ARC-300). Urine samples were treated with a 5-fold volume (w/v) of ice-cold 10% TCA and centrifuged at 12,000 rpm for 5 min. Three microliters of the supernatant were applied to a silica gel plate (Sil60 F254) with standards for the nucleoside, and analysis was conducted as described above.
Pharmacokinetic parameters of intact 125I-ITdU were calculated to fit a 2-compartment fast-intravenous-dose model by using a standard Marquardt-Levenberg least-squares algorithm written in BASIC programming language (5).
Mouse Tissue Fractionation
Nine-week-old C57BL/6 mice were purchased from Japan SLC, Inc., and held for 1 wk before the study. In a review of the previous literature, we observed that specific and significant visualization of the proliferating organ requires a greater washout and clearance of the exchangeable fraction from background tissues. Recently, it was reported that the background activity of 5-125I-iodo-2′-deoxyuridine was greatly reduced by perioral administration of KClO4 and KI (6). Therefore, to improve the clearance of background activity in this study, KI (0.2 g/L) and KClO4 (2 g/L) were given in drinking water for 24 h before the experiment. A saline solution (0.05 mL) containing 2 nmol (0.19 MBq) 125I-ITdU (radiochemical purity, 98%; specific activity, 98.9 MBq/μmol) was administered as a bolus through the tail vein. The mice were killed by blood removal from the heart under ether anesthesia at predesigned intervals of 2, 8, and 18 h. Three animals were killed at each time point. The thymus, spleen, liver, and first 5 cm of duodenum distal to the pyloric valve were removed. Tissue extraction using a quantitative acid extraction technique was performed on separate small molecules, RNA, DNA, and macromolecules as described previously (4). In brief, whole tissues were homogenized in 1 mL of ice-cold 0.2N HClO4 using a Polytron homogenizer (PT1200; Kinematica Inc.). After being cooled on ice for 10 min, the homogenates were centrifuged at 1,200g at 4°C for 10 min. The acid-soluble fraction was removed, and the radioactivity was counted using an auto-well gamma counter (ARC-300). The acid-insoluble precipitate was resuspended in 2 mL of 0.66 mg/mL type III ribonuclease (68 Kunitz U/mg; Sigma Chemical Co.) in 0.1 mol/L N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid), pH 7.6, and incubated at 37°C for 30 min, after which 0.5 mL of ice-cold 5N HClO4 was added. After cooling and centrifugation, the supernatant fluid containing RNA was removed, and the radioactivity was determined. The remaining precipitate was resuspended in 1 mL of 1N HClO4 and incubated at 90°C for 15 min. After cooling and centrifugation, the supernatant containing DNA hydrolysates was removed, and the radioactivity was determined. The pellet was washed once with 1 mL of ice-cold 1N HClO4. The remaining proteins and lipids were solubilized by 1N NaOH, and the radioactivity was determined.
RESULTS
Metabolism of 125I-ITdU in Blood In Vitro
The radioactivity distribution of 125I-ITdU in blood cells was 14%–15% of that present in whole blood, and no time-dependent change was observed (Table 1). Therefore, the radioactivity was mainly distributed in the plasma. During the time period studied, almost all of the radioactivity in plasma was found in the acid-soluble fraction. A typical radiochromatogram after 60 min of incubation (Fig. 1) had 2 radioactive peaks (peaks A and C). Peaks A and C were identified as free iodide and intact ITdU, respectively. The control experiments indicated that the injected samples contained around 3% of peak A. After 60 min of incubation, no metabolism of 125I-ITdU was observed.

Typical radiochromatogram of plasma sample after 60 min of in vitro incubation with 125I-ITdU in rat blood. Peaks A and C were characterized as 125I-iodide and 125I-ITdU, respectively.
