


Abstract
The lumped constant (LC) is a correction factor used to infer glucose metabolic rate (MRglc) from FDG metabolic rate (MRFDG). Methods: LC was determined in normal brain in 10 subjects (4 male, 6 female) by measuring regional MRglc and MRFDG independently using 1-11C-glucose and 18F-FDG with dynamic positron tomographic imaging, arterial blood sampling, and region-of-interest time-activity curve analysis with appropriate compartmental models. Results: The mean LC (±SD) for normal brain was found to be 0.89 ± 0.08. The value for cerebellum was slightly lower, 0.78 ± 0.11 (P = 0.006; 2-tailed paired t test). Conclusion: The LC values determined in this study are considerably higher than older values in the literature, probably because of methodologic differences, but agree with a recent study by Hasselbalch.
Imaging the regional metabolism of glucose in the brain as well as other organs often uses 18F-FDG. It is well known that FDG is transported across the cell membrane by the same transporters that are responsible for the facilitated transport of glucose. Once in the cytoplasm FDG is phosphorylated by hexokinase, but at that point its metabolism ceases except for very slow dephosphorylation. Thus, the accumulation rate of FDG is proportional to the metabolic rate of glucose (MRglc) because FDG is transported and phosphorylated by the same pathways as glucose. However, transport and phosphorylation of the 2 hexoses occur at somewhat different rates so a correction factor is necessary to convert FDG metabolic rate (MRFDG) to MRglc. This correction factor is the lumped constant (LC), a term originally proposed by Sokoloff et al. (1) to describe the ratio of 2-deoxyglucose (DG) metabolic rate to MRglc in the rat brain. The LC is termed “lumped” because it is made up of 6 constants. The formula for the LC, defined by Sokoloff et al., is:
 Eq. 1 where λ is the ratio of the distribution volume of FDG to that of glucose, φ is the fraction of glucose that continues down the Embden-Meyerhof pathway after being phosphorylated, Km is the Michaelis-Menten constant for phosphorylation of glucose (* indicates FDG), and Vmax is the maximum velocity for phosphorylation of glucose (* indicates FDG). The LC is used to convert MRFDG to MRglc by dividing MRFDG by the LC. Clearly, the value of the LC is critical in quantitative calculation of regional cerebral glucose metabolic rates when FDG is used as the tracer.
Eq. 1 where λ is the ratio of the distribution volume of FDG to that of glucose, φ is the fraction of glucose that continues down the Embden-Meyerhof pathway after being phosphorylated, Km is the Michaelis-Menten constant for phosphorylation of glucose (* indicates FDG), and Vmax is the maximum velocity for phosphorylation of glucose (* indicates FDG). The LC is used to convert MRFDG to MRglc by dividing MRFDG by the LC. Clearly, the value of the LC is critical in quantitative calculation of regional cerebral glucose metabolic rates when FDG is used as the tracer.
Determination of the value of the LC requires either simultaneous measurement of MRFDG and MRglc or independent measurement of each of the 6 values of the formula for the LC. A direct approach, initially proposed and used by Sokoloff et al. (1), is to measure the relative MRFDG and MRglc by determining arteriovenous (A−V) differences across the brain during steady state for both FDG and glucose. Because the metabolic rates are directly proportional to the A−V differences, the LC should be equal to the ratio of the fractional A−V differences for the 2 substances. Sokoloff et al. implemented this approach for 14C-DG in the rat and obtained a value for the LC of 0.483 ± 0.107 (1). Reivich (2), by a similar approach in humans, estimated the LC for FDG to be 0.52 ± 0.028.
Phelps et al. (3), using a 3-compartment model to describe the transport and phosphorylation of FDG, determined the kinetic parameters for FDG from dynamic PET images. They compared the calculated MRFDG for the whole brain to the average literature values for MRglc. The resultant value for the LC was 0.42 ± 0.059. Brooks et al. (4), using essentially the same approach as Phelps et al., found the value of the LC in humans to be 0.50.
Note that Sokoloff et al. (1) state that “the lumped constant is really the constant of proportionality between the steady state rates of 14C-DG and glucose phosphorylation by the brain when it is exposed to equal arterial plasma concentrations of both.” Although the approach used in this study is not a steady-state method, it is more appropriate because, when true steady state is reached, DG uptake approaches zero because of dephosphorylation.
The more direct approach for measuring the LC, described here, is to determine independently both MRFDG and MRglc using PET imaging. The method for determining MRglc is similar to the approach described by Blomqvist et al. (5), using 1-11C-glucose. MRFDG was determined using parameter optimization, similar to the approach of Huang et al. (6).
MATERIALS AND METHODS
Preparation of 18F-FDG and 1-11C-Glucose
18F-FDG was synthesized by the method of Hamacher et al. (7). The FDG yield was typically 5.2 GBq (140 mCi) at 75 min after the end of bombardment (EOB), a 65% decay-corrected radiochemical yield. The radiochemical and chemical purity of the product was measured by analytic high-performance liquid chromatography (HPLC) using an aminopropyl normal-phase column (Alltech, Deerfield, IL) with a CH3CN:H2O (90:10, v/v) mobile phase and refractive index and radioactivity detection of the effluent. Silica gel thin-layer chromatography with a CH3CN:H2O (95:5, v/v) mobile phase was also used to assess radiochemical purity, which was consistently > 99% by both analytic methods. The measured specific activity of the 18F-FDG was >740 GBq/mmol (>20 Ci/mmol) at the end of synthesis.
