


Abstract
Radioimmunotherapy (RIT) is a new treatment modality for B-cell non-Hodgkin’s lymphoma (NHL). Recent clinical trials have clearly established its efficacy in NHL patients refractory to standard chemotherapy or immunotherapy with the widely used unconjugated rituximab monoclonal antibody (mAb). The Food and Drug Administration has approved 90Y-ibritumomab tiuxetan anti-B-cell NHL mAb as the first commercially available radiolabeled antibody for cancer therapy. This comes only a few years after the introduction of rituximab into clinical practice as the first unconjugated antibody for cancer treatment, underscoring the success of both immunotherapy and RIT in the treatment of NHL. With the approval of 90Y-ibritumomab tiuxetan, and based on the results of numerous clinical trials with radiolabeled anti-B-cell NHL mAbs, RIT promises to become integral to nuclear medicine practice. In this article, the basic concepts of RIT are reviewed with important milestones in its development for B-cell NHL treatment and particular emphasis on phase II and III clinical trials establishing its efficacy in clearly defined patient populations. Finally, the prospects for the expected widespread clinical use of RIT in the management of B-cell NHL, alone or in combination with other more established therapies, are discussed. This article provides both investigative and clinical nuclear medicine physicians with a better understanding of RIT capabilities and limitations in B-cell NHL and their role as consultants in the care of NHL patients.
In February 2002, 90Y-ibritumomab tiuxetan (Zevalin; IDEC Pharmaceuticals Corp., San Diego, CA) monoclonal antibody (mAb) received final approval by the Food and Drug Administration (FDA) as the first commercially available radiolabeled antibody for cancer treatment. This approval comes only a few years after rituximab (Rituxan; IDEC Pharmaceuticals) was introduced into clinical practice as the first unconjugated antibody for cancer therapy. Interestingly, in both cases, the antibody in question is directed against B-cell non-Hodgkin’s lymphoma (NHL); rituximab is a chimeric unconjugated mAb directed against the CD20 antigen on the surface of B-cell NHL, whereas ibritumomab tiuxetan is the 90Y-labeled murine “parent” mAb of rituximab (1–4). These approvals underscore the success of both radioimmunotherapy (RIT) and immunotherapy in the treatment of hematologic malignancies in general and B-cell NHL in particular.
The promise of RIT for treatment of B-cell NHL became apparent after the 1987 report of DeNardo et al. (5) of the first patient with this disease treated with RIT. These investigators used an 131I-labeled anti-B-cell lymphoma mAb, designated Lym-1, with which they demonstrated subsequently objective responses in about half of the patients treated (5–10). Since then, numerous clinical trials have been conducted using various radiolabeled anti-B-cell lymphoma mAbs, the majority of which demonstrate remarkable antitumor activity in B-cell NHL patients at various stages of the disease (3–43).
The success of RIT in B-cell NHL treatment is, at least in part, related to properties inherent in the biology and molecular biology of this disease. These include its radiosensitivity and the abundance of well-defined target antigens on its surface, making it an attractive target for important biologic and immunologic processes, such as apoptosis, antibody-dependent cell-mediated cytotoxicity (ADCC), and complement-dependent cytotoxicity (CDC) (44–50). Yet, these properties could have been effectively exploited only through the use of appropriate antibodies capable of inducing these biologic or immunologic processes and, in the case of RIT, also by the utilization of suitable therapeutic radioisotopes to deliver cytotoxic radiation to the tumor (51–53). The identification of these appropriate antibodies and isotopes suitable for their stable labeling was the ultimate fruit of extensive basic and clinical research extending over almost 2 decades and involving several disciplines, such as immunology, radiochemistry, nuclear medicine, and medical oncology.
This article reviews the basic concepts of RIT, the milestones leading to its current emergence as a treatment option for patients with B-cell NHL, and the prospects for its clinical use in the management of this disease, alone and in combination with other treatment modalities.
CURRENT STANDARD MANAGEMENT OF B-CELL NHL
The incidence of NHL is increasing, with this disease recently afflicting 50,000–60,000 patients per year; NHL is now the fifth leading cause of cancer death in the United States (44). NHL consists of various histologic subtypes, classified using various classifications with the World Health Organization (WHO) classification currently the most widely used (44). The Ann Arbor staging system is used to assign 1 of 4 disease stages (I-IV), similar to the approach used to stage Hodgkin’s disease (44). Prognosis within a lymphoma category is determined by the cumulative number of adverse features incorporated into the International Prognostic Index (IPI), including advanced age (>60 y), advanced stage (III or IV), elevated lactate dehydrogenase (LDH) level, poor performance status (WHO >2), or >1 site of extranodal disease.
Eighty percent of lymphomas are of B-cell origin (B-cell NHLs) and have traditionally been roughly divided into low-grade (e.g., small lymphocytic, marginal zone, follicular center cell grades I and II), intermediate-grade (e.g., diffuse large cell), and high-grade (lymphoblastic, Burkitt’s) categories, each occurring in about one third of patients (44,54). The low-grade B-cell NHLs are generally remarkable for an indolent course—therefore referred to as indolent NHLs—response to many therapies, and inevitably relapse, leading to eventual death with median survivals reported as 5–15 y, heavily dependent on the IPI score at the time of presentation. The intermediate- and high-grade B-cell NHLs have a more symptomatic presentation—therefore referred to as aggressive NHLs—and frequently respond to aggressive multiagent anthracycline-containing chemotherapy, with over half of the responses being durable (44).
Symptomatic early-stage indolent NHL (i.e., stages I and II) is an infrequent scenario, comprising <10% of patients with indolent NHL and can be treated in the short run with radiotherapy. The advanced stages (i.e., stages III and IV) are treated with a variety of chemotherapeutic or immunotherapeutic regimens, including single-agent chlorambucil, cyclophosphamide, fludarabine, or rituximab. Initial response rates are generally 50%–70% with any of these single agents, with the median duration of responses ranging from 12 to 24 mo (44). Combinations of drugs such as COP (cyclophosphamide, vincristine, and prednisone), CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), and, more recently, CHOP and rituximab result in very high response rates (70%–100%) (43,55). Although the initial response rate with all of these regimens is excellent, virtually all patients relapse, with the median duration of the first response being ∼18–50 mo, followed by progressively lower response rates and shorter response duration with additional therapy (44). Furthermore, many indolent lymphomas will eventually transform into a more aggressive histology with a difficult clinical course and the disease is, therefore, considered incurable with conventional chemotherapy (44). In the last 5 y or so, rituximab immunotherapy has found widespread use for the treatment of relapsed low-grade NHLs with almost half of relapsed or refractory patients showing an objective response (OR), defined as partial or complete remission (PR or CR, respectively), for a median duration of about 1 y. Patients who are refractory to (i.e., initially nonresponding) or who relapse after chemotherapy and immunotherapy are potential candidates for novel therapies, including RIT.
Intermediate- and high-grade (i.e., aggressive) NHLs are generally treated with combinations of cytotoxic chemotherapy, such as CHOP or other anthracycline-containing combinations and, more recently, the combination of CHOP and rituximab (44,56). With these regimens, 50%–60% of patients are cured, ∼20% progress during treatment or achieve only a transient PR or stable disease with subsequent progression, whereas the other ∼30% relapse after initial CR (44,57,58). Of those who relapse, a substantial proportion of highly selected candidates can be cured with high-dose chemotherapy and stem-cell rescue (44). Overall, about 40% of patients with aggressive B-cell NHL are not cured with standard or high-dose conventional therapy. Rituximab has been shown to induce only transient partial responses in 30% of these patients (59). All of these patients are potential candidates for novel approaches such as RIT.