Blood Cell Incorporation In Vitro (n = 5)
In Vivo Kinetics of 125I-ITdU in Rats
The radioactivity distribution in blood cells at 1 min was 20% of that present in whole blood and increased slightly from 1 to 180 min (Table 2). In vivo, radioactivity was mainly distributed in plasma. Almost all of the radioactivity in plasma during the time period was found in the acid-soluble fraction. A typical radiochromatogram of 30 min after intravenous injection (Fig. 2A) showed 5 radioactive peaks (peaks A–E). Peaks A and C were identified as free 125I-iodide and intact 125I-ITdU, respectively. The other peaks were not identified. There was no 125I-IU peak during the time studied. Figure 3A shows the time course of the total radioactivity and its components in the plasma after intravenous injection of 125I-ITdU (data are expressed as mean ± SD of 5 experiments). Plasma radioactivity over the first 60 min was predominantly due to 125I-ITdU (Fig. 3A). Fifty percent of the radioactivity in plasma at 60 min after injection was intact 125I-ITdU. After 120 min, free 125I-iodide was predominant. Concentrations of free 125I-iodide, peaks B and E, gradually increased with time. The peak D concentration did not change with time.
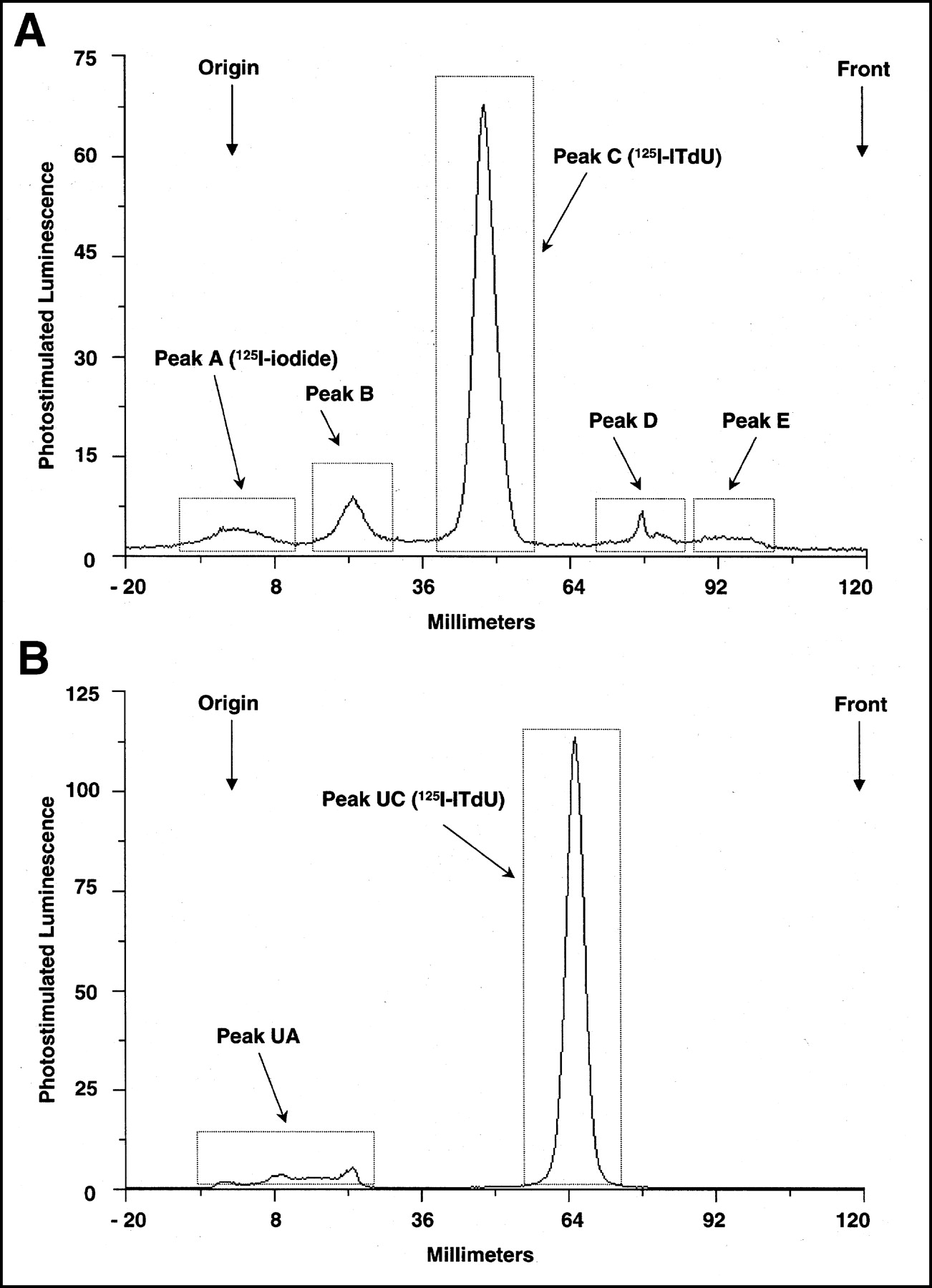
(A) Typical radiochromatogram of plasma sample 30 min after intravenous injection of 125I-ITdU in rats. Peaks A and C were characterized as 125I-iodide and 125I-ITdU, respectively. Peaks B, D, and E were not identified. There was no 125I-IU peak. (B) Typical radiochromatogram of urine sample 3.5 h after injection of 125I-ITdU in rats. Peak UC was characterized as 125I-ITdU. Peak UA was not identified. There was no 125I-IU peak in urine.
