


Abstract
The cyclam ligand (1,4,8,11-tetraazacyclotetradecane) was condensed with various azomycin-containing synthons to produce chemical compounds that could chelate radioactive metals. It was expected that these radiolabeled markers would become bound selectively to hypoxic cells on the bioreduction of their azomycin substituent. Methods: The markers were radiolabeled with 99mTc, 67Cu, or 64Cu. Their uptake and binding to tumor cells in vitro was characterized as a function of time and oxygen concentration. These data defined the hypoxia-specific factor, the ratio of the initial rate of marker binding to severely hypoxic relative to aerobic cells. In addition, the concentration of oxygen (in the equilibrium gas phase) that inhibited binding to 50% of the maximum rate was determined. The in vivo biodistribution and clearance kinetics of the favorable markers were investigated with severe combined immune deficiency mice bearing EMT-6 tumors whose radiobiologic hypoxic fraction (RHF) was ∼40%. The specific activity (percentage injected dose per gram [%ID/g]) in normal and tumor tissue and the tumor-to-blood and tumor-to-muscle ratios of the optimal markers were also measured for Dunning prostate carcinomas of anaplastic (RHF = 15%–20%) and well-differentiated (RHF < 1%) histology growing in Fischer X Copenhagen rats. Planar images were acquired with some markers from these tumor-bearing rats. Results: The tumor uptake of these cyclam-based markers is approximately 10 times higher when they are labeled with copper isotopes than when labeled with 99mTc. FC-327 and FC-334, di-azomycin-substituted cyclams, exhibited hypoxia-specific factors ≥ 7.0. The oxygen concentration that inhibited their binding to 50% of the maximal rate was ∼0.5% O2, similar to that of the radiobiologic oxygen effect. The %ID/g of 64Cu-FC-334 retained in EMT-6 tumors in mice and in the anaplastic and well-differentiated prostate tumors in rats 6 h after administration was ∼6.5, 0.4, and 0.1, respectively. Marker activity in tumor was always less than that in liver and kidney. The tumor-to-blood and tumor-to-muscle ratios of 64Cu-FC-327 and 64Cu-FC-334 activity in R3327-AT tumor-bearing rats are higher than those observed for 64Cu-di-acetyl-bis (N4-methylthiosemicarbazone) and approach those of β-d-125I-iodinated azomycin galactopyranoside, the optimal hypoxia marker of the azomycin-nucleoside class. Conclusion: These data suggest that some azomycin-cyclams exhibit good hypoxia-marking potential to tumor cells in vitro and to animal tumors of known RHF. Both PET and SPECT could be used to image tumor hypoxia with markers labeled with 64Cu and 67Cu, respectively.
Low oxygen levels in solid tumors (hypoxia) have been recognized for several years to be a potential mechanism of tumor resistance to treatment by radiation and some chemotherapeutic drugs (1,2). Hyperbaric oxygen, hypoxic radiosensitizers, perfluorohydrocarbons, pretherapy transfusion for anemia and high–linear energy transfer radiations have been investigated as agents to overcome such radioresistance (3). A metaanalysis of clinical trial data suggested that a small, but significant, benefit was achieved by some hypoxia-targeted interventions (4). When etanidazole, a hypoxia-specific radiosensitizer, was administered with standard radiochemotherapy for small cell lung cancer, a significant increase in the 5-y disease-free cure rate was realized (5). More recently, hypoxic microenvironments were shown to regulate the expression of several genes, to induce cell mutation, and to select for aggressive and metastatic phenotypes (3,6,7). Improved therapies for targeting tumor hypoxia are being developed (8). In spite of a persistent interest in the role of hypoxia in tumor characterization and treatment outcome, this property is not routinely measured today in cancer patients during their treatment planning. Potential techniques for quantifying this important property in individual tumors have been reviewed (9,10).
A promising procedure that can provide indirect evidence of low oxygen levels in the viable (potentially clonogenic) cells of solid tumors results from the bioreduction of azomycin (11). Since this technique was first defined, several variations on the original autoradiography procedure have been developed that use immunofluorescence analyses (12), magnetic resonance spectroscopy (13), and nuclear medicine detection of bound markers (14,15). Nuclear medicine techniques have the advantage of being noninvasive, able to be performed with trace (nontoxic) amounts of the marking agent, able to provide information about deep-seated as well as superficial tumors, and able to be performed on the SPECT or PET imaging equipment that is already in routine use in many cancer centers. 18F-Fluoromisonidazole (16) and 123I-iodoazomycin arabinoside (17) have been tested as hypoxia markers in pilot clinical studies. Additional agents have undergone preclinical and some clinical testing (18,19). Hypoxia marking agents labeled with 99mTc would be relatively inexpensive and find wide use (20).
Recent advances in radionuclide production and the availability of high-resolution animal imaging systems have opened additional possibilities for both PET and SPECT imaging of tumor hypoxia. We here describe the synthesis, radiolabeling, and preclinical evaluation of azomycin-substituted cyclams with tumor cells in vitro and with rodent tumors of known radiobiologic hypoxic fraction (RHF).
MATERIALS AND METHODS
Brief Description of Azomycin-Cyclam Syntheses
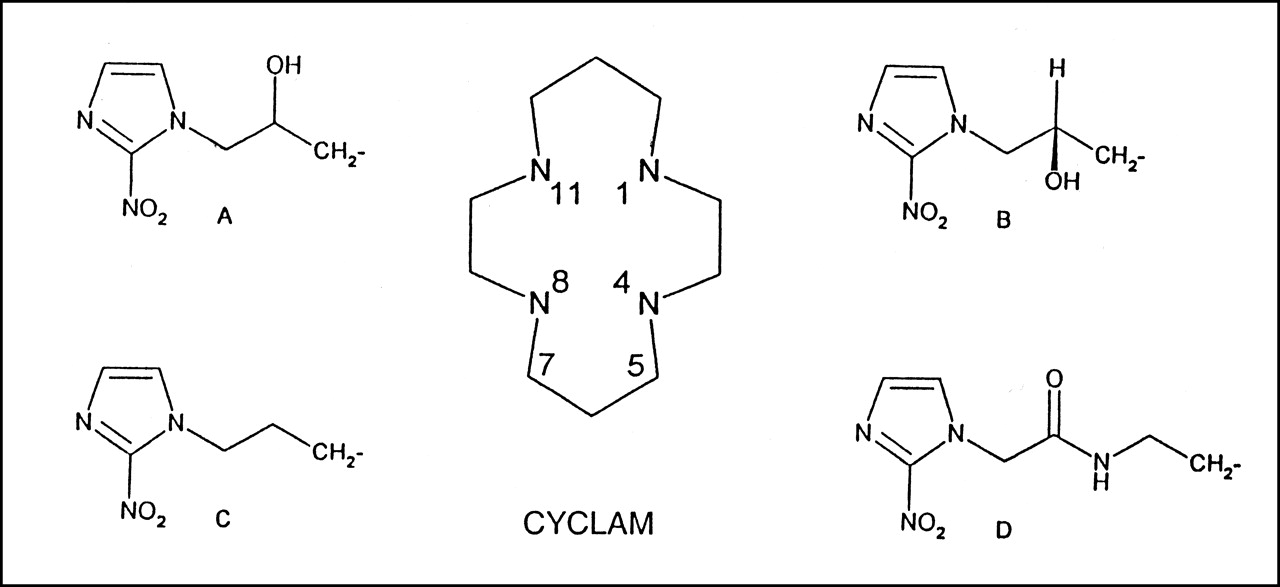
The anhydrous, alcohol-free chloroform (CHCl3) used as a solvent for most of the reactions is a product stabilized with amylenes, obtained from Sigma-Aldrich (St. Louis, MO). All other solvents and synthetic reagents were of reagent grade and purchased from either Sigma-Aldrich or Fisher Scientific (Hampton, NH). Figure 1 shows the structural components of four 2-nitroimidazole–containing synthons, and Table 1 shows the Fox Chase (FC) Cancer Center numbers for 11 potential markers that were synthesized.

(A–D) Structural components of azomycin-cyclam compounds.
Chemistry
The 4 synthons shown in Figure 1 were prepared by standard chemical procedures (21,22). The condensation of cyclam (1,4,8,11-tetraazacyclotetradecane) with the appropriate 2-nitroimidazole–derived synthon produced a mixture of mono- and polysubstituted cyclams that were separated by flash chromatography on E. Merck (Darmstadt, Germany) silica gel 60 (230–400 mesh) or by preparative thin-layer chromatography (TLC) using 1-mm Whatman (Ann Arbor, MI) 20 × 20 cm PK6F silica gel 60 or 1-mm Whatman PLK5F silica gel 150A plates. Analytic TLC was performed on E. Merck silica gel 60F plates.
After elution, the products were dissolved in a small volume of 1–2N HCl, the solution was passed through a polytetrafluoroethylene syringe filter into a 5-mL conical flask that then was placed into a beaker of absolute ethyl alcohol (EtOH), and the whole was placed into a desiccator at atmospheric pressure. Recrystallizations of the tetrahydrochlorides were performed in the same way. Compound was identified by electrospray ionization (ESI) mass spectroscopy (MSn) and nuclear magnetic resonance (NMR). A brief description of the procedures unique to realizing those compounds that were radiolabeled follows.