The synthesis of 1-11C-glucose followed the method of Shiue and Wolf (8,9) as recently modified by Link et al. (10) and Dence et al. (11). Typically, 63 GBq (1.7 Ci) 11C-cyanide at the EOB yielded 1.3–1.5 GBq (35–40 mCi) 1-11C-glucose at the end of synthesis and purification. The glucose was separated from mannose and arabinose using an Aminex HPX-87P 30 cm × 7.8 mm HPLC column (BioRad Laboratories, Hercules, CA) eluted with sterile water at 70°C with radioactivity detection. The radiochemical and chemical purity of the product was measured by analytic HPLC using another Aminex HPX-87P column at 70°C eluted with deionized water and with refractive index and radioactivity detection of the effluent.
Human Studies
Healthy volunteers (4 male, 6 female; age, 28–47 y) were recruited from the population at the University of Washington. Potential subjects were rejected if there was any history of head injury with loss of consciousness, migraine headaches, seizures, or other neurologic problems. No subjects were taking any medications at the time of the study and none was diabetic. The subjects fasted overnight before the PET study. The study was approved by the University of Washington Human Subjects Committee, and all subjects documented their informed consent.
A foam headrest was molded to the back of the head, and a thermoplastic face mask was fitted. An arterial catheter was inserted into the radial artery and was connected to an automated blood sampler (12). A venous catheter was placed in the contralateral arm for injection. Subjects were awake, but with minimal visual, auditory, or tactile stimulation during the study.
The range of activity injected was 310–363 MBq (8.4–9.8 mCi) 18F-FDG. One patient received only 130 MBq (3.5 mCi) 1-11C-glucose, but the others received 533–700 MBq (14.4–18.9 mCi). The dataset associated with the lower dose of 1-11C-glucose was examined carefully. It showed more noise than did data from the other subjects but yielded similar results for parameter estimates and therefore was included in all analyses.
Blood Sampling, Counting, and Calibration
During the PET studies arterial blood was sampled with an automated sampler (12). One-milliliter samples were obtained at 20-s intervals initially, followed by progressively longer intervals. The samples were centrifuged and 0.5 mL of plasma was pipetted into plastic tubes for counting in a 10-detector γ-scintillation counter (Cobra; Packard, Downers Grove, IL). Each sample was counted for 1 min. Sample activity was decay corrected to the time the blood sample was obtained. In addition, sample activity was converted into units of MBq/mL (27.03 μCi/mL) using calibration factors determined with sample bottles of 18F activity measured in a dose calibrator (Capintec, Ramsey, NJ). The same bottles were imaged in the tomograph to determine the tomograph calibration factor. This made it possible later to scale the PET data from counts per pixel per second to MBq/mL (27.03 μCi/mL). Thus, both the plasma and tissue data were in the same units for subsequent data analysis. Before scanning and isotope injection, a blood glucose level was drawn and analyzed by a calibrated Glucose Analyzer II (Beckman Coulter, Inc., Fullerton, CA). This analysis was repeated several times after isotope injection.
PET Imaging
The tomograph used was an Advance whole-body positron emission tomograph (General Electric Medical Systems, Milwaukee, WI) providing 35 image planes of data over a 15-cm axial field of view (13–15). The tomograph includes 18 rings of Bi4Ge3O12 detectors with 672 crystals per ring. The system sensitivity in 2-dimensional mode (axial septa in place) is 3.65 kcps/kBq/mL (135 kcps/μCi/mL) (cps = counts per second). The limiting transaxial resolution is 4.1 mm with a slice thickness of 4 mm. The maximum counting rate obtainable with a head-sized phantom is >2 million coincidence events per second.
After positioning the subject in the tomograph, an attenuation image was obtained with rotating 68Ge rod sources, for a minimum of 8 × 108 prompt coincident events. Both attenuation imaging and emission imaging were done in 2-dimensional mode with the axial septa in place. A 1-min image was acquired just before the injection of 1-11C-glucose (beginning at time t = −1 min). At t = 0 the injection began and was continued for 1 min using a syringe infusion pump (Harvard, Cambridge, MA). The image acquisition sequence, starting at t = 0 was four 15-s, four 30-s, four 1-min, four 3-min, and eight 5-min images. The same sequence was used for both 1-11C-glucose (the first study) and then for FDG, which began 90–100 min after the start of the 1-11C-glucose injection. After acquisition, the PET data were reconstructed with attenuation correction and a Hanning filter, which yielded images with a resolution of approximately 6-mm full width at half maximum. T1-weighted MR images were acquired on the same day with a Signa 1.5-T system (General Electric Medical Systems). T1-weighted images were acquired axially at an interval of 6.5 mm and were reoriented to align with the PET images using the algorithm of Woods et al. (16).
Data Analysis
Image manipulation, including image summation and region-of-interest (ROI) placement, was done on a Macintosh computer (Apple Computer, Cupertino, CA) with the program Alice (Parexel, Waltham, MA). The reconstructed FDG images were summed from 45 to 75 min to generate high-count images for placement of ROIs. Using the MR images for reference, ROIs were placed over the following areas of the brain: frontal cortex, temporal cortex, parietal cortex, occipital cortex, caudate, putamen, thalamus, cerebellum, and white matter. All of these structures were well resolved on the FDG PET studies (Fig. 1). Partial-volume effects were minimized by placing ROIs at least 5 mm from the edge of each structure. After placement of the ROIs, time-activity curves were generated for all ROIs. Subsequent manipulation of the time-activity curve data was performed with the spreadsheet program EXCEL (Microsoft, Redmond, WA). The time-activity curves were scaled to convert them to units of MBq/mL (27.03 μCi/mL). The ROI time-activity curves were not decay corrected.