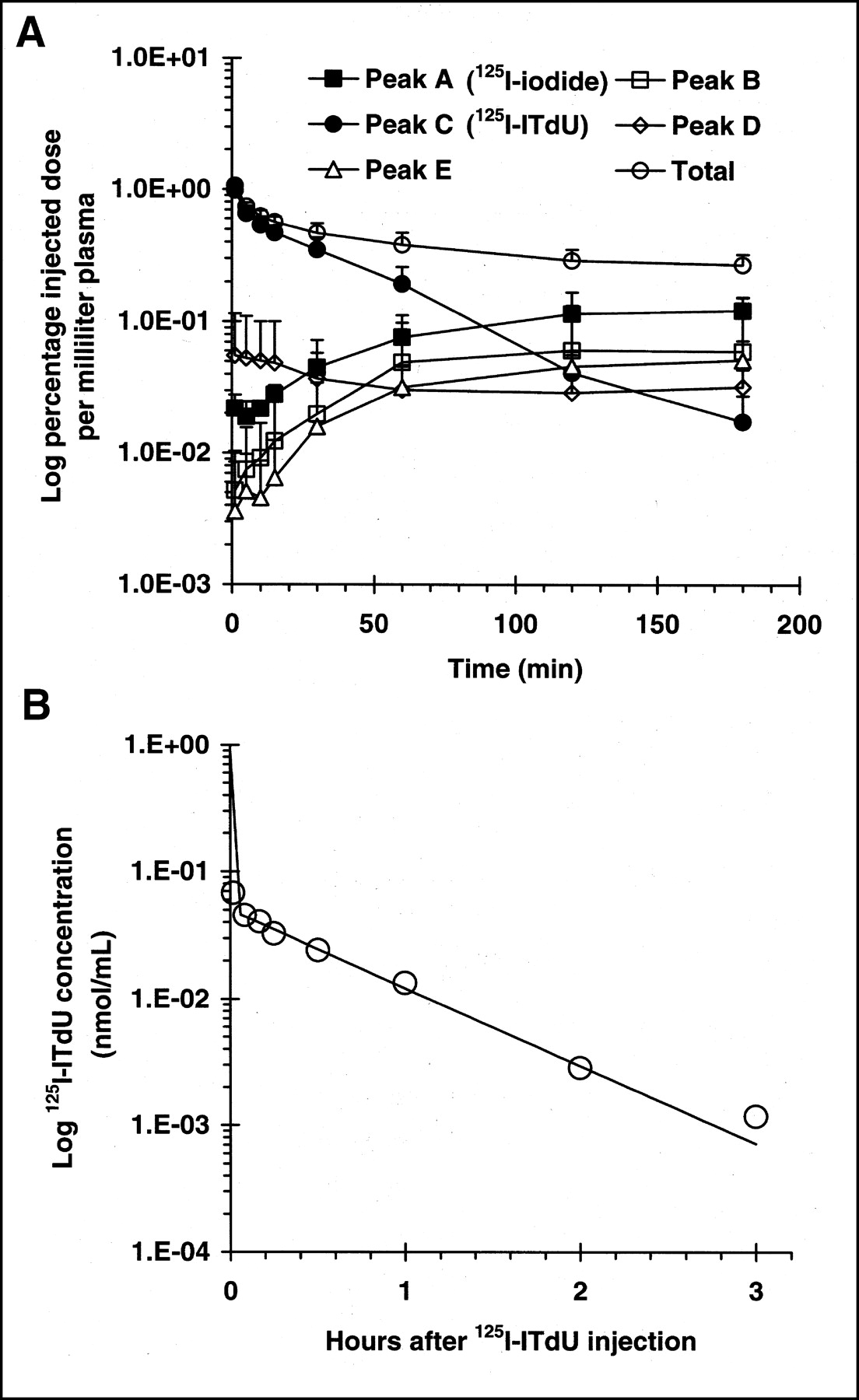
(A) Plasma levels of 125I-ITdU and its radioactive metabolites in rats in vivo after intravenous injection of 125I-ITdU. Data are expressed as mean ± SD of 5 experiments. (B) Plasma clearance curve of 125I-ITdU. Data were fitted, using standard Marquardet-Levenberg least-squares algorithm written in BASIC programming language, to biexponential curves. Half-life and fraction cleared in each component were 0.2 min (94.2%) and 29.5 min (5.8%), respectively.