BASIC CONCEPTS OF RIT
RIT is defined as a treatment modality in which cytotoxic radiation from therapeutic radioisotopes is delivered to tumors via antibodies that bind to tumor-specific or tumor-associated antigens. These antibodies often function as vehicles that carry the therapeutic radioisotopes to the tumor and have a cytotoxic effect of their own mediated by apoptosis, ADCC, and CDC (1,48–50). Furthermore, the characteristics of the antigen against which a particular antibody is targeted (i.e., the target antigen) determine to a great extent the effectiveness of both the antibody itself and the targeted radiation. Thus, the choice of antibodies, target antigens, and therapeutic radioisotopes is critical for the success of RIT. A brief overview of these important elements as well as other basic concepts of RIT follows, with special emphasis on RIT of B-cell NHL.
Antibody Production
Antibodies are immunoglobulins produced by B-lymphocytes (actually plasma cells) in response to an antigen. If a suitable animal recipient, such as a mouse, is immunized with an antigen, serum taken from the immunized host will contain a mixture of different antibodies to different epitopes of the antigen. These antibodies are produced by a heterogeneous population (i.e., different clones) of B-lymphocytes and are called polyclonal antibodies (60–62). If individual B-lymphocytes from these antibody-producing cells are isolated and cloned, each clone will produce a single species of antibody molecules, or mAbs, that bind to the same epitope. However, the production of mAbs will not be possible on a large scale because of the short survival of normal B-lymphocytes in culture. This problem was solved by the discovery of the hybridoma technique, in which the normal B-lymphocyte is immortalized by fusion with myeloma cells in culture (63). Using this technique, mice are immunized with a particular antigen that will stimulate their normal splenic B-lymphocytes to produce antibodies against this antigen, and these lymphocytes are then immortalized by fusion with myeloma cells in culture. The resulting “hybridomas” are screened subsequently using radioimmunoassay for single-cell clones capable of producing a specific antibody (i.e., an mAb) directed against the particular antigen. These selected clones can then be grown for mAb mass production either in cell culture or in the peritoneal cavity of mice (60–62). Because this mAb is produced by mouse B-lymphocytes, it is called a mouse or murine mAb. Of course, before such an mAb is used in humans, it must be tested for sterility, pyrogenicity, adventitious viruses, and general safety (64).
Most hybridomas produce IgG mAbs of various isotypes, most commonly IgG1 and IgG2. An IgG mAb has a molecular weight of 150,000 and consists of 2 identical heavy chains and 2 identical light chains comprising the constant and variable regions of the mAb (Fig. 1). The constant region determines the immune effector function of the mAb, such as the ability to fix complement and to interact with effector cells, which are important for the “naked” mAb effects due to CDC and ADCC, respectively. This constant region is the same for all IgG mAbs of the same isotype. The variable region contains the 2 antigen-binding sites that are unique for each IgG mAb and is, therefore, responsible for its specificity. Each IgG molecule can be enzymatically split into an Fc (fragment crytallizable) fragment, consisting of most of the constant region of an IgG molecule, and 2 Fab (fragment antigen binding) fragments, comprising the variable region and the smaller portion of the constant region. Each Fab fragment has only 1 antigen-binding site.
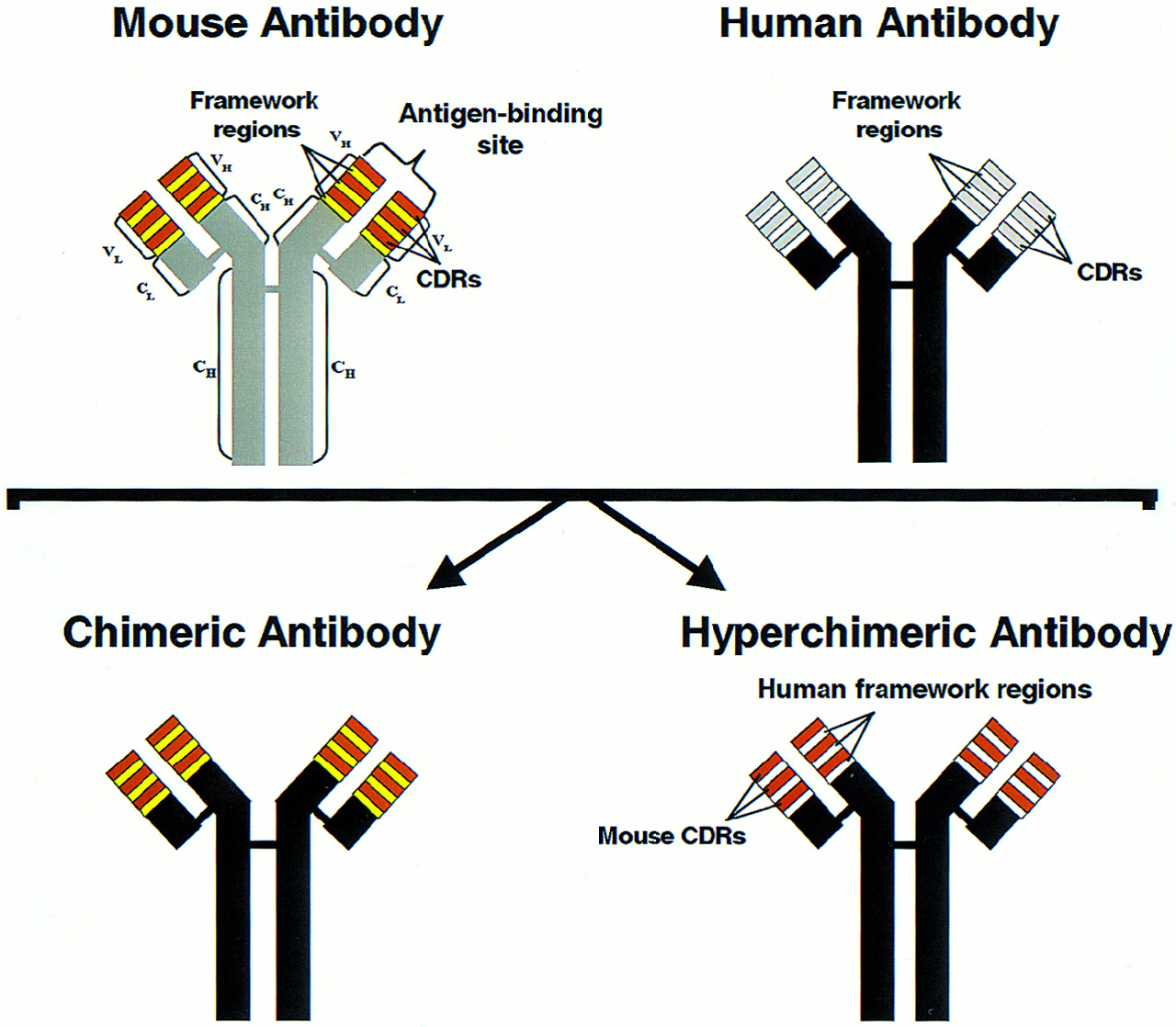
Schematic presentation of IgG antibody as well as humanized antibody production. L and H = light and heavy chains, respectively; VL and VH = variable regions of light and heavy chains, respectively; CL and CH = constant regions of light and heavy chains, respectively; CDRs = complementarity-determining regions. Chimeric antibody consists of human constant regions and murine variable regions of heavy and light chains, whereas hyperchimeric antibody consists of human constant and framework regions with murine CDRs. (Reprinted, with modifications, with permission of Ann Allergy Asthma Immunol. 1998;81:105–119; Copyright 1998.)