MRI (A) and PET (B) images at 3 levels illustrating placement of ROIs. MR images have been reoriented to align with PET images. ROI placement is shown for whole brain, frontal cortex, parietal cortex, occipital cortex, caudate, putamen, thalamus, and white matter. In each case, ROIs were also placed in adjacent slices to increase volume of tissue analyzed.
The plasma time-activity curves, also in units of MBq/mL (27.03 μ Ci/mL), were smoothed and interpolated with a program designed for the task (17) using a physiologically realistic model for the behavior of tracers in plasma. For each tissue time-activity curve, the smoothed plasma data were combined with the tissue data to create an input file appropriate for the parameter optimization program.
The parameter optimization program included appropriate models for both FDG and 1-11C-glucose. The models were set up as differential equations and were solved numerically. Each model included rate constants K1, k2, k3, and k4 as well as delay (time shift of the tissue data relative to the plasma data). A common blood volume term was applied to both tracers. Particular care was taken to test the accuracy of the interpolation of the input blood time-activity curve, the integration of the tissue activity in the model, and the integration of the output tissue data to match the integration that occurred during acquisition with the tomograph. The performance of the program was verified by testing it with data generated from an independent simulation program, STELLA (High Performance Systems, Hanover, NH). The program output exactly matched the STELLA output. Data points were weighted proportional to the square root of the product of activity times image duration. During optimization the models were reparameterized to optimize K1, volume of distribution {K1/(k2 + k3)}, k3, and k4. This reparameterization was used because it resulted more reliably in identification of the global minimum. Conventional parameterization (K1, k2, k3, k4) frequently resulted in the optimizer getting stuck in a local minimum.
The FDG model is identical to that described by Phelps et al. (3) as an extension of the original model of Sokoloff et al. (1) (Fig. 2A). The differential equations are:
 Eq. 2
Eq. 2
 Eq. 3 where Cp is the plasma activity, Ce is the extravascular FDG, and Cm is the tissue FDG-6-PO4, all as a function of time. K*1 is the rate constant describing uptake of FDG from plasma into tissue, k*2 is the rate constant for loss of FDG from tissue back into plasma, k* 3 is the rate constant for phosphorylation of FDG by hexokinase, and k*4 is the rate constant associated with dephosphorylation of FDG-6-PO4 to FDG.
Eq. 3 where Cp is the plasma activity, Ce is the extravascular FDG, and Cm is the tissue FDG-6-PO4, all as a function of time. K*1 is the rate constant describing uptake of FDG from plasma into tissue, k*2 is the rate constant for loss of FDG from tissue back into plasma, k* 3 is the rate constant for phosphorylation of FDG by hexokinase, and k*4 is the rate constant associated with dephosphorylation of FDG-6-PO4 to FDG.
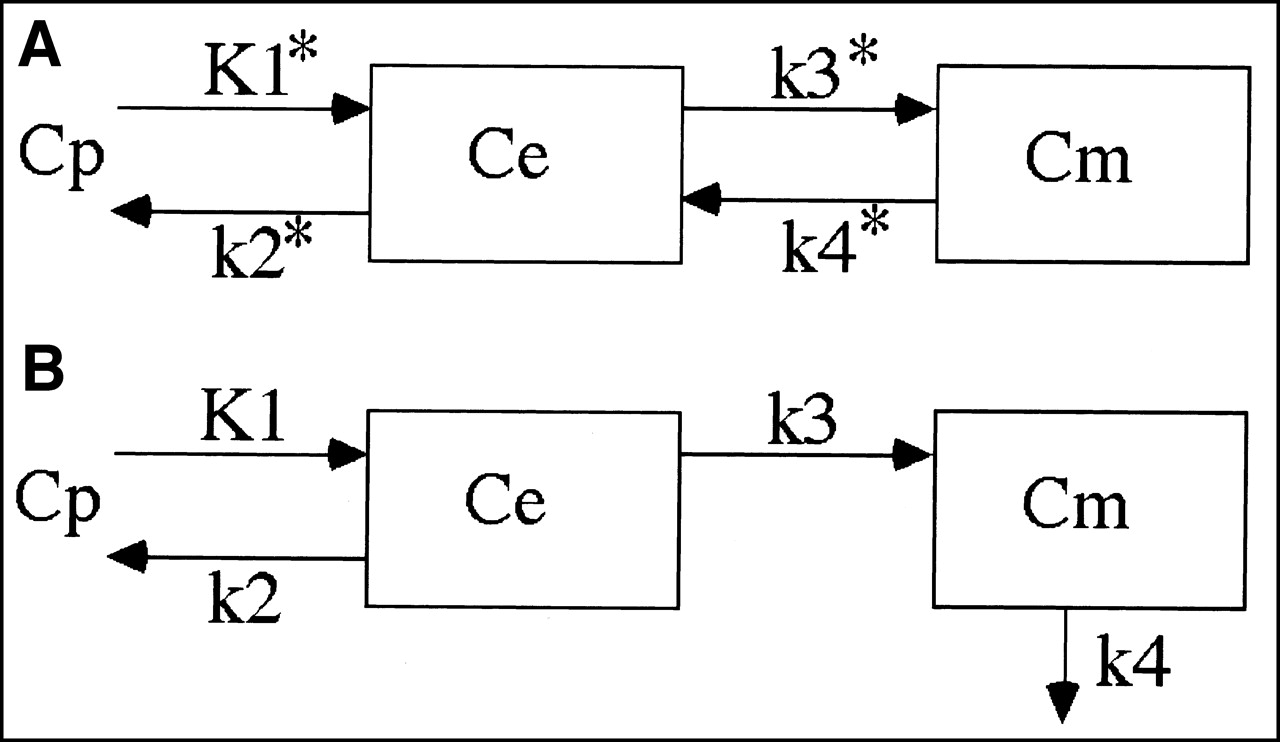
Models for describing kinetic behavior of FDG (A) and 1-11C-glucose (B). In addition to parameters shown, kinetic models include blood volume term, which was constrained to be identical for both tracers, and delay terms to allow temporal shifting of tissue activity relative to blood activity. This was used to deal with different arrival times at brain and blood sampler that arise from delays in arterial transit and in blood sampler tubing.