1,4,8,11-Tetra[2-Hydroxy-3-(2-Nitroimidazol-1-yl)Propyl]-1,4, 8,11-Tetraazacyclotetradecane (FC-316). Cyclam (98%), 50.2 mg (0.246 mmol), was added to a solution of 187.9 mg (1.11 mmol) of 1-(2,3-epoxypropyl)-2-nitroimidazole in 1.0 mL of CHCl3. A clear yellow solution resulted. It was allowed to stand at 22°C for 10 d. A TLC plate developed with CHCl3:absolute EtOH:concentrated NH4OH (3:6:1) showed 3 components, the largest at RF = 0.92 and 2 of equal size at RF = 0.35 and 0.27. The reaction mixture was diluted with 1 mL of the solvent, and the solution was applied to 4 Whatman PLK5F silica gel 150A preparative plates. The plates were developed in the above solvent system. The upper zone was eluted from the first 2 plates, and the product converted to the tetrahydrochloride. After drying over P2O5 in vacuo, the yellow solid weighed 9.9 mg. Plates 3 and 4 gave 14.4 mg of product with identical ESI MSn data.
1-[2-Hydroxy-3-(2-Nitroimidazol-1-yl)Propyl]-1,4,8,11-Tetraazacyclotetradecane (FC-323). Cyclam (98%), 1.88 g (9.4 mmol), was dissolved in 12 mL of methyl alcohol (MeOH). A solution of 1-(2,3-epoxypropyl)-2-nitroimidazole, 366.8 mg (2.17 mmol) in 3.0 mL of MeOH, was added dropwise with stirring. After 20 min at 25°C, a trace of white solid appeared. The flask was wrapped in aluminum foil and allowed to stand at room temperature for 4 d.
A 5.0-mL aliquot of the reaction mixture was evaporated on a film evaporator and the residue dried in a desiccator over CaCl2 to give 0.677 g of a solid. It was stirred under reflux with 10 mL of CH2Cl2 for 35 min. The mixture was filtered through sintered glass, and the clear yellow filtrate was evaporated to dryness. After further drying in vacuo over CaCl2, the pale yellow solid weighed 0.439 g. It was dissolved in 1.0 mL of absolute EtOH and applied to a 30 × 153 mm silica gel column. The column was developed with CHCl3:absolute EtOH:NH4OH (4:5:1); 20 mL fractions were collected.
The product from fractions 7–19 weighed 114.0 mg. TLC indicated 2 slow-moving components. This material was rechromatographed in the same flash chromatography system. Fractions 8–11 yielded 60.5 mg of a yellow syrup. This was dissolved in 820 μL of 1N HCl. On standing in a beaker of absolute EtOH, some crystals and some yellow globules separated. The slightly cloudy mother liquor was transferred to another flask, and crystallization gave 44.1 mg of cream-colored crystals after drying over CaCl2.
1-[3-(2-Nitroimidazol-1-yl)Propyl]-1,4,8,11-Tetraazacyclotetradecane (FC-325). Cyclam (98%), 51.3 mg (0.251 mmol), and 3-(2-nitroimidazol-1-yl)propyl bromide, 117 mg (0.50 mmol), were dissolved in 5.0 mL of CHCl3. The solution was stirred under reflux in a bath at 65°C–67°C for 28 h, then cooled and mixed with 1 mL of 1.0N NaOH, and the CHCl3 layer was separated, washed with water, and concentrated to 5 mL. This solution was applied to 3 silica gel 60 preparative plates, which were developed in CHCl3:absolute EtOH:concentrated NH4OH (3:6:1). The lowest fluorescent quenching zone was removed and eluted from all 3 plates. This light yellow oily product, 63.3 mg, yielded 20.5 mg of the tetrahydrochloride of the monosubstituted compound.
The 1,4 (FC-335); 1,8 (FC-334); and 1,11 (FC-327) Di-3-(2-Nitroimidazol-1-yl)Propyl-1,4,8,11-Tetraazacyclotetradecanes. For FC-335, cyclam (98%), 102.5 mg (0.5 mmol), and 1-(3-bromopropyl)-2-nitroimidazole, 240 mg (1.025 mmol), were heated under reflux in 15 mL CHCl3 for 72 h in a bath at 65°C–67°C. The solvent then was removed by passing a slow stream of N2 through a still head in the flask. The residue was partitioned between 15 mL of CHCl3 and 2.0 mL 1N NaOH. The CHCl3 layer was washed with water, and the solvent was evaporated to give 541 mg of clear yellow oil, which was dissolved in ∼1 mL of CHCl3 and applied to a 153 × 20 mm column of silica gel. The column was developed with a mixture consisting of CHCl3:absolute EtOH:concentrated NH4OH (3:6:1). Ten-milliliter fractions were collected. Fractions 3–5 gave 226 mg of a yellow syrup that showed 4 components on TLC. This product, dissolved in 5 mL of CHCl3, was treated with 1.0 g (4.60 mmol) di-tert-butyldicarbonate in 1 mL of the same solvent. After 4 d at 21°C, the CHCl3 was evaporated in a stream of N2 at 60°C. Unreacted di-tert-butyldicarbonate was removed by extraction with 10 mL of hexane. The deep yellow residue was dissolved in 1.5 mL CHCl3 and applied to 3 preparative silica gel 150A plates. The plates were developed in CHCl3:absolute EtOH (either 85:15 or 90:10). Zone 3 gave 8.2 mg of material, which was partitioned between CHCl3 and 0.25N HCl. After washing with water, the CHCl3 was evaporated to give 5.1 mg of product, which was warmed with MeOH. An insoluble white solid was separated, and the MeOH was evaporated to yield 2.9 mg of a yellow resin, which was dissolved in trifluoroacetic acid. After 3 h at 22°C, the solvent was evaporated to give a yellow resin, which was converted to the tetrahydrochloride. Small cream-colored crystals were obtained in a yield of 1.9 mg. The 1,4 orientation was assigned by NMR.
Elution of zone 4 gave 22.7 mg of yellow resin, and elution of zone 5 gave 31.9 mg of clear yellow resin. Both products were submitted for NMR. After evaporation of the DCl3 from the returned solutions, the residues were dissolved in 0.5 mL trifluoroacetic acid. After 90 min at 23°C, the trifluoroacetic acid was evaporated in a stream of N2 at 60°C–65°C, and the residues converted to the hydrochlorides.
For FC-334, the tetrahydrochloride obtained from zone 4, 16.6 mg, was very fine yellow crystals. The 1,8 orientation was established by NMR.
For FC-327, the product from zone 5, 13.6 mg, was clumps of light yellow needles. The 1,11 orientation was assigned by NMR.
Fractions 10–21 from the column yielded an additional 25.9 mg of the tetrahydrochloride of the monosubstituted compound, which was purified and identified as FC-325.
1,4,8-Tri[3-(2-Nitroimidazol-1-yl)Propyl]-1,4,8,11-Tetraazacyclotetradecane (FC-328).
The zone corresponding to RF = 0.65 was removed from 2 of the plates used in the preparation of FC-325 and eluted with the developing solvent. On conversion to the tetrahydrochloride, 28.0 mg of material was obtained that ESI MSn showed to be a mixture of 56% disubstituted and 44% trisubstituted cyclams. This material was converted to the base and combined with 49.2 mg of similar material from a parallel experiment. The combined products in CHCl3 were applied to 2 preparative silica gel 60 plates that were developed with CHCl3:absolute EtOH:(C2H5)2NH (3:6:1). Although an analytic plate had shown a separation between the di- and trisubstituted compounds, a useful separation was not seen on the preparative plates. The material was eluted from the plate. The yellow resinous residue weighed 20.6 mg. It was dissolved in 1.0 mL CHCl3. A solution of 30.0 mg (0.136 mmol) of di-tert-butyldicarbonate was added, and the mixture was allowed to stand 5 d at room temperature. It was then applied to a preparative silica gel 150A plate, which was developed in CHCl3:MeOH (90:10). The zone corresponding to RF = 0.61 was eluted. The clear yellow resin obtained weighed 10.6 mg. This product was dissolved in 1.0 mL trifluoroacetic acid. After 3 h at 25°C, the acid was evaporated and the residue converted to the tetrahydrochloride. The product, 8.7 mg, was identified as the trisubstituted cyclam.
1-[2-(2-Nitroimidazol-1-yl)Acetamido]Ethyl-1,4,8,11-Tetraazacyclotetradecane (FC-339). Methanesulfonic anhydride (97%), 429.1 mg (2.46 mmol), was dissolved in 5 mL of CH3CN (dried over 0.3-nm sieves). This solution was stirred in an ice-bath while 360 μL (261 mg, 2.58 mmol) of triethylamine was added dropwise. N-(2-Hydroxyethyl)-2-nitro-1H-(imidazol-1-yl)acetamide, 491 mg (2.29 mmol), was added gradually while stirring continued at 24°C for 1 h. Much, but not all, of the solid was in solution. After 3 d the reaction mixture was slowly added to a stirred solution of 536 mg (2.62 mmol) of cyclam (98%) in 15 mL of CHCl3. The small amount of solid dissolved. The flask was wrapped in aluminum foil, and stirring continued for 4 d. The solvent was evaporated, the residue was dissolved in 35 mL of 1N HCl, and the solution was extracted with 4 × 25 mL of CHCl3. The aqueous phase was placed in a crystallizing dish. After evaporation of the water, the dish contained a layer of long white needles (cyclam·4HCl). There was a thick yellow syrup underneath. This was dissolved in 5 mL of water, and the solution was made basic with 5N NaOH and extracted with 3 × 5 mL of CHCl3. Evaporation of the solvent caused small white crystals to separate. When the volume reached ∼4 mL, the yellow liquid was applied to 2 silica gel 150A preparative plates, which were developed in CHCl3:absolute EtOH:concentrated NH4OH (3:6:1). The lowest zones were removed and eluted with the developing solvent. Evaporation gave 6.7 mg of clear yellow residue. Conversion to the tetrahydrochloride gave a white solid.