Humanized mAbs
Patients receiving murine antibodies, particularly in high amounts, may form human antibodies against these foreign proteins or human antimouse antibodies (HAMA), which usually occur within 2–3 wk after the first mAb administration and within hours or days after a repeated administration. Even after repeated mAb administrations, HAMA formation usually does not cause significant adverse reactions, although flu-like symptoms or mild-to-moderate anaphylactic reactions (e.g., urticaria, bronchospasm) may occur, which are easily treatable with antihistamines or corticosteroids (35,65). A more common problem is that HAMA can seriously alter the mAb pharmacokinetics through the formation of large murine antibody/HAMA complexes that are quickly removed by the reticuloendothelial system (RES), primarily in the liver, spleen, and bone marrow. Consequently, the circulating free murine antibody level will be greatly reduced, resulting in severe impairment of tumor targeting. Moreover, when these antibodies are labeled with 131I, free iodine is quickly released from the RES and localizes subsequently to normal thyroid, resulting in substantially increased radiation dose to this organ (66). On the other hand, antibodies labeled with radiometals, such as 90Y, will demonstrate high retention of isotope in RES organs, again with substantially increased radiation dose to these organs, which is particularly critical in the case of the bone marrow.
With the advent of genetic engineering, it is now possible to produce humanized mAbs that are considerably less immunogenic (i.e., capable of inducing anti-antibody responses) than murine mAbs (Fig. 1). The first-generation humanized mAbs were chimeric mAbs produced by cloning the variable region genes of a murine mAb into a human expression vector containing the appropriate human constant region, resulting in a murine/human chimera or chimeric mAb (67). Because only the variable region of the chimeric mAb is of murine origin, it is considerably less immunogenic than murine mAb. Second-generation humanized mAbs are produced using a technique known as complementarity-determining region (CDR) grafting and, therefore, often called humanized CDR-grafted mAbs (19,24). Here, antigen-binding sites, which are formed by 3 CDRs of the heavy chain and 3 CDRs of the light chain are excised from cells producing murine mAb and grafted into the DNA coding for the human antibody framework. The second-generation or hyperchimeric mAbs contain less murine protein than the first-generation chimeric mAbs because only the antigen-binding CDRs rather than the entire variable domain of the murine mAb are grafted and, hence, are less immunogenic than the first-generation chimeric mAbs (67). Nevertheless, both the first- and second-generation chimeric mAbs induce anti-antibody responses, albeit in a very small fraction of patients compared with murine mAbs (3,4,19,24,26–29). These antibodies are called human antichimeric antibodies (HACA) (3,4,25–28) or human antihuman antibodies (HAHA), a term used to describe anti-antibody responses to the second-generation CDR-grafted hyperchimeric mAbs (19,24). Anti-antibody responses to radiolabeled mAbs used in RIT of B-cell NHL will be discussed in more detail after the review of the clinical trials conducted using these mAbs. However, humanized chimeric or hyperchimeric mAbs generally possess a much more potent immune effector function (i.e., are much more capable of inducing ADCC and CDC) compared with their murine parents and, therefore, are presumably more effective antitumor agents (1,2,48,49).
Tumor-Specific or Tumor-Associated Antigens as Antibody Targets
The specificity of a particular antibody used in RIT of cancer is determined primarily by the choice of the target antigen. Ideally, this antigen should be specific for the tumor cells with no or very minimal expression on normal cells. However, in practice, most of the target antigens, including those on B-cell NHLs, are actually tumor associated rather than tumor specific. This means that they are also expressed on the surface of normal cells of certain organs or tissues (depending on the particular antigen), albeit usually on a substantially lower percentage of these cells or in a lower antigen number per cell (44,45). Consequently, the antibodies directed against these antigens are only relatively rather than absolutely tumor specific.
Table 1 shows the most important target antigens on B-cell NHLs and the names of reported unconjugated mAbs against these antigens with their known radiolabeled counterparts (1–43, 68–72). As can be seen, most of these antigens are the so-called “CD” (cluster of differentiation) antigens, such as CD19, CD20, CD21, CD22, CD37, and CD52 with the HLA-DR antigens representing another group of important target antigens for anti-B-cell lymphoma mAbs. All of these antigens are also expressed on the surface of normal B-lymphocytes in the blood, spleen, lymph nodes, and bone marrow and on some of their precursors in the marrow (44,45). The latter depends on the particular antigen. For example, the CD20 antigen, against which the unconjugated rituximab, 111In/90Y-ibritumomab tiuxetan, and 131I-tositumomab mAbs are directed, is expressed only on the pre-B-cells in the marrow but not on the stem cells or progenitor cells (pre-pre-B-cells) (44).
Commonly Targeted Antigens on B-Cell NHL with Corresponding mAbs
One direct consequence of the expression of these target antigens on normal B-cells and their precursors is the common use of relatively high unlabeled or unconjugated antibody protein doses either before or together with the administration of the various radiolabeled anti-B-cell NHL mAbs. The rationale behind this is to block the target antigens on the normal B-cells and their precursors, commonly referred to as the antigenic sink, thereby achieving a more favorable tumor targeting. Interestingly, this blockage also affects, albeit to a lesser extent, even malignant B-cells that may be circulating in the blood or reside in the involved spleen or bone marrow, because these sites are easily accessible to the infused unlabeled mAb. Nevertheless, this partial blockage is accepted to achieve a more favorable targeting of the other less-accessible lymphoma sites, particularly bulky tumors. Because the various mAbs used for RIT differ with respect to their specificity and the number of their target antigens on the surface of normal and malignant B-cells, variable amounts of unlabeled mAb are given with each of the radiolabeled mAbs used to achieve optimal tumor targeting (3–43). For example, only 5 mg of unconjugated Lym-1 are usually given before radiolabeled Lym-1 is given, except in patients with circulating malignant cells (21). In contrast, higher amounts of unlabeled anti-CD20 mAbs, mostly in the range of 200–700 mg, are given with each radiolabeled mAb administration (3,4,16–18,23,26–38). Thus, the recommended dose of unlabeled mAb infused before radiolabeled ibritumomab tiuxetan, chosen to be the chimeric rituximab, is 250 mg/m2 or about 430 mg in a 70-kg man (3,4,26–29). With 131I-tositumomab (Bexxar; Corixa Corp., South San Francisco, CA), the 131I-labeled anti-CD20 mAb, the recommended dose of unlabeled mAb (i.e., tositumomab) is similar (450 mg) (16,17,30–35,38).
The CD20 antigen is characterized by its high frequency of expression (i.e., >90%) on B-cell NHLs, fairly homogeneous expression on the cell surface of these tumors, with generally high antigen number (i.e., >100,000) per cell (44,45). Another important characteristic of this antigen is that it is slowly internalizing (i.e., transported into the tumor cell after antibody binding) compared with rapidly internalizing antigens, such as CD19 or CD22 (45). Thus, mAbs directed against CD20 are considered slowly internalizing, whereas those directed against CD19 or CD22 are rapidly internalizing. As discussed below, this has important implications with respect to the choice of the radioisotope used to radiolabel these mAbs.
Therapeutic Radioisotopes Used for RIT and Antibody Radiolabeling
Several therapeutic radioisotopes for radiolabeling the various mAbs used in RIT are available (51–53). Table 2 lists some of these isotopes with their physical properties, advantages, and disadvantages.
Radioisotopes Used in RIT
Most of these isotopes emit β-particles, which represent the only or principal source of therapeutic radiation delivered by these isotopes. These particles are especially suitable for RIT because of their relatively long range in tissues (up to of several millimeters), thereby affecting (and potentially killing) even tumor cells that are not targeted directly by the antibody. This is especially important because of the common finding of heterogeneous antibody distribution within tumors, particularly bulky ones, due to factors such as heterogeneous antigen distribution, tumor vascularization, and permeability of tumor vessels (73,74). α-Particles are another type of radiation that has been used for RIT of cancer. In contrast to β-particles, these have only a short range in tissues of usually <100 μm. However, their high linear-energy transfer (LET) ionization makes them severalfold more effective than β-particles because they can deliver lethal radiation to tumor cells by a single hit (51–53). Because of their short range in tissues, α-particles are more suitable for treatment of easily accessible tumors, such as leukemic cells in the blood or bone marrow. These cells have to possess a fairly homogeneous antigen expression on their surface because every, or nearly every, tumor cell needs to be targeted with the antibody carrying the α-emitting isotope. One such isotope is 211As (75). 213Bi is another α-emitting isotope that has been used successfully for treatment of leukemia (76).