The 1-11C-glucose model (Fig. 2B) is similar to the model proposed by Blomqvist et al. (5). It is like the FDG model, except in the way k4 is handled. Once glucose is phosphorylated to glucose-6-PO4 it continues to be metabolized and is assumed not to be dephosphorylated. There are several different metabolic pathways that glucose-6-PO4 can enter. Although it is possible to set up a model with multiple pathways and delays, the large number of required parameters would make such a model intractable. Thus, the k4 term represented a compromise, grouping together all of the possibilities for loss of label from tissue associated with the metabolism of glucose. This includes loss primarily as lactate and CO2 but also loss of any other labeled metabolic product. Blomqvist et al. discussed the potential limitations of this approach.
The differential equations for the 1-11C-glucose model are:
 Eq. 4
Eq. 4
 Eq. 5 In both models, radioactive decay is included. Thus, it was essential that the tissue data were not decay corrected before analysis. The plasma data were decay corrected to the time each blood sample was obtained. To account for 1-11C-glucose activity that was still present at the time the FDG was injected, the calculated glucose activity was extrapolated and added to the calculated FDG activity.
Eq. 5 In both models, radioactive decay is included. Thus, it was essential that the tissue data were not decay corrected before analysis. The plasma data were decay corrected to the time each blood sample was obtained. To account for 1-11C-glucose activity that was still present at the time the FDG was injected, the calculated glucose activity was extrapolated and added to the calculated FDG activity.
Because glucose is metabolized in the rest of the body, as well as in the brain, there is a steady accumulation of metabolites in the plasma. We showed previously that the appearance rate of ionic metabolites is linear (in terms of fraction of total activity in plasma) and reached 18% at 60 min (18). Metabolites were not determined in this study. The concept of linear appearance of metabolites, passing through 18% at 60 min, was incorporated into the analysis model. It was assumed that these metabolites remained in the blood, thus contributing to total tissue activity, but there was no uptake of metabolites into brain.
Two possible constraints to the model were explored. The first required the ratio K1/k2 to be identical for both hexoses. Gjedde and Diemer (19) have argued that, because the glucose transporter is symmetric, the ratio K1/ k2 then should be the same for all hexoses. The second constraint was to set k*4 equal to zero. This allowed us to compare our results with those of Hasselbalch et al. (20). Both of these constraints were tested for plausibility by comparing the quality of the fits with and without the 2 different constraints.
In addition, these constraints were statistically evaluated using a likelihood ratio hypothesis testing procedure that compares the sums of squares of weighted residuals obtained from the unconstrained model fit with the corresponding sums of squares for the constrained model fits. If the hypothesized constraint was valid for a given dataset, then the distribution of the differences between these sums of squares should have a χ2 distribution with 1 degree of freedom. For a given region the values of χ 2 were summed across the 10 independent patient datasets to obtain an overall χ2 statistic with 10 degrees of freedom for evaluation of the hypothesized constraint for each region. Because the different regions for a given patient are not independent, due to the spatial correlation of reconstructed PET data, it would not be appropriate to sum χ2 statistics across regions to obtain an overall test. Instead, a Bonferroni correction was used that makes the significance level (αT) for the evaluation of the region-specific χ2 statistics be the desired significance level (α) for the combined evaluation of all regions, divided by the number of regions (i.e., αT = α/ 10). Thus, the probability value for the test with the Bonferroni correction is 10 times the maximum probability value for the test of the hypothesis in the separate regions.
RESULTS
The average plasma glucose concentration in the 10 subjects during the PET scans was 4.98 mmol/L (range, 4.46–5.55 mmol/L) and did not change significantly over the course of the combined imaging study. Typical tissue time-activity curves for several tissues, along with model output, are shown in Figure 3. Note that the quality of the data was excellent (i.e., low noise) and that the model output accurately fit the observed data. The overall quality of the fits is shown in Figure 4A, which shows the residual error after fitting for all 100 datasets (10 subjects, each with 10 tissue time-activity curves). There is little pattern associated with the residual errors, consistent with the hypothesis that the models are appropriate descriptors of the behavior of glucose and FDG.

Typical datasets from both radiopharmaceutical studies in 4 different regions in 4 different subjects. In each case, 1-11C-glucose was injected at t = 0 and FDG was injected at t ≈ 90 min. •, Tissue activity; solid line, model output after parameters of model have been optimized to achieve best fit.