1,11-Di[3-(2-Nitroimidazol-1-yl)Propyl]-1,4,8,11-Tetraazacyclotetradecane-5,7-Dione (FC-343).1,4,8,11-Tetraazacyclotetradecane-5,7-dione, 22.3 mg (0.102 mmol); 3-(2-nitroimidazol-1-yl) propyl bromide, 50 mg (0.214 mmol); and N,N-diisopropylethylamine, 41 μL (30.4 mg, 0.234 mmol), were dissolved in 500 mL of dimethyl sulfoxide (stored over 0.4-nm molecular sieves). The clear yellow solution was stirred at 22°C for 4 d. TLC indicated little reaction. The solution was heated in a bath at 65°C in an N2 atmosphere for 25 h. TLC in CHCl3:MeOH (91:9) showed ultraviolet absorbing zones at RF = 0.13 (very weak), 0.28 (strong), 0.34 (moderate), and 0.59 (strong). The entire reaction mixture was applied to 2 silica gel 60 preparative plates, which were developed in the above solvent system. The zone corresponding to RF = 0.28 on the TLC plate was split: 11.0 mg in the lower and 13.2 mg in the upper part. On analytic TLC in CHCl3:absolute EtOH:concentrated NH4OH (3:6:1), the 0.28 zones each showed a single strong spot at RF = 0.82, with several I2-positive spots below and at the origin. The 0.28 material was dissolved in 0.2 mL of the NH4OH-containing solvent mixture and applied to a silica gel 60 preparative plate, which was developed in the same solvent system. The zone at RF = 0.89 gave 20.6 mg of light yellow resin, which was converted to 10.1 mg of the dihydrochloride, obtained as pale cream-colored crystals.
ESI Mass Spectrometry
Typically, 0.4–1 μg of purified cyclam derivatives was dissolved in 1 mL of 50% MeOH containing 1% acetic acid. Nano-ESI was with a Protana (Odense, Denmark) source and New Objective (Woburn, MA) metal-coated EconoTips containing 1 μL of the analyte solution with 0.5–1.0 atmosphere of back pressure. A tip potential of +850–1,000 V was used for ESI, and mass spectra were recorded with an LCQ Quadrupole ion trap mass spectrometer (Thermo Finnigan, San Jose, CA) using default target values, a heated capillary temperature of 200°C, and ion optics parameters automatically optimized for the monosubstituted cyclam, FC-325, at a mass-to-charge ratio of 354.4. High-resolution spectra were recorded to verify the charge states of the analyte ions in each case. External mass calibration and instrument tuning was with a mixture of caffeine, a peptide with the sequence MRFA, and Ultramark 1621 (Lancaster Synthesis Inc., Windham, NH) as per the manufacturer’s recommendation. MSn spectra were obtained using between 20% and 30% collision energy. The ESI MSn characteristics (fingerprints) of these azomycin-cyclams are shown in Table 2.
ESI MS Analyses of Nitroimidazole-Derivitized Cyclams
NMR Spectroscopy of FC-334 and FC-327
NMR spectra were recorded at 25°C on a Bruker (Billerica, MA) DMX 600 spectrometer equipped with a 5-mm x,y,z-shielded pulsed-field-gradient triple-resonance probe. Proton spectra were recorded in D2O with solvent presaturation and a recycle delay of 10 s. Carbon spectra were recorded at 150 MHz with broadband proton decoupling using the GARP sequence (23) gated on during acquisition and off during the 10-s relaxation delay. P-Dioxane (67.4 ppm) and trimethylsilyl propionate (0 ppm) were used as external chemical shift standards for carbon and proton spectra, respectively.
Additional hypoxic markers were characterized in our in vitro tumor cell and animal tumor models for quantitative comparisons. 3H-fluoromisonidazole was obtained from K. Krohn at the University of Washington. 14C-2-(2-nitro-1H-imidazol-l-yl)-N-(2,2,3,3,3,- pentafluoropropyl acetamide) was provided by C. Koch of the University of Pennsylvania. 99mTc-4,9-Diaza-3,3,10,10-tetramethyldodecan-2,11-dione dioxime (Prognox; Nycomed Amersham plc, Amersham, U.K.) was synthesized locally and radiolabeled from published procedures (24). Di-acetyl-bis (N4-methylthiosemicarbazone) (ATSM) was synthesized locally by published procedures and radiolabeled with 64Cu (25).
Radiolabeling of Azomycin-Cyclams
Approximately 150 mg of stannous tartarate was placed into a 4-mL screw-capped vial containing ∼3 mL deaerated distilled water and a small stirring bar. The vial was stirred in a boiling water bath for 15 min and then cooled, and its contents were passed through a 0.22-μm filter. A final concentration of ∼6 × 10−4 mol/L was achieved.
99mTc. The specific radiolabeling conditions for each cyclam marker are given in Table 3. In general, the marker to be labeled was dissolved in deaerated distilled water. The pH of the solution was adjusted with 0.1N NaOH to 10.0–10.5. An aliquot of saturated stannous tartarate was added, followed by the 99mTcO4− generator eluate. The labeling mixture was heated for specific times and temperatures (Table 3) and then passed through a mini-column containing ∼0.4 g AG 1 × 8 anion exchange resin (chloride form). The final volume was adjusted to ∼3.0 mL with deaerated distilled water. The final marker solution was assayed for total radioactivity and chemical content by a combination of γ-counting, high-performance liquid chromatography (HPLC), electrophoresis, and, in some cases, TLC analyses.
99mTc Labeling of Cyclams
99mTc labeling of azomycin-cyclams should be performed at a pH > 10.0 to produce maximum chelation of the metal (26). Unfortunately, some of the cyclam markers with multiple substituents have limited solubility at this high pH. These compounds were labeled at pH 7.0–7.5, and only ∼88% of the available technetium became incorporated. For those compounds that could be labeled at the higher pH range, technetium incorporation was essentially quantitative (26). The mini-column procedure removes most unreacted TcO4−
64Cu and 67Cu. The radiolabeling conditions were customized to each cyclam marker. In general, the marker was dissolved in distilled water, and the radioactive CuCl2 solution, in 0.5 mL distilled water, was added. The pH of the solution was adjusted to 6.0–7.0 with 0.1N NaOH. The labeling solution was heated for the times and temperatures specified in Table 4. In some instances, 0.9 equivalent of carrier (nonradioactive) CuCl2 was added halfway through the labeling procedure. The pH was readjusted to 6.0–7.0 at that time. The volume of the labeling solution was adjusted to ∼3.0 mL after the reaction was complete. The final marker solution was analyzed for total radioactivity and chemical content by a combination of γ-counting, HPLC, electrophoresis, and, in some cases, TLC.
64Cu Labeling of Cyclams
In contrast to labeling with 99mTc, the pH of the reaction mixture must be held at 6.0–7.0 to prevent the precipitation of Cu(OH)2. In addition, phosphate buffers should be avoided because they will precipitate cupric phosphate. Copper ions, at the no-carrier-added level (∼1 pmol/L) when subjected to HPLC analyses, are affected by traces of unidentified species that influence partitioning between the column substrate and mobile phase, producing variable analytic peaks and retention times. The addition of 0.9 equivalent of carrier CuCl2 to the analytic solution was used to raise the copper concentration to the μmol/L level and obviate these effects. Thus, the level of chelated marker is increased from the pmol/L to the μmol/L range, and reproducible analyses of radiochemical purity were achieved.
In Vitro Test System with Human Tumor Cells
The uptake of radiolabeled markers into DU-145 prostate tumor cells in vitro was used to define their hypoxia specificity and the oxygen concentration that inhibited this process. The ratio of uptake of marker into cells that were severely hypoxic relative to aerobic cells was defined as the hypoxia-specific factor (HSF) (27). The oxygen concentration that reduced marker binding to 50% of the maximum value ([O2]50%) was also determined.
DU-145 tumor cells in the exponential growth phase were trypsinized from 75-cm2 flasks and pooled in spinner minimal essential medium (Ca2+-free) + 5% fetal bovine serum at a concentration of 3–5 × 105 cells per milliliter. The glass incubation flasks contained both an entrance port and an exit port for, respectively, passing gas through and extracting samples from the flask. Cell samples (5 mL) from the same population were dispersed into 5–6 different stirring chambers that were connected by PharMed (Miami, FL) tubing to tanks of premixed (analyzed for oxygen content) gasses (Airgas, Radnor, PA). The cells were magnetically stirred (250 rpm) at 37°C, with a different gas mixture flowing to each chamber at 1 L/min for 45 min before the addition of the radiolabeled marker in microliter volumes. Cell samples were removed through the sampling port every 30–60 min for up to 3–4 h and diluted into a ×10 volume of cold phosphate-buffered saline. These samples were centrifuged at 5,000g for 5 min to pellet the cells, the supernatant was removed, the cell pellets were resuspended in 0.6 mL of cold phosphate-buffered saline, and the suspensions were passed through Millipore (Bedford, MA) Durapore membrane filters (0.65-μm pore size), on which the radiolabeled cells collected. The filters were washed twice with cold phosphate-buffered saline and placed into counting tubes. The total radioactivity was determined with a Packard (Downers Grove, IL) Cobra II γ-spectrometer. The total radioactivity was normalized to the number of cells recovered in each sample. The specific radioactivity (dpm/106 cells) of marker uptake into cells in vitro was computed. Linear regressions through marker levels in cells at early times defined uptake rates, and the ratio of the rate of uptake under severe hypoxia, relative to the rate in air, defined the HSF. These rates were plotted versus the log [O2], and the [O2]50% was extrapolated.