Regardless of the type of radiation emitted by the various radioisotopes used for RIT, they can be grossly divided into 2 categories on the basis of their chemical characteristics: halogens (e.g., 131I and 211As) and radiometals (90Y, 186Re, 188Re, 67Cu, and 213Bi). In general, halogens, 131I in particular, can be labeled easily and directly using the IODO-GEN (Pierce, Rockford, IL) or chloramine-T methods (77,78), whereas radiometals require chelators such as benzyl-diethylenetriaminepentaacetic acid, [N-[2-bis(carboxymethyl)amino]-3-(p-isothiocyanatophenyl)-propyl]-[N-[2-bis(carboxymethyl)amino]-2-(methyl)-ethyl]glycine (tiuxetan), or 1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid (DOTA), which are conjugated to the antibodies to allow stable antibody labeling (79,80). The use of suboptimal, unstable chelators will invariably result in leaching of the isotope from the antibody (79–82).
Of the isotopes listed in Table 2, 131I and 90Y are by far the most commonly used in the various RIT trials. Because 131I has traditionally been the conventional and most commonly used isotope in radionuclide therapy (i.e., for treatment of patients with hyperthyroidism and thyroid cancer), it is understandable why most of the antibodies used in the initial RIT trials, including those in B-cell NHL, were labeled with this radioisotope. 131I has some advantages, which include its low cost, the relatively simple radioiodination methods available for antibody labeling, and the ability to obtain images of the organ and tumor biodistribution with 131I-labeled mAbs, before (through the use of a tracer dose) or after RIT, because of the γ-emission (in addition to β-emission) of this isotope. This latter advantage is offset, however, by the increased radiation exposure to the treating personnel or relatives, which requires more stringent radiation precautions or, in the case of administration of high amounts of radioactivity, patient isolation.
Another important disadvantage of 131I is related to the effect of dehalogenation, which inevitably occurs after catabolism of conventionally labeled radioiodinated mAbs in various normal organs (e.g., liver, spleen, bone marrow, and kidneys) and, albeit to a variable degree, also in tumor cells (83). The free iodine released requires protection for the thyroid and, particularly when high doses of 131I-labeled mAbs are administered, can result in hypothyroidism despite thyroid blockade (18,36,37). A more significant problem is seen when rapidly internalizing mAbs (e.g., anti-CD19 and CD22 mAbs) are labeled with 131I (24,83). In this case, the rapid internalization, and, hence, the usually associated rapid intracellular degradation of the radiolabeled mAb in the lysosomes, result in a substantial fraction of 131I being liberated from tumor cells. Consequently, there is a significant decrease in the residence time of radioiodine and, hence, the radiation absorbed dose in the tumor. Therefore, these mAbs are much better suited for radiolabeling with radiometals, such as 90Y, because these remain in the intracellular compartment (bound to the stable chelator) after mAb internalization and subsequent catabolism (24,84). Slowly internalizing mAbs are much less susceptible to the effects of intratumoral dehalogenation, although significant dehalogenation has been observed with some slowly internalizing mAbs, such as those against carcinoembryonic antigen or HLA-DR; therefore, it is possible that, even for these mAbs, radiometals may offer an advantage compared with radioiodine (85–87). However, both radioiodine and radiometals are suitable labels for slowly internalizing mAbs, explaining why both 131I and radiometals are used for radiolabeling the various anti-CD20 mAbs used in clinical RIT trials (3,4,16–18,23,26–38,40–43).
90Y is now being used increasingly to radiolabel various mAbs, including both rapidly and slowly internalizing ones, a development that has been greatly facilitated by the current availability of stable chelators. With their use, the problem of significant leaching of free 90Y from the mAb with subsequent localization in bone, and, hence, increased bone marrow toxicity, has been largely eliminated (3,4,22–24,26–29,39,40,43). The requirement for a chelator for antibody labeling with 90Y may be considered by some a disadvantage because this may not always be available at the labeling laboratory or institution and because it must first be conjugated to the “naked” mAb before the labeling procedure. However, because mAbs already conjugated with a suitable chelator can be made available, even commercially (e.g., ibritutomab tiuxetan), this requirement is only a relative disadvantage.
90Y is a pure β-emitter, and, hence, imaging cannot be performed with this isotope when standard doses of 90Y-labeled mAbs are given. Bremsstrahlung imaging is possible when very high amounts of radioactivity are given with considerable concentration in organs and tumors, which is not the case in most clinical situations (88). It also results in low-quality images. Thus, a chemical surrogate for 90Y that emits γ-radiation must be used to determine the organ and tumor biodistribution and dosimetry with 90Y-mAbs. Luckily, 111In, the isotope used frequently in nuclear medicine imaging, represents such a surrogate that for all practical purposes can predict these parameters for the corresponding 90Y-mAbs. Here again, with the availability of corresponding 111In-mAbs to virtually all 90Y-mAbs used for RIT (e.g., 111In/90Y-ibritumomab tiuxetan), the need for 111In is only a minor disadvantage. On the other hand, because 90Y is a pure β-emitter, there is negligible radiation exposure to treating personnel or relatives with this isotope and outpatient therapy can, therefore, be performed even with very high doses of 90Y (89).
Other important advantages of 90Y are related to the longer pathlength of its β-particles compared with those of 131I, which is particularly advantageous in tumors with heterogeneous antibody distribution, as is the case in most tumors. Moreover, because 90Y is not a halogen, it can be used to label both rapidly and slowly internalizing mAbs with a clear advantage compared with 131I with the former and a potential advantage with the latter (24,82,83,85–87). Other isotopes used for RIT of B-cell NHLs are 186Re and 67Cu, but clinical trials using these isotopes have been quite limited to date (21,25). Their advantages and disadvantages for RIT are summarized in Table 2.
Nonmyeloablative and Myeloablative RIT
Two distinct approaches of RIT are used to deliver cytocidal radioactivity to tumors, including B-cell NHLs: the nonmyeloablative or low-dose RIT approach and the myeloablative or high-dose RIT approach (90,91). In the nonmyeloablative RIT approach, the radionuclide dose given does not result in bone marrow ablation. Myelosuppression is usually the only significant and dose-limiting toxicity ([DLT] i.e., the toxicity determining the maximum tolerated dose [MTD]). This toxicity usually occurs 2–3 wk after therapy with the nadir reached at about 4–8 wk and full recovery usually before 12 wk after RIT. Only minimal nonhematologic toxicity is usually observed with this approach, including patients with B-cell NHLs treated with the various radiolabeled anti-B-cell NHL mAbs (3–10,12–17,19–35,38–40). In myeloablative RIT, considerably higher amounts of radioactivity are administered, which are very likely or almost certain to result in ablation of marrow (11,18,19,36,37,39,92). This approach, therefore, requires a hematopoietic, usually autologous, stem-cell transplant (HSCT) with peripheral blood stem cells or bone marrow. With this approach, significant nonhematologic or “second-organ” toxicity is observed, occurring within 1–2 mo after therapy with recovery usually completed within a few weeks, in addition to severe hematologic toxicity rescued with HSCT. The most commonly observed nonhematologic toxicity is gastrointestinal, hepatic, and, at the highest doses, also cardiopulmonary, with the latter in addition to gastrointestinal toxicity usually representing the DLT (11,18,19,36,37,92). As described below, this is also true with myeloablative doses of radiolabeled anti-B-cell NHL mAbs (11,18,19,36,37).