Plots of residual errors after optimized fits of all datasets (10 subjects, 10 regions, 52 times). (A) Optimizations done with no model constraints. (B) Optimizations done with constraint that K1/k2 be the same for both hexoses. (C) Optimizations done with constraint that k*4 = 0. Vertical axis is SDs for fit with no constraints. Thus, 67% of data in A is within 1 SD. Horizontal positions of points have been slightly blurred to allow display of all points.
1-11C-Glucose and 18F-FDG metabolic rates, along with the ratio (i.e., the LC), are shown in Table 1.
Metabolic Rates and LC Estimates ± SD
The mean LC (±SD) calculated for normal brain using PET imaging of 1-11C-glucose and 18F-FDG is 0.89 ± 0.08 and agrees with our studies on contralateral brain in glioma patients (18). The value for cerebellum (0.78 ± 0.11) is slightly lower than that for the rest of the brain, and the value for white matter (0.92 ± 0.12) is slightly higher. The cerebellar value was compared with the average gray matter value (all cortex values averaged) using a paired t test and found to be significantly lower (P = 0.0006) for the 2-tailed test. The corresponding probability value for white matter was 0.15. Calculated LC values for all subjects, grouped by region, are shown in Figure 5.
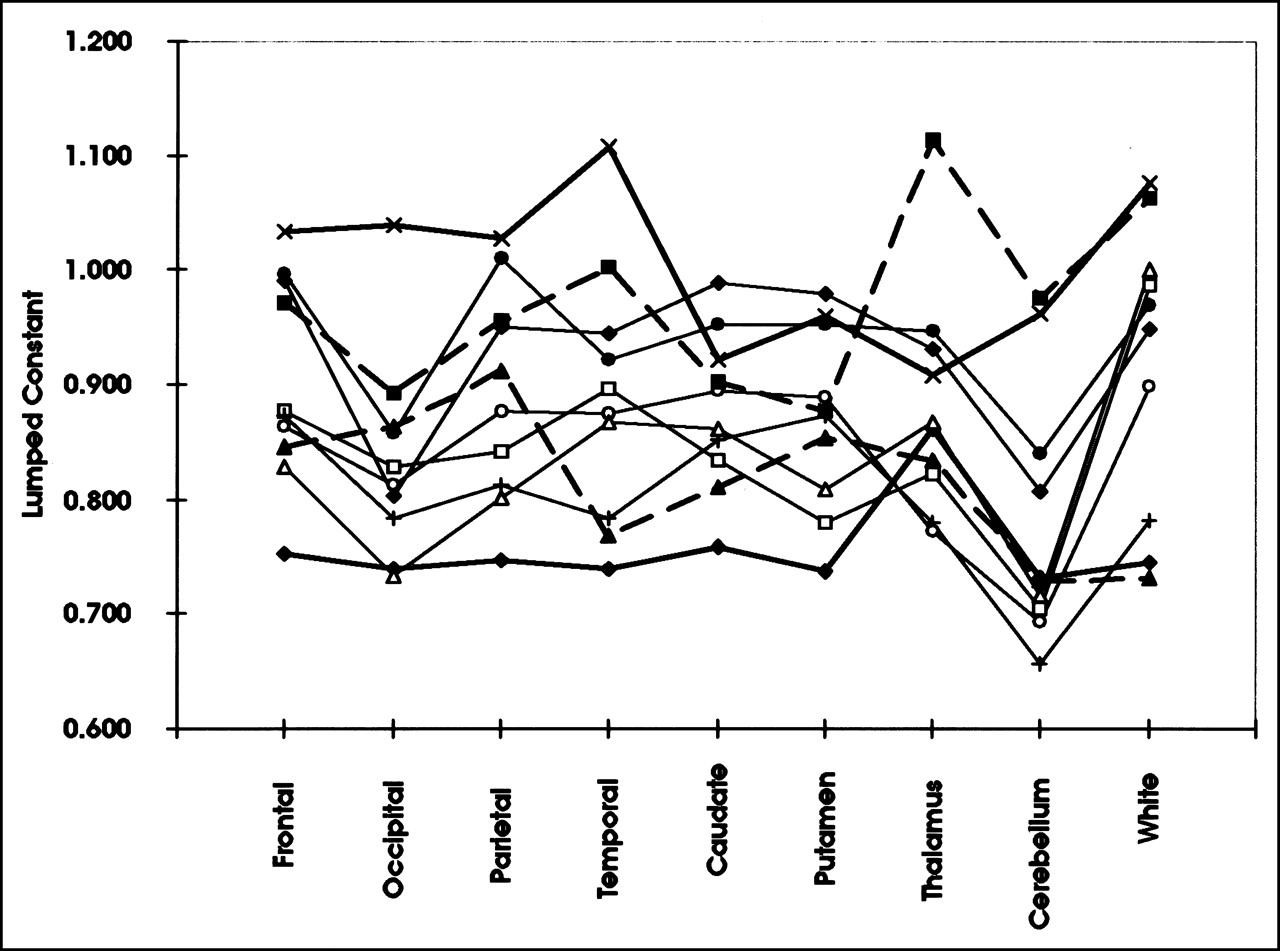
Calculated LC by region for individual subjects. Individual subjects are associated with each different line and symbol. Note that some subjects have consistently higher LC values than other subjects.