In Vivo Rodent Tumors with Known RHF
Preliminary measurements of marker avidity to EMT-6 tumors growing in severe combined immune deficiency (SCID) mice were obtained in the following manner. EMT-6 mouse tumor cells growing in exponential culture were trypsinized, counted, and resuspended in sterile physiologic saline at 5 × 106 cells per milliliter. Aliquots (0.02 mL) were injected subcutaneously on the upper backs of 12- to 15-wk-old SCID mice. In vivo/in vitro assays of cells released from EMT-6 tumors irradiated in air-breathing and asphyxiated mice yielded an RHF of ∼40%. Usually, tumors of 0.2–0.8 g appeared within 10–12 d of the cell implantation. At that time, 200 kBq per mouse of a specific marker was injected intravenously into 25–30 tumor-bearing animals that were divided into 5–6 groups of 5 animals each. At various times the animals were killed and their tissues were sampled, weighed, and analyzed for radioactivity. The percentage injected dose per gram (%ID/g) of marker in each tissue (mean values, n = 5) and its biodistribution and clearance kinetics were determined from plots of %ID/g versus time. Marker activity in tumor tissue (T) was expressed as the ratio to that in blood (T/B) and in muscle (T/M).
For promising markers, their uptake into Dunning rat prostate carcinomas growing in Fischer X Copenhagen rats was also measured. Two variants of this tumor model were used: the R3327-AT line, which is rapidly growing, anaplastic, and has an RHF of 15%–20%, and the R3327-H line, which is slow growing, well differentiated, and has an RHF of <1% (28). For marker biodistribution and clearance kinetics, 500 kBq of marker per rat were administered intravenously. The animals were killed at various times, and their tissues were sampled, weighed, and analyzed for radioactivity. The %ID/g in each tissue was computed, and clearance kinetics were determined from time plots. Comparisons of marker uptake into tumors were made at 5–6 h after their administration.
67Cu was available from Brookhaven National Laboratory (Ipswich, NY) on only 2 occasions, early in the project. On those occasions, 5 MBq of 67Cu-labeled marker were administered to tumor-bearing rats and whole-body planar images were acquired with a Picker (Cleveland, OH) 2000 XP DoubleHead imaging system, 5–6 h after marker administration.
RESULTS
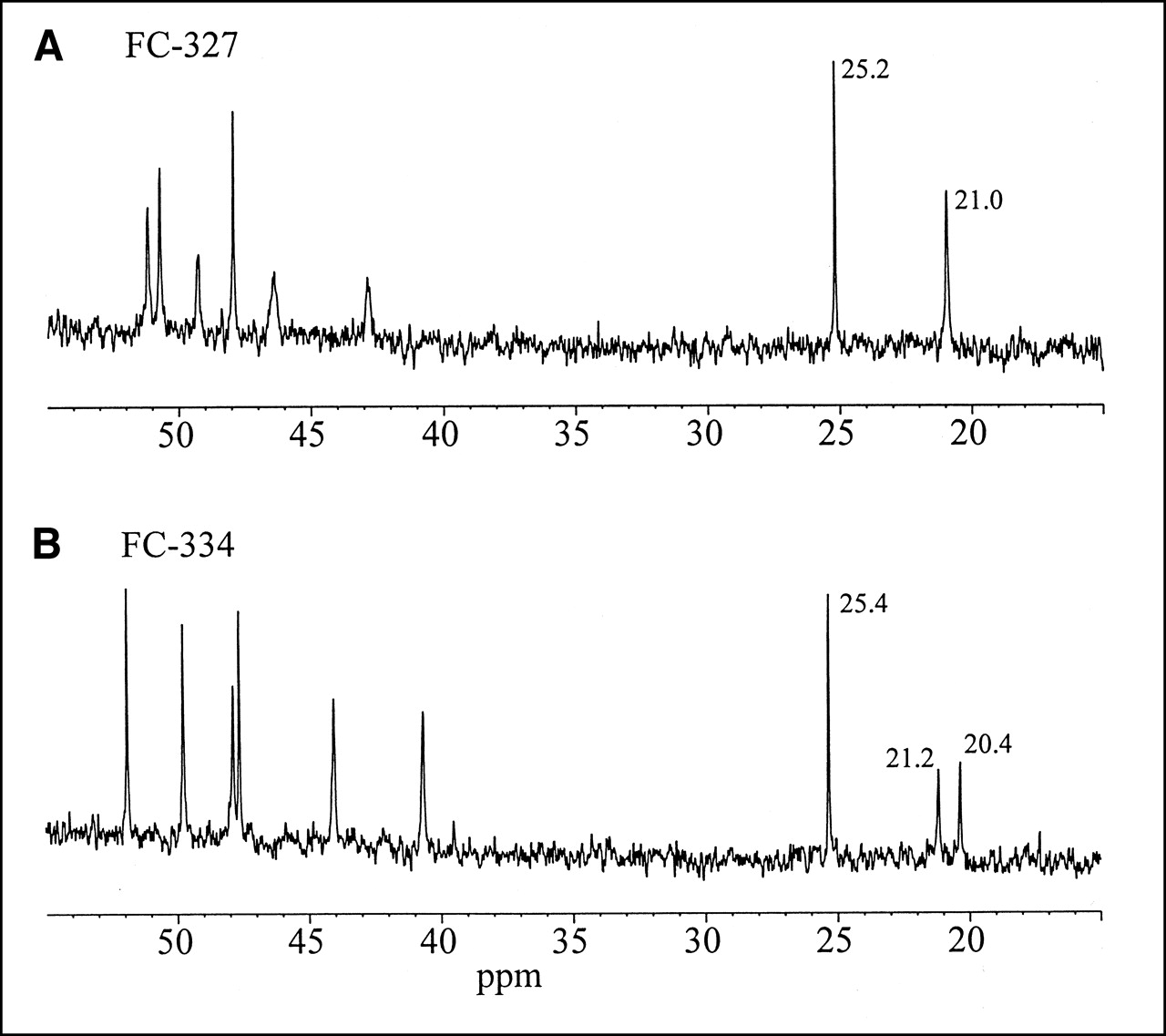
The 2 azomycin cyclams, FC-327 and FC-334, are isomers having substituents on the cyclam ring in the 1,11 and the 1,8 orientations (Table 1), respectively. The ESI MSn spectra reproducibly distinguish these compounds on the basis of the relative intensities of product ions 413.5 and 294.2 and the presence or absence of product ions 280.3, 287.3, and 334.3 (Table 2). These spectra, although diagnostic of the isomer differences, were not useful for assigning the actual orientations of the substituents. For this we used NMR spectroscopy. The symmetry caused by the 1,8 substituents is greater than that caused by the 1,11 substituents and is expected to be reflected in both the carbon and proton NMR spectra of these compounds, particularly for the most upfield resonances corresponding to the central methylene carbons (C6 and C13) of the trimethylene chains within the cyclam ring. Symmetry arguments predict chemical shift equivalence between C2 and C9, C3 and C10, C5 and C12, C6 and C13, and C7 and C14 for the 1,8 disubstituted cyclam. Chemical shift equivalence between C2 and C10, C3 and C9, and C5 and C7 is expected for the 1,11 disubstituted cyclam; however, the symmetry involving C6 and C13 in this case is broken. Chemical shift equivalence between all corresponding carbons of the 2 side chain substitutions is expected in both cases. Figure 2 shows upfield regions of 13C-NMR spectra of compounds FC-327 and FC-334. The three 2-nitroimidazole ring carbon resonances were observed further downfield between 128 and 145 ppm. Resonances between 40 and 52 ppm were not specifically assigned but collectively belong to the expected 6 pairs of degenerate N-linked methylene carbons. The resonances at 25.2 and 25.4 ppm are assigned to the degenerate central carbons of the trimethylene linkers to the nitroimidazole side chains. The resonance at 21 ppm in FC-327 is assigned to the degenerate C6 and C13, whereas the symmetry is clearly broken in FC-334, as evidenced by the appearance of 2 resonances at approximately half intensity at 21.2 and 20.4. Hence, FC-327 is assigned to the 1,8 disubstituted cyclam and FC-324 is assigned to the 1,11 disubstituted cyclam. The symmetry between H213 and H26 is also observed in proton spectra of FC-327 as a multiplet resonance at 2.18 ppm. This resonance is split into 2 multiplets at 2.33 and 2.21 ppm when the symmetry between these sites is broken in the 1,11 disubstituted cyclam. FC-334 consistently gave higher-resolution NMR spectra than did FC-327.

13C-NMR spectra of FC-327 (A) and FC-334 (B).