Dosimetry of Radiolabeled Antibodies as RIT Agents
The dosimetric approaches used to calculate the total-body, normal organs (including the red marrow), and tumor radiation absorbed doses from radiolabeled antibodies used in RIT are similar to those for other radiopharmaceuticals. These have been discussed extensively in a previous continuing education article in this journal (93) and in numerous publications on this subject (93–107). Briefly, generally planar anterior and posterior gamma-camera images of the entire body or the chest, abdomen, pelvis, and any other tumor-containing region are obtained on at least 3 or 4 occasions after a tracer injection of radiolabeled mAb, given 1 wk before therapy. In the case of 90Y-mAb, the tracer dose is labeled with 111In (as 90Y surrogate) to allow γ-imaging. Regions of interest (ROIs) are then drawn around the organs and tumors, with the appropriate background ROIs, and their “actual” content of radioactivity is determined using an activity quantification technique for the gamma camera based on either the geometric mean method, a modification thereof, or the buildup factor methodology (95,96). The organ and tumor time-activity data are then fit to either an exponential function or by a trapezoidal modeling method and then integrated to obtain the cumulated activity. The cumulated activity in the red marrow is calculated either using a blood-derived method, assuming a red marrow-to-blood activity concentration ratio of 0.2–0.4, or an imaging-based method based on a well-defined ROI in the lumbar or sacral marrow (99–102,108). The cumulated activity in the total body is calculated using either whole-body scanning or γ-probe (i.e., thyroid or hand-held probe) counts obtained on at least 3 occasions over a 1-wk period (103). The normal organ, tumor, red marrow, and total-body residence times (τ), defined as the area under the curve of cumulated activity divided by the activity injected, are then determined, and the radiation absorbed doses are calculated by multiplying τ by the appropriate S factors, according to one of the dosimetry programs of the MIRD Committee (104–106). The weights of normal organs needed for calculating the S factors are estimated using either CT or the standard human weights given by the MIRD Committee, whereas tumor masses are usually determined by CT.
RIT Dosing Methods
Principally, 2 RIT dosing methods can be used to determine the radioactive amount of radiolabeled mAb to be prescribed for patients treated using the nonmyeloablative or myeloablative approach (90,91). One of these methods is dosimetry-based—that is, the radioactive amount given is based on a prescribed radiation dose to the critical dose-limiting organ: the red marrow or the total body (as marrow surrogate) in the case of nonmyeloablative RIT (12,16,17,24,30–35) or the critical dose-limiting second organ (e.g., lungs, liver, or kidneys) in the case of myeloablative RIT (11,18,36,37,92). Using this approach, the radioactivity (in MBq) to be administered to an individual patient is calculated by dividing the prescribed radiation dose (in cGy) by the anticipated radiation dose to the critical organ per MBq of administered activity (i.e., in cGy/MBq) determined by a pretherapy tracer study. Using this approach, it is assumed that the radiation dose to the critical organ per MBq of administered activity will be very similar for both the tracer and the therapy doses, which has been confirmed using various 131I-labeled mAbs, with imaging both before and after therapy (16,17,24,30–35,38). Because the mAb pharmacokinetics, and, hence, red marrow, total-body, and organ radiation doses, are unique to each patient and are influenced by factors such as patient size, body mass, and tumor burden, this method is considered patient specific (11,16,17,24,30–35,38). An important example of the utilization of this method in nonmyeloablative RIT is the approach used to determine the patient-specific radioactive amount of 131I-tositumomab based on a prescribed total-body dose, which is used here as a surrogate for the red marrow dose (16,17,30–35,38). An example of the use of the dosimetry-based method for myeloablative RIT is the approach used to determine the patient-specific radioactive amount of 131I-anti-CD20 (1F5 and B1) and 131I-anti-CD37 (MB-1) mAbs based on a prescribed radiation absorbed dose to the critical second organs (11,18,36,37).
The other dosing method is based on fixed amounts of radioactivity or amounts adjusted to the body weight (i.e., MBq/kg) or body surface area (i.e., MBq/m2) (3–10,13–15,19–23,25,26–29,39). This method does not require a pretherapy tracer study to calculate the therapy dose, although a tracer study is usually still performed to determine tumor targeting or altered mAb biodistribution in normal organs. One important example of the use of this method in nonmyeloablative RIT trials is the approach used to determine the radioactive amount of 90Y-ibritumomab tiuxetan, which is based on the patient’s body weight (3,4,26–29).
Advantages of RIT Compared with Immunotherapy
The approach of conjugating the antibodies with suitable therapeutic radioisotopes distinguishes RIT from immunotherapy either with unconjugated (naked) antibodies or with those conjugated to toxins, so-called immunotoxins (60). Treatment with antibodies conjugated to radioisotopes (RIT) rather than with immunotoxins or naked antibodies results in several distinct advantages. One obvious advantage, at least compared with treatment with unconjugated anti-B-cell NHL mAbs, is that with RIT, both biologic and immunologic mechanisms of the antitumor effects of these mAbs, such as apoptosis, ADCC, and CDC, along with the effects of targeted radiation play a role in the antitumor responses observed (3–43). Another important advantage is that with RIT, there is no need to target every tumor cell with a particular antibody to cause an antitumor effect at the cellular level because even antigen-negative nontargeted cells can be irradiated and potentially killed by radiation from targeted neighboring cells. This is not the case with unconjugated antibodies or immunotoxins, where each particular tumor cell must be targeted with the antibody for the antitumor effect to occur at the cellular level. Consequently, if the antigen in not expressed on each or nearly each tumor cell, it is unlikely that a complete regression of tumor mass will occur with unconjugated antibodies or immunotoxins. This factor may, at least in part, explain the generally higher fraction of CRs among the ORs (i.e., both CRs and PRs), using even nonmyeloablative doses of radiolabeled mAbs compared with similar unconjugated mAbs (2–4,16,17,20–35,38,39). Another potential advantage of RIT is that its mode of radiation delivery at the low-dose rate of usually <20 cGy/h may make it more effective than the high-dose rate external-beam radiation (XRT) on a gray-by-gray basis, the so-called inverse-dose-rate effect. This is presumably related to the effects of apoptosis on lymphoma cells and to cell cycle redistribution with cell accumulation in the radiosensitive G2-M phase of the cell cycle (46,47).
RIT AS A SINGLE MODALITY FOR B-CELL NHL
Pilot and Phase I/II Trials
Numerous pilot and phase I or phase I/II RIT trials have been reported using various radiolabeled mAbs directed against B-cell NHL (3–25). These trials were designed to investigate the targeting potential, pharmacokinetics, and dosimetry of the various radiolabeled mAbs or to determine their toxicity profile and MTD in previously treated B-cell NHL patients (3–25). As expected, the mAbs used in these trials recognized different antigens present on B-lymphocytes and differed in their binding affinities, immunoreactivities, isotypes, internalization rates, and number of antigenic sites on normal and malignant B-cells. The majority of these mAbs are directed against the CD antigens such as CD37 (MB-1 mAb) (11,12,18), CD21 (OKB7 mAb) (14), CD20 (B1, IF5, and 2B8 mAbs) (3,4,16–18,23), and CD22 (LL2 mAb) (15,19,20,24,25). In contrast, the Lym-1 mAb, the first mAb used in RIT trials of lymphoma patients, is directed against the HLA-DR antigen (5–10,21,22). Because of the initial lack of stable chelators for mAb labeling with radiometals, most mAbs were labeled with 131I (5–19). However, with the availability of stable chelators, 90Y-, 186Re-, or 67Cu-labeled mAbs have been used increasingly as a more practical or potentially more effective therapy (3,4,20–25).