Blood Cell Incorporation After Intravenous Injection of 125I-ITdU (n = 5)
The total radioactivity in the plasma as a function of time after injection was characterized by an initial rapid decline in the concentration of the isotope, followed by a very slow elimination phase (Fig. 3A). The concentration of intact 125I-ITdU in plasma was well described by a classical 2-compartment model (Fig. 3B) in which there is a major rapid distributive phase (A = 0.8 ± 0.1; α = 225.0 ± 0.2 h−1) and a much slower postdistributive phase (B = 0.1 ± 0.0; β = 1.4 ± 0.1 h−1) (A and B represent the mean ± SD of 5 experiments of 125I-ITdU plasma concentrations [nmol/mL]; α and β represent the mean ± SD of 5 experiments of rate constants for A and B, respectively).
The urine samples collected at 3.5 h after injection were also analyzed by TLC. The following data are expressed as mean ± SD of 5 experiments. During the 3.5-h period after injection, 51.5% ± 6.1% of the injected radioactivity was excreted by the kidneys. A typical radiochromatogram (Fig. 2B) had 2 radioactive peaks (peaks UA and UC). Peak UC was identified as intact 125I-ITdU. Peak UA (14.2% ± 2.1%) might be free 125I-iodide but was not identified. Urine radioactivity over the 3.5 h was predominantly due to 125I-ITdU (85.5% ± 2.1%). There was no 125I-IU peak in the urine or in the blood.
Mouse Biodistribution and Tissue Fractionation
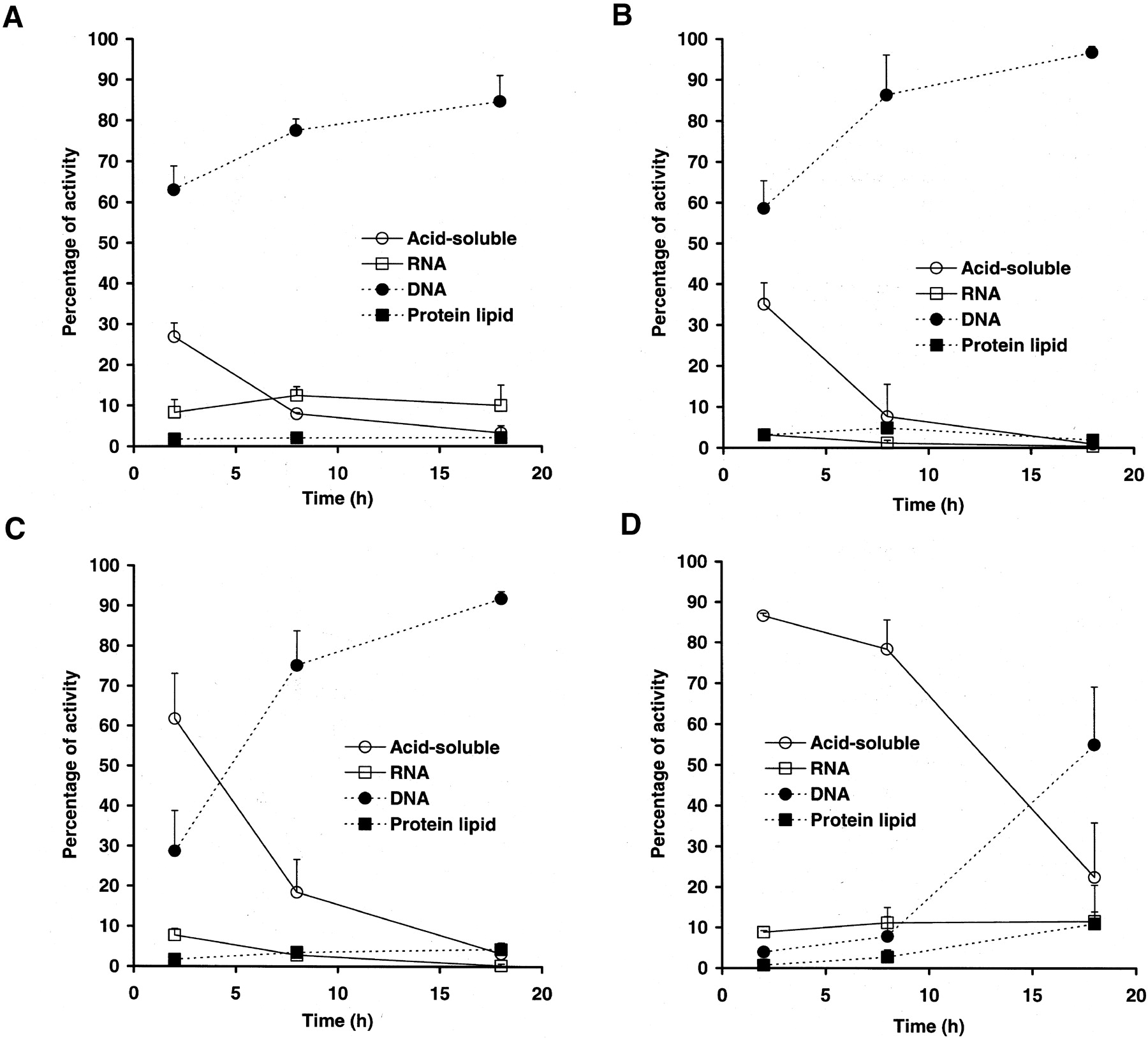
The time-course study of the tissue fractionation of highly proliferating tissues showed that >70% of the radioactivity was distributed in the DNA fraction at 8 h after injection in these tissues (Fig. 4A–4C). However, a significant amount of acid-soluble radioactivity remained at 2 h after injection. The duodenum showed the most rapid incorporation into DNA, followed by that in the thymus and that in the spleen. The lowest proliferating organ, the liver, showed no incorporation into the DNA at 8 h after injection (Fig. 4D). However, >50% of the radioactivity was distributed in the DNA fraction at 18 h after injection (Fig. 4D).

Time-dependent radioactivity distribution in each fraction in organs of mice. Data are expressed as mean ± SD of 3 experiments. (A) Duodenum. (B) Thymus. (C) Spleen. (D) Liver.
DISCUSSION
This in vitro metabolism study showed that 125I-ITdU was not catabolized in rat blood, a result that is in accordance with the previously obtained in vitro enzymatic degradation assay (4). The in vivo blood metabolite analysis also showed a high stability of 125I-ITdU over the first distribution phase. There was no 125I-IU formation. Moreover, urine metabolite analysis showed that urine radioactivity over the 3.5 h was predominantly due to 125I-ITdU (86%). These data support the high stability of the glycosidic bond