Two modeling constraints were explored as part of the data analysis: (a) K1/k2 was constrained to be the same for both glucose and FDG. This had the effect of reducing the number of free parameters by 1, which makes the optimizer converge more easily. In general, it is desirable to define the system with a minimum number of free parameters but the model must still be able to adequately fit the data. (b) k*4 was constrained to be equal to zero.
The effect of these constraints is shown in Figures 4B and 4C. Note that the residual errors with the K1/k2 constraint are worse than without the constraint. The structure in the residual errors suggests that the model tends to overshoot sometimes and undershoot at other times.
The quality of the constrained fit versus the unconstrained fit was tested with the Bonferroni-corrected χ2 statistic. The probability value for either constraint is much less than 0.0001, making it statistically unlikely that either constraint is valid. This data analysis scheme was undertaken to allow us to compare the resulting estimates of the LC with those from Hasselbalch et al. (20), who set k*4 to zero. The k*4 = 0 constraint resulted in a reduction of the estimated MRFDG from 23.0 ± 2.2 mmol/100 g/min to 19.6 ± 2.9 mmol/100 g/min, and an average brain LC from 0.89 ± 0.08 to 0.77 ± 0.065.
DISCUSSION
The models used to analyze the FDG and 1-11C-glucose PET data are critical for valid determination of the LC. For FDG the model was the 4-parameter (3) extension of the original 3-parameter model of Sokoloff et al. (1). Two additional parameters were included in the analysis: (a) delay between blood sampling and tissue imaging times to account for differences in arrival time of blood at the radial artery sampling site and the carotid arteries and (b) blood volume to account for activity in the vasculature of the tissue region. These are well-accepted, valid additions to the model (21). The model computations were validated against independently simulated data and shown to be correct. Because this model is the most widely used and validated model in PET imaging, we accept it for analysis of FDG data to calculate MRFDG.
The 1-11C-glucose model was similar to that proposed and validated by Blomqvist et al. (5). As with the FDG model, delay and blood volume were included. Our implementation differed from the model of Blomqvist et al. in how we approached loss of metabolites from tissue. We propose that the rate of loss is proportional to the amount of metabolized tracer in the tissue. This is a broad assumption, in that glucose metabolism occurs through several pathways and loss of label occurs as CO2 from the pentose shunt and tricarboxylic acid cycle, as lactate from glycolysis, and as other minor metabolites. Blomqvist et al. measured this loss term explicitly by determining cerebral blood flow and the A−V difference for metabolites. This explicit approach is more invasive and is not feasible for regional analysis. We used a linear loss term, directly proportional to the amount of phosphorylated tracer in the tissue. The resultant cerebral MRglc estimates were essentially identical to those of Blomqvist et al., suggesting that our simpler approach to dealing with tracer loss was reasonable.
The glucose model must also be corrected for circulating metabolites. In previous work we measured the rate of appearance of metabolites in peripheral plasma after injection of 1-11C-glucose (18). We found, as did Blomqvist et al. (5), that the fraction of plasma radioactivity ascribable to metabolites increased linearly, reaching 18% at 60 min. This concept was built into the model, with the assumption that the metabolites contribute to activity in the blood but that they do not cross the blood-brain barrier. One weakness with this concept is that CO2 can cross the blood-brain barrier, but at 60 min only 1%–2% of the plasma activity was 11C-CO2, so it should have minimal effect on the parameter estimates.
The only common parameter between the 2 models is blood volume. We explored the feasibility of constraining the ratio of K1/k2 to be identical for both hexoses, but the residual errors and statistical examination do not support this constraint.
The rate constants for FDG and 1-11C-glucose are compared against literature values in Table 2. FDG K*1 and k*3 in this study are slightly higher, whereas k*2 and k* 4 are similar to reported values. The overall measure of MRFDG,
 , is also somewhat higher. For 1-11C-glucose there are fewer literature values, but K1 is similar, k2 and k3 are lower, and k4 is similar to values reported by Blomqvist et al. (5). The resulting metabolic rate,
, is also somewhat higher. For 1-11C-glucose there are fewer literature values, but K1 is similar, k2 and k3 are lower, and k4 is similar to values reported by Blomqvist et al. (5). The resulting metabolic rate,
 , is slightly higher. There is a general trend toward higher gray matter FDG metabolic rates in more recent studies compared with older studies. This is likely due to improved resolution in modern tomographs and therefore less partial-volume effect. This would be particularly true for gray matter because the cortical thickness is similar to the resolution of most tomographs. Another reason for the different values in this study compared with older values is that most early studies did not include k*4. Finally, many of the early studies used impure FDG contaminated with fluorodeoxymannose (FDM), which would result in lower estimates for K*1 and k*3 (22).
, is slightly higher. There is a general trend toward higher gray matter FDG metabolic rates in more recent studies compared with older studies. This is likely due to improved resolution in modern tomographs and therefore less partial-volume effect. This would be particularly true for gray matter because the cortical thickness is similar to the resolution of most tomographs. Another reason for the different values in this study compared with older values is that most early studies did not include k*4. Finally, many of the early studies used impure FDG contaminated with fluorodeoxymannose (FDM), which would result in lower estimates for K*1 and k*3 (22).