Interestingly, several of the initially used mAbs were abandoned subsequently in favor of more optimal ones or second-generation humanized chimeric or hyperchimeric mAbs. For example, Press et al. (11,18) and Kaminski et al. (12) initially investigated the 131I-MB-1 anti-CD37 mAb as a candidate for RIT of B-cell NHL, but this was later abandoned, because of its considerable cross-reactivity with a wide range of B-cell precursors, in favor of 131I-labeled anti-CD20 mAbs with a more restricted cross-reactivity (16–18). The OKB7 mAb initially investigated by Sheinberg et al. (14) was not further developed for clinical use. Goldenberg et al. (15) and Vose et al. (20) used a murine 131I-labeled anti-CD22 LL2 mAb, but this was later replaced by the humanized hyperchimeric 90Y-labeled LL2 (90Y-hLL2 or epratuzumab) because of the known rapid internalization of LL2, expectedly reduced immunogenicity, and potentially greater immune effector function of the humanized compared with the murine mAbs (19,24,25). Thus, many of the conducted pilot or phase I trials have enabled the recognition of ideal candidate mAbs (labeled with either 131I or 90Y) for further drug development as RIT agents in patients with B-cell NHL. These were then the subject of formal phase I studies, with the primary goal of determining their MTD, followed by definitive phase II and III trials to formally determine their efficacy in well-defined patient populations.
It is noteworthy that both the nonmyeloablative and the myeloablative approaches of RIT were used even in the initial trials and that both the dosimetry-based method (11,12,16–18,24) and dosing based on fixed amounts of radioactivity or radioactive amounts adjusted to the body weight or surface area were used for RIT dosing (3–10,13–15,20–23,25). The specific dosing method used in the dose-escalation process to determine the MTD of a particular mAb was usually maintained in the subsequently performed phase II and III trials with the same mAb (3,4,11,16–18,26–38).
The selection criteria for B-cell NHL patients entered into the pilot or phase I/II trials were quite similar among the various trials: All required prior treatment with standard chemotherapy and most of them adhered to the standard eligibility criteria for investigational therapy trials. The latter included normal renal and hepatic function, no surgery or chemotherapy for at least 4 wk before RIT, a white blood cell count of ≥3,000 per mm3, or an absolute neutrophil count (ANC) of ≥1,500 per mm3 and a platelet count of ≥100,000 per mm3. Initially, some trials did not require bone marrow biopsy to determine eligibility and, therefore, some patients with extensive marrow involvement were treated (6,14,15,21). Subsequently, however, nearly all of the trials required that the degree of lymphomatous marrow involvement does not exceed 25% of the hematopoietic marrow elements, based on unilateral or bilateral marrow biopsies (3,4,11–13,16–20,22–25). These criteria remained, with minor modifications, through the subsequently performed phase II and III trials and should apply for the clinical use of RIT in the nonmyeloablative setting using the now commercially available 90Y-ibritumomab tiuxetan mAb. Clinical trials of myeloablative RIT required additional criteria such as normal cardiac and pulmonary function and availability of an adequate number of bone marrow mononuclear cells or CD34+ peripheral blood stem cells (11,18,19,39).
Although multiple mAbs, labeled with various isotopes, have been tested in the many trials of RIT of B-cell NHL, the single-dose nonmyeloablative or myeloablative MTD has been reported for only a few mAbs (3,4,17,18,22). Most notable are 131I-tositumomab, for which both the nonmyeloablative and the myeloablative MTDs are now known (17,18), and 90Y-ibritumomab tiuxetan, for which only the nonmyeloablative MTD has been reported to date (3,4). Interestingly, whereas Press et al. (18) reported the myeloablative MTD for “131I-anti-CD20 (1F5 and B1) and anti-CD37 (MB-1) mAbs,” all used in the same dose-escalation trial, all of the patients treated at the higher dose levels received 131I-B1 (tositumomab). However, because these investigators used a dose-escalation scheme based on escalating radiation doses to critical dose-limiting second organs rather than escalating amounts of radioactivity, it might be argued that the determined MTD may in fact have been quite similar for the different mAbs used. Taking this even further, one might also argue that this MTD may be quite similar for the various anti-B-cell NHL mAbs used, at least when labeled with 131I, provided that there are no major differences in the microdistribution of delivered radiation dose (i.e., differences at the “microdosimetric” level). This may be a reasonable assumption for virtually all currently used anti-B-cell lymphoma mAbs. On the other hand, the dosing methods used in the phase I dose-escalation trials designed to determine the nonmyeloablative MTDs of 131I-tositumomab and 90Y-ibritumomab tiuxetan mAbs were considerably different: A dosimetry-based approach based on total-body radiation dose, used as a surrogate for the red marrow dose, was used for the former, whereas dosing based simply on body weight was used for the latter (3,4,17). Subsequently, the same dosing approaches unique for each of these 2 mAbs were used in the phase II and III nonmyeloablative RIT trials with these mAbs, and the dosing approach used for 90Y-ibritumomab tiuxetan is now also used in the standard clinical practice (26–35,38,109). It is very important to emphasize, however, that the use of the total-body dose as a surrogate for the red marrow dose appears reasonable only in the case of 131I-labeled mAbs. This is because, with 131I-labeled mAbs, there is usually a moderate correlation between the total-body dose and the red marrow dose, at least when calculated on the basis of blood, and because the total-body dose (actually, remainder of the body dose) contributes significantly (i.e., 40%–50%) to the red marrow dose (107). The latter is related to the penetrating γ-radiation of 131I. In contrast, with 90Y-labeled mAbs, poor correlation is expected between the total-body dose and the imaging or blood-based red marrow dose because most of the residualizing 90Y label is retained in the body independent of the amount of cumulated activity in the red marrow. Moreover, because of the nonpenetrating radiation of 90Y, the total-body dose contributes only little (i.e., <10%) to the red marrow dose. For all of these reasons, the total-body dose of 75 cGy found to be the MTD for 131I-tositumomab could not be used as the MTD for 90Y-labeled mAbs (e.g., 90Y-ibritumomab tiuxetan).
The final results of the phase I trial with nonmyeloablative doses of 131I-tositumomab were reported by Kaminski et al. (17). Thirty-four patients were entered, 18 with low-grade NHL and 16 with intermediate-grade NHL. Twenty-one patients had chemotherapy-resistant NHL and 14 had large tumor burdens (i.e., >500 g). The administered 131I-tositumomab doses were based on escalating radiation doses to the total body predicted by a pretherapy tracer study (185 MBq [5 mCi]; 15 mg). The same amount of unlabeled antibody protein ranging from 150 to 700 mg in the majority of patients was preinfused 1 h before the labeled tracer or therapy dose to improve the 131I-tositumomab biodistribution. Dose escalation was started at a radiation dose of 25 cGy to the total body, increasing in increments of 10 cGy. The MTD was then determined to be 75 cGy to the total body in patients who had not undergone a prior marrow transplant but appeared to be lower in patients with a prior bone marrow transplant (see below). The radioactive doses given ranged from 1.26 to 5.56 GBq (34–161 mCi) 131I-tositumomab. Twenty-two of 28 evaluable patients (79%) responded, with 14 (50%) having a CR. The median CR duration was >13 mo, with 8 patients remaining disease free at the time of the published report.