An analysis of the ESI MSn spectra for contaminants (Table 2) indicated that all the azomycin-substituted cyclams had a chemical purity ≥ 95%, with the exceptions of FC-316, whose chemical purity was ≥90%, and FC-325, whose chemical purity was ≥92%. The radiochemical purity of the 99mTc- and copper-labeled markers was determined by HPLC to be ≥95%.
99mTc was available each day to this investigation from our Department of Nuclear Medicine. 67Cu was available on only 2 occasions early in our studies, but 64Cu became available from the Mallinckrodt Institute of Radiology (St. Louis, MO) during the second half of the study. Consequently, all the potential markers were radiolabeled with 99mTc and either 67Cu or 64Cu for in vitro tumor cell uptake characterization. None of the 8 azomycin-cyclams that were labeled with 99mTc exhibited a significant hypoxia-dependent uptake into tumor cells in vitro. Only the 5 markers that showed the best HSF and [O2]50% values when labeled with radioactive copper proceeded to testing with tumors in vivo. Relative animal tumor marking potential was determined by radioactivity analyses of necropsy tissue specimens. Rat tumor images were acquired with only 67Cu-FC-316, the marker that was being characterized at the time when 67Cu was available.
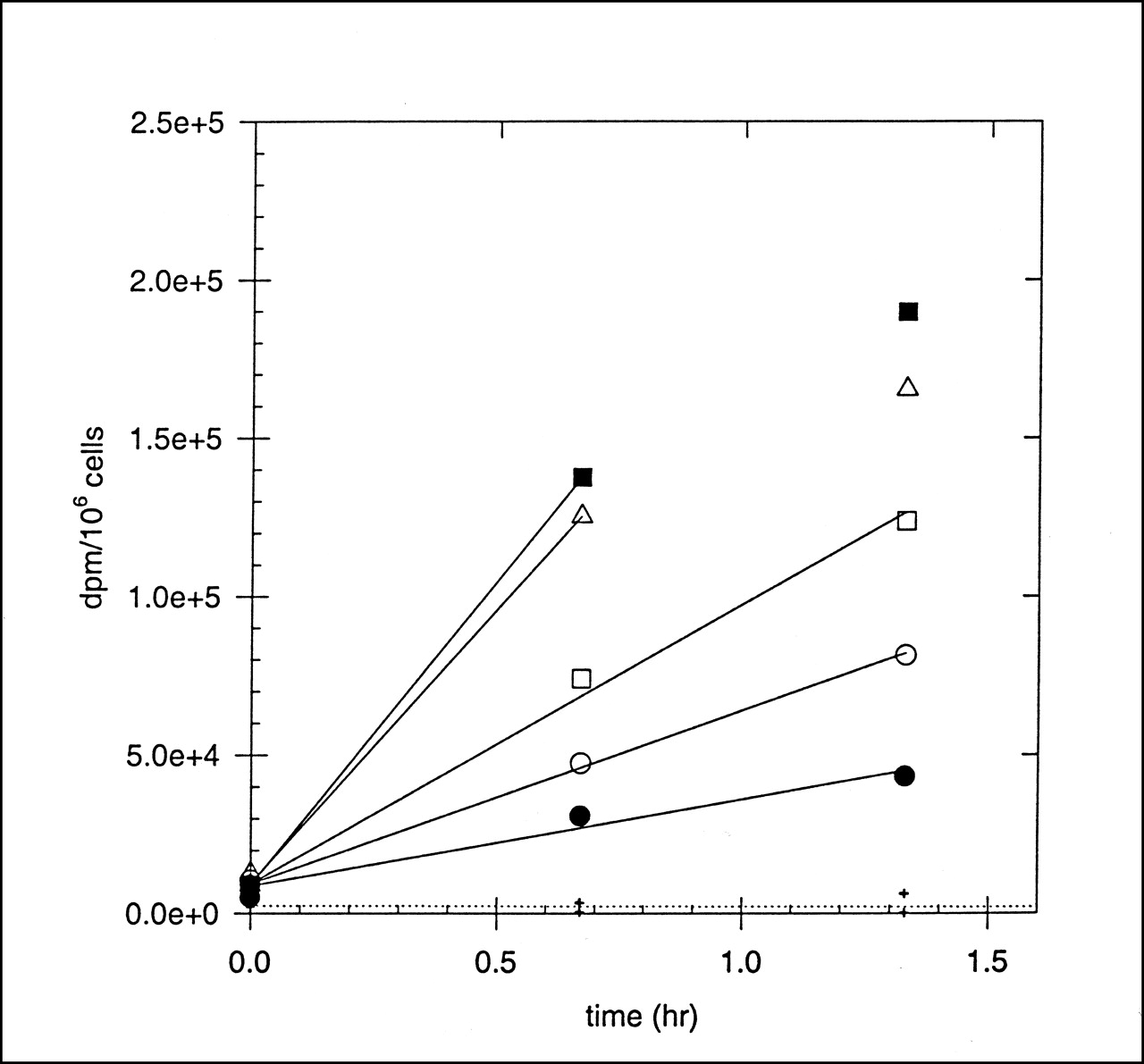
The kinetics of hypoxia marker uptake into human prostate carcinoma (DU-145) cells was measured as a function of oxygen concentration. Figure 3 shows data from an experiment in which the kinetics of FC-327 uptake into tumor cells incubated in different oxygen environments was determined. The lines are linear best-fits through the first 2 or 3 time points, and it is apparent that the rate of marker uptake is higher into severely hypoxic cells than into aerobic cells. In this experiment, the HSF was ∼8.0. For each marker investigated, the binding rates were normalized to that of aerobic cells (usually the minimum rate measured), and these were plotted against the oxygen concentration (percentage) in the gas phase. Figure 4 shows a composite plot of binding rates for the cyclam markers FC-316, FC-327, and FC-334. For comparison, the oxygen dependency of uptake of radioactive Cu+2 ions, 64Cu-cyclam and 64Cu-ATSM, is also shown. Copper ions alone were found to have a time- and oxygen-dependent uptake into these cells, but with an HSF of only ∼3. The uptake of 64Cu-cyclam into cells showed no dependence on [O2]. For the azomycin-cyclam markers shown in Figure 3, the [O2]50% was 0.3%–0.5%, the same concentration range that produces 50% of the radiosensitizing O2 effect. 64Cu-ATSM also showed good hypoxia-dependent marking of these tumor cells, with an HSF and [O2]50% of ∼5 and 0.52, respectively. The HSF and [O2]50% for these and other hypoxic markers that have been characterized in this system are shown in Table 5. Of those markers that are labeled with radiolabeled metals, FC-334, FC-327, FC-316, and 64Cu-ATSM exhibit acceptable HSF values with oxygen dependencies of marker inhibition that match the radiobiologic O2 effect. Although the HSF for 99mTc-HL-91 is equally good, it binds mainly to cells that are severely hypoxic (29). This is also a potential limitation of 18F-EF5 (30). None of the metal-labeled hypoxia markers exhibit HSF values in the high range of 25–40 observed for misonidazole and the azomycin nucleosides (31).
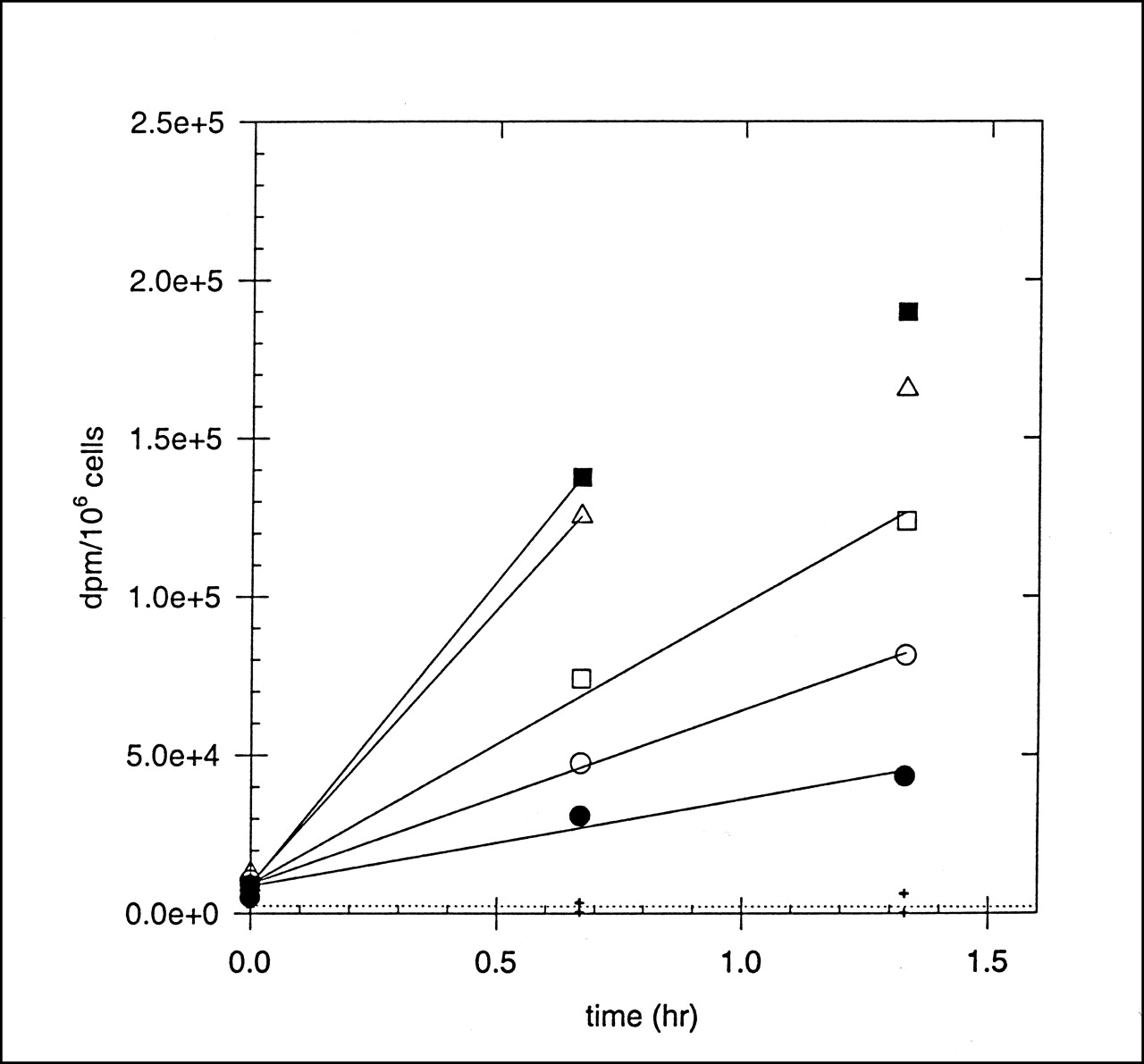
Uptake of 64Cu-FC-327 into DU-145 prostate cancer cells at various times as function of O2 concentration in gas phase. Crosses indicate radioactive background that adhered to filters. Lines are linear regressions through early time points, before saturation. • = air; ○ =1% O2; □ = 0.3% O2; ▵ = 0.1% O2; ▪ = nitrogen.