of 125I-ITdU in vivo in rats. From these results, we considered that the in vitro glycosidic bond stability data would directly apply to the in vivo condition. However, the percentage of a small number of metabolites (<0.1% injected dose per milliliter of plasma), which might be mainly due to 125I-iodide, increased with time in the second slow-clearance phase in plasma. This small increase of metabolite concentration in plasma cannot be ignored due to the relatively slow clearance of metabolites. Because radioiodinated ITdU might be the only chemical entity known to undergo metabolic trapping in proliferating tissues, the elevated background levels produced by slow clearance of metabolites might result in the masking of radioiodinated ITdU uptake during scintigraphic evaluation. Therefore, the prolonged retention of radioiodinated ITdU or acceleration of metabolite elimination would be very useful in the application of radioiodinated ITdU as a practical diagnostic agent. The mechanism of or enzymes responsible for the metabolite production are unknown. Therefore, it is difficult to design chemically modified ITdU analogs that would be resistant to this metabolism, though such an approach seems promising. Another practical approach to prolong the retention of radioiodinated ITdU is to combine it with competitive inhibitors of tubular radioiodinated ITdU secretion, such as cimetidine. Lu et al. (7) and Borbath et al. (8) showed that cimetidine, an inhibitor of the organic cation secretory system, inhibited the secretion of 5-76Br-bromo-2′-fluoro-2′-deoxyuridine and increased the standardized uptake value in the proliferating organ without affecting incorporation into DNA. Approaches for the acceleration of metabolite elimination are difficult, given that 3 metabolites remain unidentified. If the radiolabeled metabolite is mainly free 125I-iodine, perioral administration of KClO4 and KI might be effective for reducing the background levels. Dupertuis et al. pointed out that gastric secretion and intestinal reabsorption of free radioiodine have been shown to constitute an iodine reservoir that competes with and can delay renal elimination (6). They showed that stomach secretion and whole-body retention of free radioiodide were markedly reduced 4 h after injection in mice that were preadministered KClO4 and KI (6). In any case, further validation studies of organic cation inhibitor combinations or acceleration of metabolite elimination should be conducted to potentiate radioiodinated ITdU as a practical imaging agent.