The results of the completed phase I/II trial with nonmyeloablative doses of 90Y-ibritumomab tiuxetan mAb were reported by Witzig et al. (3) and Wiseman et al. (4). Eligible patients had relapsed or refractory CD20+ B-cell low-grade, intermediate-grade, or mantle-cell NHL. There was no limit on bulky disease, and 59% had at least 1 mass of ≥5 cm. Dose escalation was started at a dose of 7.4 MBq/kg (0.2 mCi/kg) of 90Y-ibritumomab tiuxetan, increasing in increments of 3.7 MBq/kg (0.1 mCi/kg) until the MTD was determined. The treatment scheme used in this trial is shown in Figure 2. As can be seen, the chimeric rituximab (250 mg/m2) was chosen as the unlabeled mAb preinfused within 4 h before the murine radiolabeled ibritumomab tiuxetan mAb to improve its biodistribution. mAb pharmacokinetics, dosimetry, and tumor targeting were determined on the basis of a pretherapy 111In-ibritumomab tiuxetan (111In-Zevalin) study (185 MBq [5 mCi]; 1.6 mg) preceded by a rituximab dose of 250 mg/m2, the same dose used before administering therapy with 90Y-ibritumomab (90Y-Zevalin). Figure 3 shows an example of imaging with 111In-ibritumomab tiuxetan demonstrating the expected whole-body and normal organ biodistribution in addition to tumor targeting in a patient with B-cell NHL.
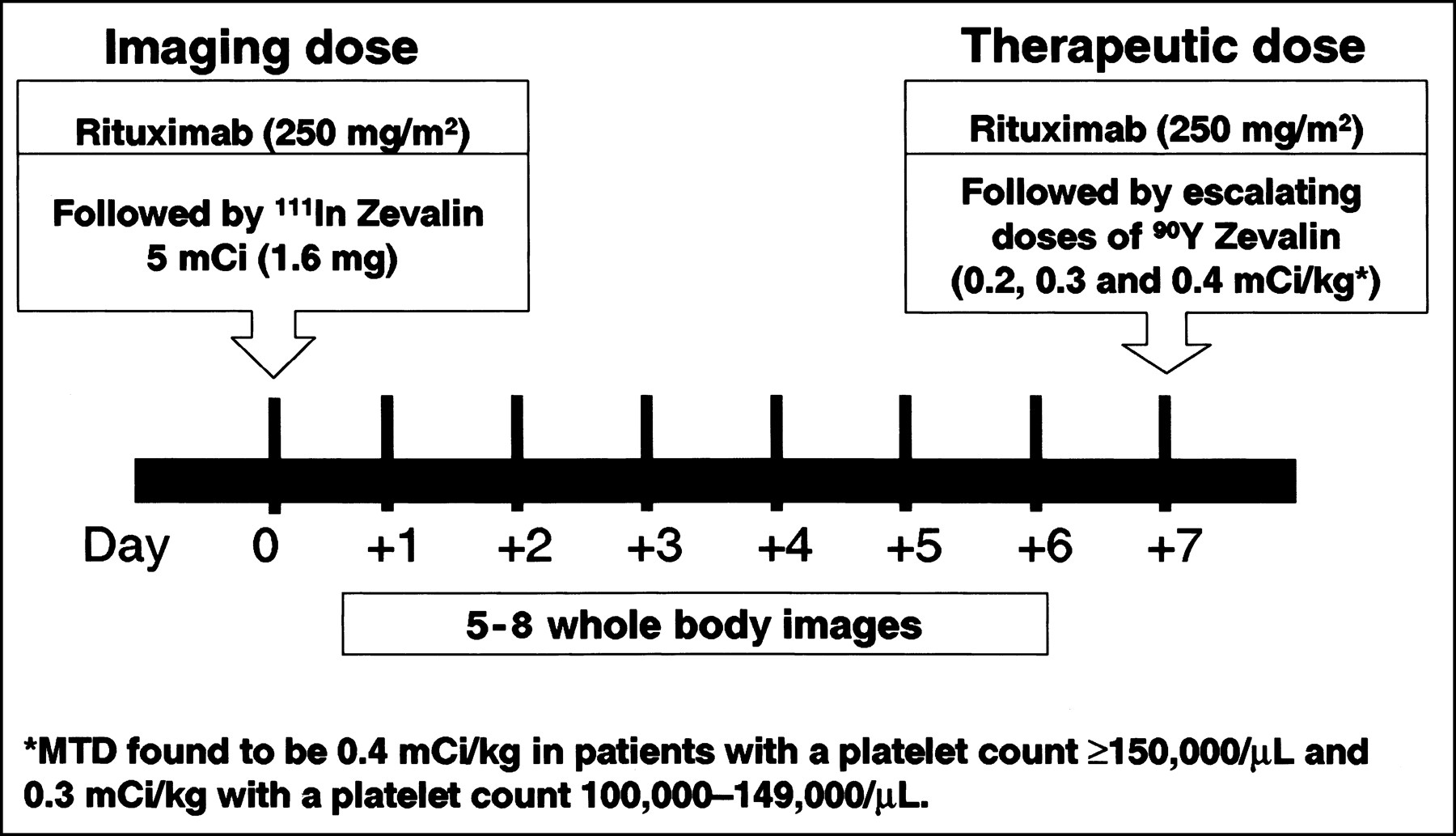
Treatment schema used in phase I/II trial with 90Y-ibritumomab tiuxetan (90Y-Zevalin). Similar scheme is used in clinical practice except that already determined MTD of 90Y-ibritumomab tiuxetan is now given for therapy and only 2 or 3 whole-body images are required to ascertain acceptable (i.e., unaltered) radiolabeled ibritumomab tiuxetan biodistribution (see Fig. 3).
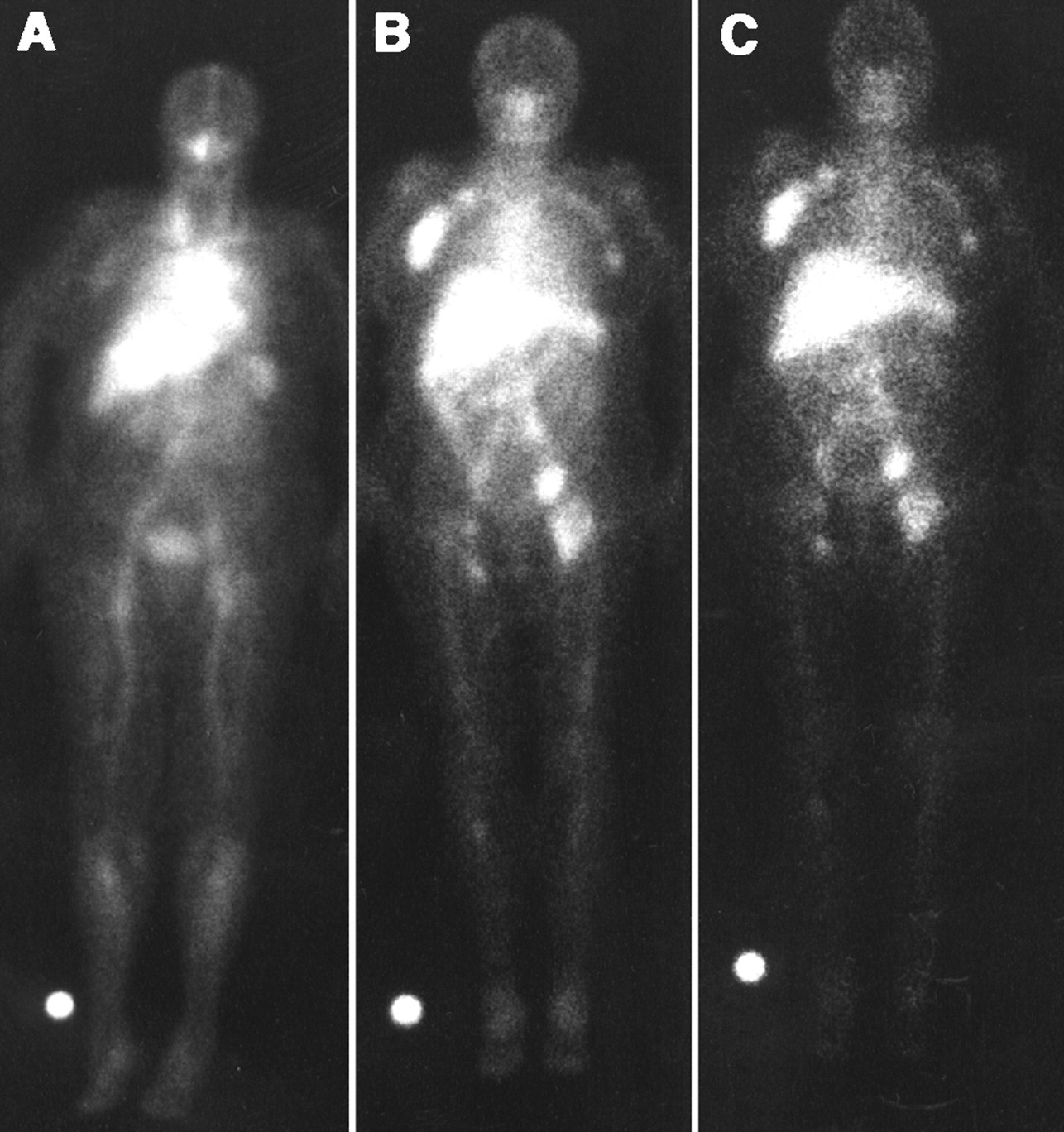
Anterior whole-body images obtained at 4 (A), 63 (B), and 140 (C) h after injection of 185 MBq (5 mCi; 1.6 mg) 111In-ibritumomab tiuxetan. Scan shows expected acceptable biodistribution of mAb at various time points and targeting of axillary, paraaortic, bilateral iliac, and femoral lymphadenopathy (courtesy of Gregory Wiseman from the Mayo Clinic Foundation, Rochester, MN).