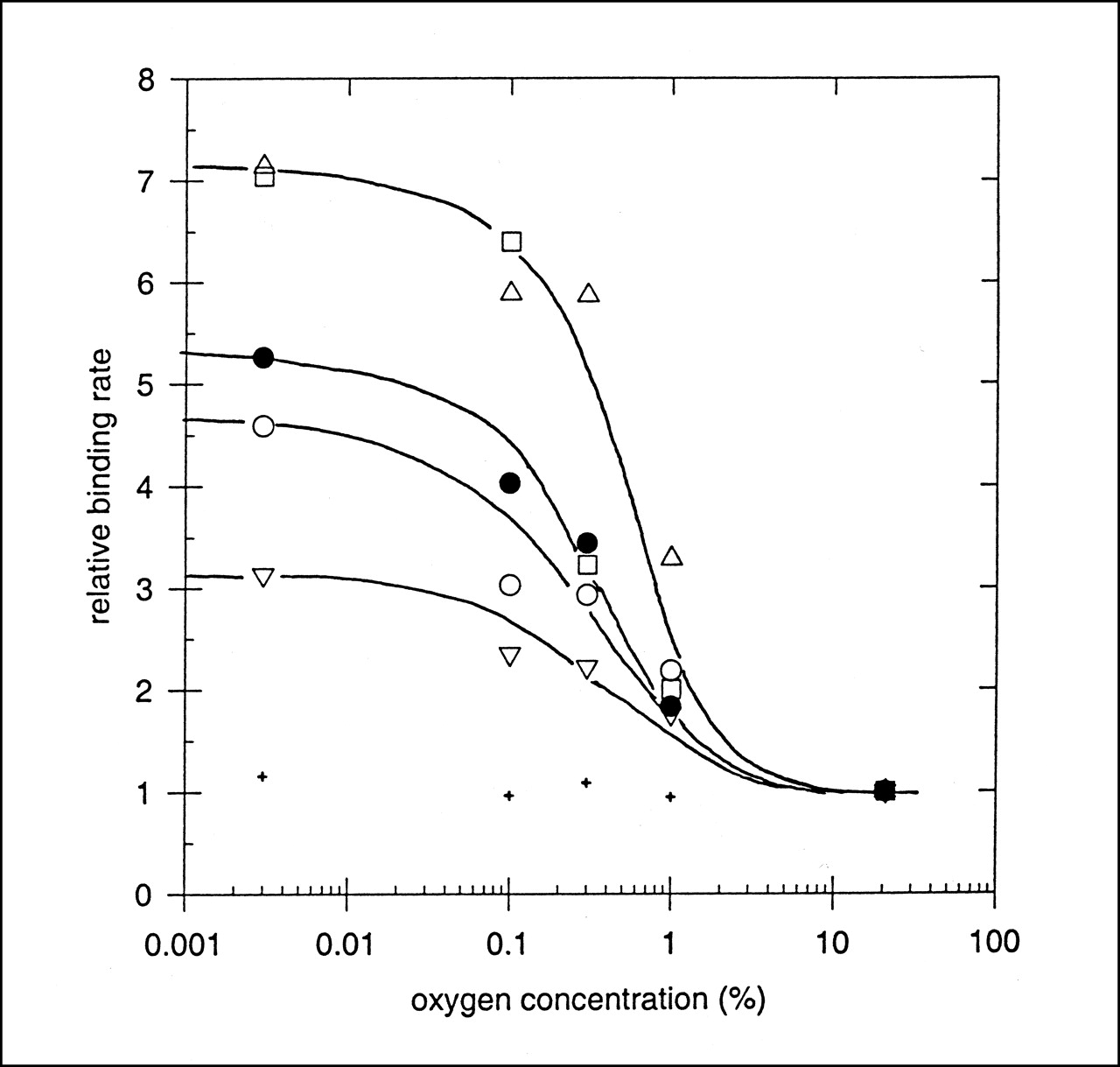
Linear binding rates (Fig. 3) of various hypoxia markers to DU-145 cells as function of O2 concentration in gas phase, normalized to their binding rate in air. + = 64Cu-cyclam; ▿ = 64Cu-CuCl2; ○ = 67Cu-FC-316; • = 64Cu-ATSM; □ = 64Cu-FC-327; ▵ = 64Cu-FC-334.
Measures of Hypoxia Marking of Tumor Cells In Vitro and in EMT-6 Tumors Growing in SCID Mice
The optimal markers (FC-327 and FC-334) were radiolabeled with 64Cu and injected into EMT-6 tumor-bearing mice. Their biodistribution to and clearance from blood, liver, kidney, muscle, and tumor tissues was measured. Figure 5A shows a plot of the logarithm of specific activity (%ID/g*), with SEs that are the size of the symbol. Tissue sample weights were normalized to the weight of a standard 20-g mouse (*). This marker becomes distributed very rapidly to the various tissues at unique concentrations. In some studies, tissues acquired at 30 min after administration also showed these characteristic tissue levels of hypoxia marker. After 1 h for kidney, blood, and muscle and after 4 h for liver, the levels of FC-334 were cleared with an exponential half-life of 7–10 h. The pharmacokinetics of this azomycin-cyclam in these tumor-bearing mice are qualitatively similar to those measured for 99mTc-HL-91 (Fig. 5B). The amount of FC-334 associated with EMT-6 tumor tissue increased by a small but significant amount over the first 6 h. Similar distribution and clearance kinetics were observed for FC-327. If the uptake of this hypoxia marker into tumors is a measure of their individual RHF, the mechanism by which tissue oxygen concentration is “sensed” is extremely rapid and probably unrelated to the bioreduction of azomycin, an enzyme-catalyzed process whose kinetics have been well characterized (32). The reduction potential, net charge, or some other property of the metal substituents of these markers probably governs their tissue distribution. The ratios of specific activity in tumor (T) relative to that in blood (B) and in muscle (M) at 5–6 h after administration were also used to compare the relative tumor avidity of the markers (Table 5). These values were highest for 64Cu-labeled FC-327 and FC-334.