The biologic activity of ITdU is dependent on an ability to interact with the intracellular metabolic pathways of the pyrimidine metabolism. This necessitates transport across membranes into the cell cytoplasm. Findings in our study suggested that influx of 125I-ITdU into rat peripheral blood cells was evident in vitro and in vivo. These data indicated that the nucleoside transport mechanism appeared to remain unaffected by structural modification at the 4′-thio substitution.
In accordance with findings in our previous in vitro cell fractionation studies, the time-course study of the tissue fractionation also showed that >70% of the radioactivity was distributed in the DNA fraction in proliferating tissues at 8 h after injection. These in vivo data indicate that proliferating tissue accumulations of 125I-ITdU reflect the tissue DNA synthesis activity. The low-proliferating organ, the liver, did not show any DNA incorporation at 8 h after injection. However, >50% of the radioactivity was distributed in the DNA fraction at 18 h after injection. The radioactivity accumulations in the liver at 18 h after injection were 100 times lower than those in the duodenum (data not shown). Therefore, these low levels of DNA-incorporated activity might derive from background DNA syntheses, such as unscheduled DNA synthesis or mitochondrial DNA synthesis.
Nevertheless, this study provides some useful information related to the in vivo behavior of radioiodine-labeled ITdU in rodents; however, one must take into account that metabolism in humans, especially in human tumors, can be significantly different due to differences in species. Therefore, imaging human tumor with 123I-ITdU and SPECT needs to be done to better understand this as a tumor proliferation imaging agent.
CONCLUSION
Pharmacokinetics and metabolite analysis of radioiodinated ITdU were investigated in rodents. 125I-ITdU is less susceptible against catabolism and readily incorporated into DNA in proliferating tissues in vivo. Therefore, radioiodinated ITdU may be of use in measuring DNA synthesis with SPECT and PET. Further validation studies to potentiate radioiodinated ITdU as a practical imaging agent are warranted.
Footnotes
Received Dec. 3, 2002; revision accepted Jun. 13, 2003.
For correspondence or reprints contact: Yasuhisa Fujibayashi, PhD, DMSc, Biomedical Imaging Research Center, Fukui Medical University, 23 Shimoaizuki, Matsuoka-cho, Yoshida-gun, Fukui, 910-1193, Japan.
E-mail: yfuji{at}fmsrsa.fukui-med.ac.jp
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Evaluation of 4'-[Methyl-11C]Thiothymidine in a Rodent Tumor and Inflammation Model
- Preferential Tumor Targeting and Selective Tumor Cell Cytotoxicity of 5-[131/125I]Iodo-4'-Thio-2'-Deoxyuridine
- 123I-ITdU-Mediated Nanoirradiation of DNA Efficiently Induces Cell Kill in HL60 Leukemia Cells and in Doxorubicin-, {beta}-, or {gamma}-Radiation-Resistant Cell Lines
- Evaluation of 4'-[Methyl-14C]Thiothymidine for In Vivo DNA Synthesis Imaging