Kinetic Rate Constants for FDG and 1-11C-Glucose from Different Human Studies
The only previous work in which both 1-11C-glucose imaging and FDG imaging were done in the same healthy subjects was by Blomqvist et al. (23). The underlying hypothesis in that study was that the difference in behavior between FDG and 1-11C-glucose (corrected only for 11CO2 loss) was due to nonoxidative glucose consumption. A critical underlying assumption was that the true LC for FDG in normal brain was 0.52, taken from Reivich et al. (2). Given this assumption, Blomqvist et al. found, in the resting brain, that MRglc was linearly related to MRFDG with a slope of 0.65. This either could be interpreted as evidence for substantial nonoxidative glycolysis in the normal resting brain or it could also be that the real LC is higher; 0.52/0.65 = 0.80. This value is very nearly the same result as ours and that of Hasselbalch et al. (20). Because there is unlikely to be significant net nonoxidative glycolysis in normal resting brain, we favor the interpretation that the LC is higher. Lactate production in the normal resting human brain averages 0.112 mol/mol of glucose consumed (24). This means that approximately 5.6% of the glucose is metabolized by glycolysis and the 94.4% is by oxidative metabolism. This low fractional lactate production by the normal resting brain suggests that the more likely explanation of the results of Blomqvist et al. is a higher LC. If glycolysis is neglected, the calculated LC is 0.80 for the subjects in the study of Blomqvist et al. This assumption may not be valid in activated brain, where nonoxidative glucose metabolism can be significant (25).
Previous approaches that have been used to estimate the FDG LC can be categorized as follows: (a) comparison of PET measures of FDG uptake with literature values for glucose metabolism; (b) comparison of PET measures of FDG uptake with Fick measures of glucose uptake in the same subjects; (c) comparison of PET measures of FDG uptake with glucose metabolism inferred from oxygen metabolism determined using 15O-oxygen; (d) derivation of the LC from kinetic parameters of FDG, given certain assumptions regarding FDG and glucose kinetics; and (e) determination of arteriovenous extraction fractions for FDG and glucose.
There are different limitations associated with each of these approaches. In general, the approach of comparing the PET FDG study with some measure of glucose metabolism in the same subjects is likely to be more convincing, although this approach has been used only by Lammertsma et al. (21) (approach c above) and Hasselbalch et al. (20,26) (approach b above).
The first attempt to estimate the FDG LC in the human brain was by Phelps et al. (3), who compared FDG uptake determined with PET against literature values of cerebral MRglc. MRglc was not measured in the same subjects as MRFDG. Furthermore, the literature value (mean ± SD) for MRglc, 29.7 ± 4.25 μmol/100 g/min, was probably too high. More recent measures of global cerebral MRglc are lower: 22.8 ± 4.1 μ mol/100 g/min (20) and 26.4 ± 1.9 μmol/100 g/min (5). These values, when used with the FDG data of Phelps et al., yield LC values of 0.55 and 0.47, respectively, and if the FDG parameters K*1, k*2, and k*3 are combined with K1, k2, and k3 from Blomqvist et al. (5), then the calculated LC would be 0.63.
Brooks et al. (4) used a similar approach, comparing MRFDG from dynamic PET imaging against literature values of MRglc. The resulting value for the FDG LC was 0.50. They used the same literature value as that of Phelps et al. (3) for MRglc and so their study had the same limitations.
Gjedde et al. (27) addressed the LC question using several assumptions regarding values for the brain water volume, the phosphorylation ratio (k*3/ k3), and the Michaelis constant for glucose transport. The values chosen were selected with the assumption that the whole-brain FDG LC was 0.42 (3). Because of this assumption, the value of the LC is really not addressed, although they were able to show that the LC became relatively elevated in regions associated with cerebral infarcts in some patients, presumably due to increased nonoxidative glycolysis. All of these early measurements are compromised by comparing FDG measurements in one group of subjects with glucose measurements in another group.
A more direct approach to the FDG LC was reported by Reivich et al. (2), who measured the whole-brain A−V difference for both FDG and glucose. Extraction fractions were calculated and the ratio, 0.52, was reported as the LC. One reason for a discrepancy between their value and this work probably relates to the effect of k*4, which causes the extraction fraction of FDG to decrease with time. In addition, their FDG was contaminated with up to 20% FDM, for which the extraction fraction is lower than that for FDG (22). This would lower the measured extraction fraction and underestimate LC.
More recently, Hasselbalch et al. (26) examined the feasibility of estimating the LC from A−V differences, essentially using the approach of Reivich et al. (2), and calculated an LC value of 0.48. However, they point out that the extraction fraction of FDG steadily declines, indicating that true steady state is never reached. When they used a bolus injection method, the calculated value for LC ranged between 0.75 and 0.87.
Lammertsma et al. (21) studied 3 subjects with FDG and with 15O-oxygen. The 15O study was analyzed in a well-validated manner to estimate regional cerebral oxygen metabolism (MRO2). Glucose metabolism was estimated from the MRO2 by assuming a stoichiometric uptake ratio for MRO2/ MRglc of 5.6. The FDG study was analyzed in several ways. The most sophisticated methods estimated the 4 parameters of the FDG model (K*1, k*2, k* 3, k*4) to calculate metabolic rate. They estimated an FDG LC of 0.75 ± 0.08 (±SD). The FDG used in their initial studies was also contaminated with FDM. To examine the impact of FDM contamination on the LC they repeated the study with FDG contaminated with < 5% FDM in 1 subject and found that the LC decreased from 0.78 to 0.64. This was different from the study of Wienhard et al. (22), which showed the LC for FDM was approximately 20% lower than that for FDG. Overall, the results of the study by Lammertsma et al. support the concept that the FDG LC is higher than the early values of 0.43–0.52. Frackowiak et al. (28), in a study similar to that of Lammertsma et al., using FDG and 15O-oxygen, estimated the LC in normal brain to be 0.75.