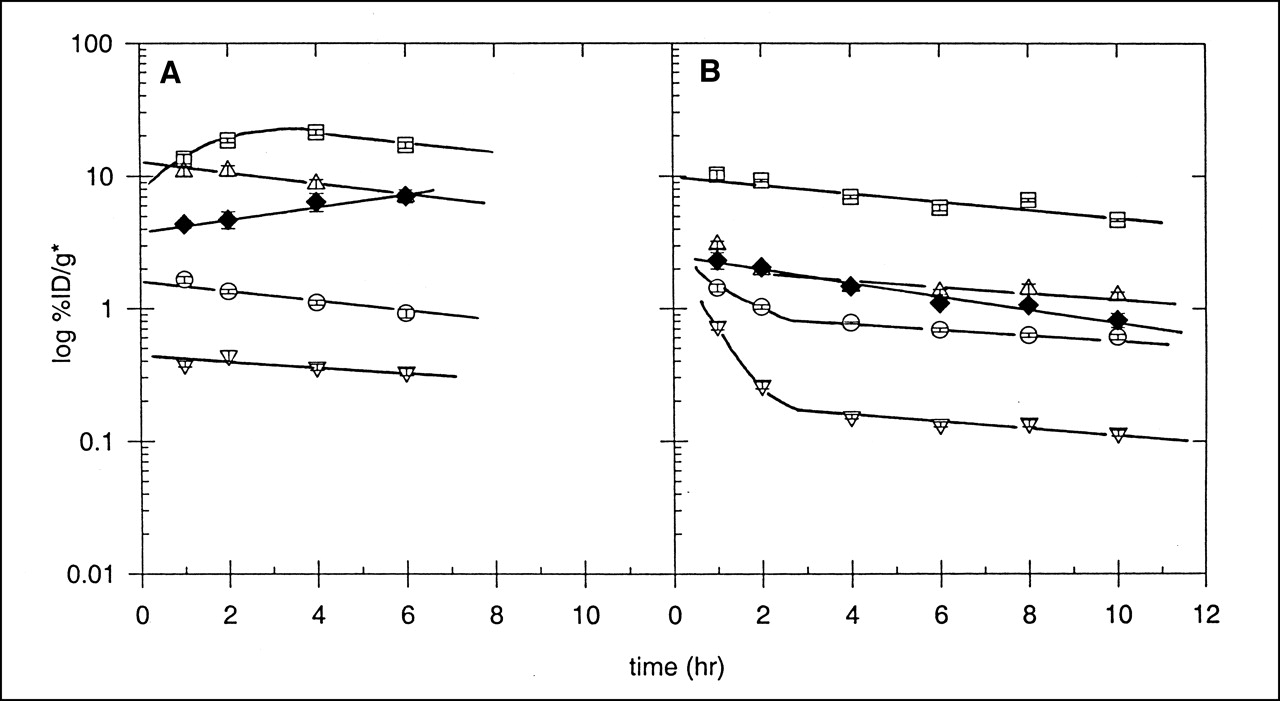
(A) Specific activity (%ID/g*) ± SE (n = 5) of 64Cu-FC-334 in liver (□), kidney (▵), EMT-6 tumor (♦), blood (○), and muscle (▿) at various times after intravenous administration to EMT-6 tumor-bearing SCID mice. (B) Specific activity (%ID/g*) ± SE (n = 5) of 99mTc-HL-91 in liver (□), kidney (▵), EMT-6 tumor (♦), blood (○), and muscle (▿) at various times after intravenous administration to tumor-bearing SCID mice.
More extensive studies of hypoxia marker avidity to rat prostate R3327-AT and R3327-H carcinomas growing in Fischer X Copenhagen rats were undertaken. These tumors exhibit RHFs of ∼20% and <1%, respectively (28). Table 6 shows the %ID/g* delivered to tumors and the T/B and T/M values at 5–6 h after marker administration of the optimal azomycin-cyclams and for 64Cu-ATSM, 64Cu-CuCl2, 99mTc-HL-91, and β-d-125I-iodinated azomycin galactopyranoside (β-d-IAZGP) (33). Tissue activity was normalized to a standard 300-g rat (*). Animals with slow-growing R3327-H tumors were not available on some days when the 64Cu was delivered. Nevertheless, the data show that the amount of azomycin-cyclam marker and ATSM retained in the less-perfused anaplastic tumor (R3327-AT) is approximately 2–3 times that retained in the better-perfused tumor (R3327-H). Also, the T/B and T/M values of FC-316, FC-334, and ATSM were larger for animals with R3327-AT tumors that exhibit the larger RHF. When FC-327 and FC-334 were radiolabeled with 99mTc, the amount of marker found in tumors 6 h after administration was ∼10 times lower than when they were labeled with 64Cu (Table 6). These data strongly suggest that the radiometal has the dominant effect in the biodistribution and uptake of these markers into tumors. This is also true for the delivery and retention of these markers into normal tissues. In contrast, 99mTc-HL-91 was retained in tumor and the normal tissues at higher specific activities than were the 99mTc-labeled azomycin cyclams. Although the %ID/g of β-d-IAZGP in these rat tumors 6 h after administration is lower, its relative specificity to the hypoxic R3327-AT tumors is the highest of all hypoxia markers we have investigated. These data show that gross tumor marking should be distinguished from the selective marking of a hypoxic microenvironment when one is evaluating potential radiodiagnostics of this tumor phenotype and radioresistance.
Specificity of Hypoxic Markers for Dunning Rat Prostate Carcinomas
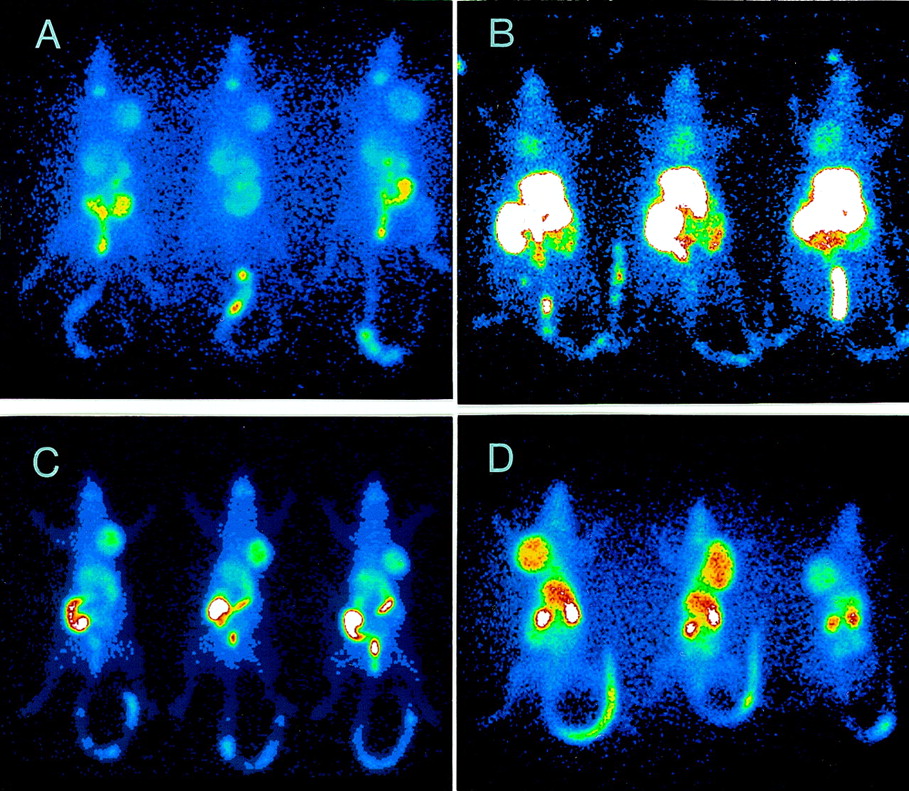
The ability of FC-316 and other markers to identify hypoxic microenvironments in R3327-AT tumors was also investigated by planar imaging procedures. Figure 6 shows whole-body planar images of tumor-bearing rats, acquired 24 h after the administration of ∼5 MBq 123I-β-d-IAZGP and at 6 h after the administration of ∼5 MBq 99mTc-FC-325, 99mTc-HL-91, and 67Cu-FC-316. The tumors can be visualized with each marker, and 67Cu-FC-316 shows the highest tumor activity relative to gastrointestinal and liver radioactivity. This result is consistent with the high %ID/g* found in these tumors at these times (Table 6). 99mTc-FC-325 is found mainly in the liver and gastrointestinal tract at this time. The radioactivity associated with marker undergoing gastrointestinal excretion may be a limitation for hypoxia markers, especially in inquiries about tumors of the gastrointestinal and urinary tracts. To conclude that the tumor image in Figure 6D is of a tumor-hypoxic microenvironment will require validation by some independent measure of tumor oxygenation. Such correlative studies are difficult because of the lack of a gold standard for measuring this tumor property. Unfortunately, 67Cu was not available at the time that FC-327 and FC-334 were available for testing.
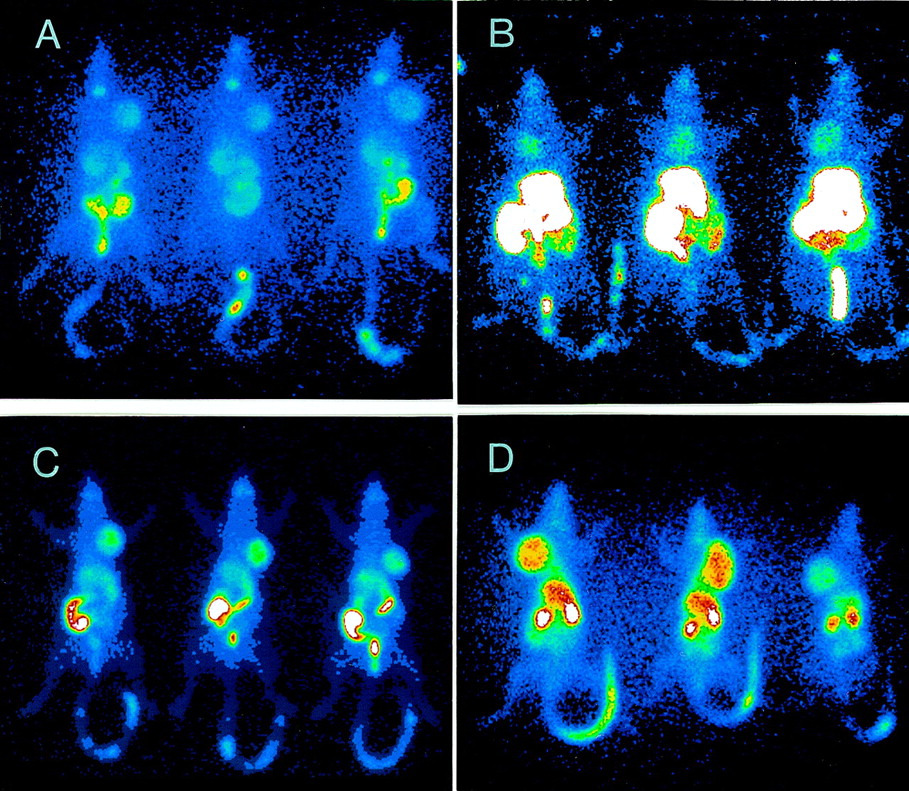
Planar images of R3327-AT tumor-bearing rats (3 per group) 24 h after administration of 123I-β-d-IAZGP (A) and 5–6 h after administration of 99mTc-FC-325 (B), 99mTc-HL-91 (C), and 67Cu-FC-316 (D). Images in panels A and B were acquired at ×1.42 zoom, and images in panels C and D were acquired at ×1.33 zoom.