Hasselbalch et al. (20) estimated the FDG LC by measuring FDG uptake with PET imaging compared against Fick determination of whole-brain glucose uptake. Their study emphasized careful determination of cerebral blood flow using the 133Xe equilibrium method and found that global cerebral blood flow was somewhat lower than that reported previously. This had the effect of yielding lower glucose uptake rates so that the resulting LC was 0.81 ± 0.15. The only significant methodologic limitation with their approach was that they used the 3-parameter model for FDG, ignoring the impact of dephosphorylation. Although there is controversy regarding k*4, we have found consistently better fits of the model output to the FDG data when k*4 was included (Fig. 4). We also found the MRFDG was consistently underestimated when k* 4 was neglected. If k*4 had not been included in the analysis of the data reported here, then the average LC would have been 0.75 ± 0.11, very close to that reported by Hasselbalch et al.
A higher value for the LC, similar to the value we report, is supported by results from other species. Crane et al. (29) studied the kinetics of FDG in the rat brain and predicted the FDG LC should be 0.89. They measured the hexose utilization index (HUI), a close approximation to the LC, and found it to be 0.85 ± 0.16 (±SD). The HUI value of DG is 0.52, 40% lower than the HUI for FDG. This emphasizes that the 2 hexoses have different transport and phosphorylation properties (30). Collins et al. (31) compared 14C-DG with 6-14C-glucose in the rat with autoradiography. Animals were killed 10 min after injection of the tracers and the uptake resulted in a calculated LC for DG of 0.95.
The value of 0.89 ± 0.08 for the LC in normal human brain is consistent with the work of several investigators and with our LC of 0.86 in contralateral brain in 40 glioma-bearing subjects (18). The only lingering doubt relates to the performance of the glucose model. As Blomqvist et al. (23) commented, the model probably does not adequately account for nonoxidative glycolysis, with rapid loss of the label as lactate. This may account for the variation seen in the calculated LC values between individuals and between different regions of the brain. A less likely explanation is that the ratio of hexokinase isozymes is different in different parts of the brain. Although type I hexokinase is dominate in normal brain, some type II has been detected (32), and a shift in their proportion could conceivably cause a small shift in the LC but this is quite unlikely. A shift in mitochondrial binding of hexokinase is another mechanism that can increase the LC (33).
If a small amount of nonoxidative glycolysis occurs in normal brain and the glucose model fails to account for it, then the data can be interpreted differently. If we assume nonoxidative glycolysis accounts for the increased LC in some subjects, the lowest LC seen should be in subjects with minimal nonoxidative glycolysis. This would establish a lower bound for the LC of approximately 0.75. This value agrees well with those of Lammertsma et al. (21) (0.75 ± 0.08) and Hasselbalch et al. (20) (0.81 ± 0.15).
CONCLUSION
What, then, is the most likely value for the LC? It is certainly higher than the widely used value of 0.52 and, on the basis of this study, it may be as high as 0.89. Taking the results of Lammertsma et al. (21) and Hasselbalch et al. (20) into account, the most likely value seems to be approximately 0.80. We suggest that this value be used when MRglc is calculated from MRFDG in studies of normal brain. As we reported earlier (18), the LC for brain tumor varies so much that no LC can be assigned, and so studies with FDG of tissues other than normal brain should refer to the result only as MRFDG.
Acknowledgments
The authors thank Barbara Lewellen, Scott Freeman, and Aaron Charlop for technical assistance with PET imaging and chemistry and Kenneth Maravilla and Vernon Terry for assistance with MRI. This research was supported by National Institutes of Health grant CA42045
Footnotes
Received Nov. 9, 2001; revision accepted Apr. 26, 2002.
For correspondence or reprints contact: Michael M. Graham, PhD, MD, Department of Radiology, University of Iowa, 3863 JPP, 200 Hawkins Dr., Iowa City, IA 52242.
E-mail: michael-m-graham{at}uiowa.edu
REFERENCES
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Mapping 18F-FDG Kinetics Together with Patient-Specific Bootstrap Assessment of Uncertainties: An Illustration with Data from a PET/CT Scanner with a Long Axial Field of View
- Peripheral insulin resistance attenuates cerebral glucose metabolism and impairs working memory in healthy adults
- Non-invasive assessment of stimulation-specific changes in cerebral glucose metabolism with functional PET
- High-temporal resolution functional PET/MRI reveals coupling between human metabolic and hemodynamic brain response
- Principles of Tracer Kinetic Analysis in Oncology, Part I: Principles and Overview of Methodology
- Communication consumes 35 times more energy than computation in the human cortex, but both costs are needed to predict synapse number
- Does 2-FDG PET Accurately Reflect Quantitative In Vivo Glucose Utilization?
- A 3-Month Aerobic Training Program Improves Brain Energy Metabolism in Mild Alzheimers Disease: Preliminary Results from a Neuroimaging Study
- Quantification of Task-Specific Glucose Metabolism with Constant Infusion of 18F-FDG
- What Is in a Number? The FDG Lumped Constant in the Rat Brain
- Correlation of Regional Metabolic Rates of Glucose with Glasgow Coma Scale After Traumatic Brain Injury