DISCUSSION
The synthetic procedures for producing, purifying, and chemically characterizing these novel azomycin-cyclams and those used for their radiolabeling are described. Most of the potential hypoxia markers were produced in small quantities of 10–50 mg. Approximately 1–3 mg of each marker were radiolabeled and consumed on each day of these studies. 64Cu and 67Cu were produced at the Mallinckrodt Institute of Radiology and the Brookhaven National Laboratory, respectively, and delivered to the FC Cancer Center on the morning of a specific experiment. 64Cu could be obtained reliably every second week, but 67Cu was available only twice during the early part of this investigation. Usually, multiple experiments, with tumor cells in vitro and with either mouse or rat tumors, were coordinated to maximize the information gained from a single radiolabeling procedure. Although the chemical procedures for the described syntheses are relatively standard, the chromatography techniques for compound separation, purification, and characterization were laborious. If a hypoxia marker of the azomycin-cyclam class warrants clinical evaluation, intermediates are available that should permit a more efficient synthetic route. Furthermore, identification of the isomers of the optimal disubstituted compounds could exploit ESI MS3 analyses that showed unique product ion profiles (Table 2).
The in vitro binding studies indicate that FC-327 and FC-334 are the optimal hypoxia marking agents of this novel class. They exhibit the highest HSFs of the copper-labeled markers tested, to date. Furthermore, their oxygen dependency of binding inhibition occurs over the same O2 concentration range as the radiobiologic oxygen effect. This feature bodes well for marking those cells that determine tumor radioresistance. The data in Table 5 indicate that both 99mTc-HL-91 and 18F-EF5 bind to cells of lower oxygen concentrations, those that are relatively anoxic. It is thought that cells of intermediate oxygen concentration are those that will limit the radiocurability of cancer (34). The HSFs of misonidazole (32), fluoromisonidazole (31), and the iodinated azomycin-nucleosides (31) are significantly higher than those of the metal-labeled markers described in this study. The optimal markers of tumor hypoxia and radioresistance should exhibit a high HSF value and bind to those cells that are responsible for limiting tumor cures by radiotherapy. Of these cyclam-based markers, the di-azomycin substituted cyclams (FC-327 and FC-334) exhibit the optimal hypoxia marking properties defined by the in vitro cell assay.
The pharmacology of radiodiagnostic agents of tissue hypoxia is a major determinant of their ability to mark hypoxic cells in solid tumors in vivo (31). Only those azomycin-cyclam markers that showed good hypoxia marking characteristics in vitro were further characterized in mice and rats in which tumors of known RHF were growing. Their biodistribution to and clearance from both tumor and normal tissues were measured with EMT-6 tumor-bearing SCID mice. The establishment of unique levels of radioactivity in most tissues was prompt, within 30 min. This distribution phase was followed by clearance rates with 7- to 10-h half-lives from most tissues. The level of radiolabeled FC-334 in tumor tissue increased slightly between 1 and 6 h after administration. Similar pharmacokinetics were measured for 64Cu-ATSM in this mouse tumor system but with different activities unique to each tissue. These kinetics of hypoxia marker distribution and clearance are qualitatively and quantitatively distinct from those of misonidazole and β-d-IAZGP (35,31). Those markers become rapidly distributed to all animal tissues at approximately the same concentration (within a factor of 2), bind to cells of low oxygen tension over several minutes to hours, and are excreted biphasically by renal and then hepatobiliary mechanisms. Consequently, their maximal T/B and T/M values are observed after 3 and 6 h after their administration to mice and rats, respectively (31,36). The proportion of β-d-IAZGP (or any of the azomycin-based markers) bound to tumor in a hypoxic microenvironment 1 h after administration to EMT-6 tumor-bearing mice is difficult to quantify because it is a minority of the total delivered dose. It could be that the rapid excretion phase of FC-334 occurs within 10 min for the low concentration administered and that the data in Figure 5A depict only the second clearance phase. Two phases of clearance are suggested for 99mTc-HL-91 from blood and muscle (Fig. 5B). In the studies in which tissue was sampled 30 min after administration, the widely different levels of radioactivity in different tissues, characteristic of each metal-labeled marker, were already established. If hypoxia marker does not distribute to all tissue at an approximately equal concentration, its retention in tumor will be determined by both the oxygen and the marker concentrations. This would complicate any quantitative analyses. It remains to be shown that the uptake of FC-334 into individual rodent tumors is a strong predictor of their individual radiosensitivity.
The Dunning rat prostate carcinoma model has been useful for hypoxia marker testing. The %ID/g* and T/B and T/M values of 64Cu-FC-334 are ∼2–3 times higher, on average, in the R3327-AT tumors relative to the R3327-H tumors. Although this specificity is in the correct direction of increased tumor hypoxia, it is a relatively small difference to predict for the ×20 difference in RHF. Somewhat smaller differences in relative tumor marking were observed for 67Cu-FC-316 and 64Cu-ATSM. When FC-327 or FC-334 was radiolabeled with 99mTc, it was retained in these tumors, 5–6 h after administration, at ∼10 times lower specific activities with no specificity for the relatively hypoxic R3327-AT tumors. In this tumor model, β-d-IAZGP shows a 4–5 times higher avidity to R3327-AT tumors relative to R3327-H tumors. Although the %ID/g* of β-d-IAZGP in mouse and rat tumors is 5–10 times less than that of some metal-labeled markers, it appears to be more specific for marking viable cells in hypoxic tumors.
How, then, should the effectiveness of hypoxia marking of these and other agents be measured and validated? Correlation between marker avidity and 31P NMR spectra of tissue energetic states (37), Eppendorf microelectrode measurements of partial pressure of oxygen (pO2) (36), Comet assays of DNA damage (38), and hypoxia-specific molecular expression assays (39,40) have been attempted. The NMR and molecular expression assays measure factors that may or may not be directly related to the oxygenation status that predicts for the radioresistance of the whole tumor. The Eppendorf microelectrodes measure pO2 levels in both viable and necrotic regions of animal tumors and yield relatively low values of median pO2 for both the R3327-AT and the R3327-H (36). The Comet assay measures radiation-induced damage to the DNA of tumor cells that may or may not be clonogenic. Consequently, the correlation of hypoxia marker uptake with tumor radioresistance will probably require other assays. To date, the most informative assay has been the in vivo/in vitro measure of tumor cell viability after radiation doses that are large enough to inform about the hypoxia portion of the in vivo tumor cell survival curve (33). Several other studies have used vasoactive drugs and the breathing of different gas mixtures to modulate the RHF of tumor-bearing animals. When radiolabeled hypoxic markers are administered to such animals, their biodistribution to and clearance from tumor and other normal tissues can also be modulated significantly (36). This complicates the quantification of tumor RHF by these pharmacologic agents. The validation of FC-327 and FC-334 as markers of tumor hypoxia by these radiobiologic procedures requires additional studies.
CONCLUSION
This research has identified some azomycin-cyclams as potentially useful agents for determining the presence of hypoxic cells in solid tumors. Because the optimal markers (FC-327 and FC-334) can be radiolabeled with either 64Cu or 67Cu, they are amenable to clinical inquiry by PET and SPECT, respectively. The current resolution of clinical SPECT may be adequate to develop grades of tumor hypoxia with 3–4 levels. Such information would facilitate the identification of subgroups of patients for whom hypoxia-targeted therapy is indicated. For the “painting” of additional radiation dose to the radioresistant subvolumes of individual tumors, maps of tumor hypoxia with high spatial resolution (at least 0.5 cm) are required. Currently, only PET can produce tomographic images of this spatial resolution.
Acknowledgments
Dr. Subbarayan Murugesan, a 1998 visiting scientist in the laboratory of one of the authors, was involved in several early discussions. Renuka V. Iyer, Paul T. Haynes, and Donna Mosley assisted with the animal imaging studies, and Pat Bateman assisted with preparing the manuscript. This study was supported by National Institutes of Health grants CA06927 and CA55893 and by an appropriation from the Commonwealth of Pennsylvania. 64Cu was provided by the Mallinckrodt Institute of Radiology, where its production for research is subsidized by National Institutes of Health grant R24 CA86307.
Footnotes
Received Sep. 6, 2001; revision accepted Feb. 19, 2002.
For correspondence or reprints contact: J. Donald Chapman, PhD, Department of Radiation Oncology, Fox Chase Cancer Center, 7701 Burholme Ave., Philadelphia, PA 19111.
E-mail: JD_Chapman{at}fccc.edu
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.