Although the administered 90Y-ibritumomab tiuxetan dose was not based on dosimetry, the investigators had to ensure that, on the basis of the pretherapy 111In-ibritumomab tiuxetan study, critical threshold doses of 300 cGy to the marrow and 2,000 cGy to any other nontumor organ (particularly, the lungs, liver, or kidneys) were not exceeded when administering their body weight-based doses of up to 14.8 MBq/kg (0.4 mCi/kg). This condition was met in all but 1 patient entered into the trial, in whom local clinical site dosimetry showed an imaging-based red marrow dose of >300 cGy; this patient therefore was not treated with 90Y-ibritumomab tiuxetan. Interestingly, the estimated red marrow dose based on blood or imaging determined subsequently by centralized dosimetry showed that the red marrow dose was <300 cGy in all patients. The MTD was determined to be 14.8 MBq/kg (0.4 mCi/kg) in patients with a baseline platelet count of ≥150,000 per mm3 and 11.1 MBq/kg (0.3 mCi/kg) for patients with a baseline platelet count of 100,000–149,000 per mm3. Fifteen patients were treated in the phase I portion and 35 patients were treated in the phase II portion of the trial, of whom 10 were treated at the 11.1-MBq/kg (0.3 mCi/kg) dose level and 25 were treated at the 14.8-MBq/kg (0.4 mCi/kg) dose level. Because similar responses were seen in the phase I and II portions of the trials (PR and CR were seen apparently independent of the dose given), the 15 patients treated in the phase I portion of the trial were combined with those enrolled in the phase II portion for an intent-to-treat analysis. The overall response rate (ORR) for all of the 51 patients entered into the trial (1 patient was not treated) was 67% (25% CR; 41% PR). The ORR for low-grade disease (n = 34) was 82% (27% CR; 56% PR; 95% confidence interval, 54–80), whereas that for intermediate-grade disease (n = 14) was 43% (29% CR; 14% PR). None of the 3 patients with mantle-cell NHL responded. Responses occurred in patients with bulky disease (≥7 cm; 41%) and splenomegaly (50%). Kaplan-Meier estimates of the time to disease progression (TTP) in responders and the duration of response (DR) were 12.7 mo (95% confidence interval, 10.2–17.8) and 11.6 mo (95% confidence interval, 10.2–17.8), respectively. Adverse events were primarily hematologic and correlated with the baseline extent of marrow NHL and the baseline platelet count, and only 1 patient (2%) developed an anti-antibody response (HACA/HAMA).
The results of the phase I trial with myeloablative doses of 131I-anti-CD20 (1F5 and B1) and 131I-anti-CD37 (MB-1) mAbs combined with autologous HSCT were reported by Press et al. (18). As stated earlier, these investigators used a dosimetry-based approach to determine the MTD of these mAbs. The administered radioactive amount of 131I-labeled mAb was based on escalating radiation doses to critical second organs (i.e., lungs, liver, and kidneys). More specifically, the radioactivity given to an individual patient was calculated by dividing the prescribed radiation dose (in cGy) by the anticipated radiation dose to the critical organ per MBq of administered activity (i.e., cGy/MBq) determined by a pretherapy tracer study with 185–370 MBq (5–10 mCi) 131I. The same amount of unlabeled mAb protein (2.5 mg/kg for B1 or 1F5 and 10 mg/kg for MB-1) was preinfused 1 h before the labeled tracer or therapy infusion to improve the mAb biodistribution. Dose escalation was started at a radiation dose of 1,000 cGy to the critical organ (found to be the lungs in most patients), increasing to 1,500, 1,675, 2,075, 2,375, 2,725, and 3,075 cGy. A unique feature of this trial was that only patients with favorable biodistribution, defined as those with a tumor-to-critical organ radiation absorbed dose ratio of >1.0 for all tumors, as determined by a pretherapy tracer study, were treated subsequently. Favorable distribution was achieved in 24 of the 43 patients initially considered, of whom 19 were treated subsequently, primarily with 131I-B1. The investigators found that the MTD was 2,725 cGy to the lungs (an average of ∼11.48 GBq/m2 [∼310 mCi/m2] 131I-B1). Cardiopulmonary or gastrointestinal toxicity was the principal second-organ and DLT. Other nonhematologic toxicities (e.g., hepatic) were also observed, but these were not dose limiting. Of the 19 patients treated, 18 (95%) had an OR (PR or CR) with 16 (84%) achieving a CR. Two patients had a PR and 1 had a minor response (25%–50% regression of tumor). Nine patients remained in continuous CR for 3–53 mo at the time of the report.
A detailed review of the particular results of the numerous pilot or phase I/II trials of nonmyeloablative and myeloablative RIT in B-cell NHL, which were considerably different in design and in the type of the radiolabeled mAb used, is beyond the scope of this article. Nevertheless, the most important findings of these trials may be summarized in a few points: First, transient myelosuppression was found to be the only significant and DLT in the nonmyeloablative RIT trials regardless of the radiolabeled mAb used (3–10,12–17,19–25), whereas cardiopulmonary or gastrointestinal toxicity was most significant and DLT in the myeloablative trials using 131I-anti-CD20 and CD37 mAbs (11,18). Second, to date, the nonmyeloablative single-dose MTD has been reported for 2 mAbs that either are in an advanced stage of development (i.e., 131I-tositumomab or Bexxar) or are already commercially approved (i.e., 90Y-ibritumomab tiuxetan or Zevalin) (3,4,17). The MTD of the former was found to be 75 cGy to the total body, whereas that of the latter was 14.8 MBq/kg (0.4 mCi/kg) in patients with a platelet count of ≥150,000 per mm3 (11.1 MBq/kg [0.3 mCi/kg] in patients with a platelet count of 100,000–149,000 per mm3) (3,4,17). The single-dose MTD determined for 90Y-Lym-1 mAb was found to be 370 MBq/m2 (10 mCi/m2) (22). Although, to date, no single-dose MTD has been reported for the other 131I- or 90Y-labeled anti-B-cell NHL mAbs, doses of up to 1.66–3.7 GBq/m2 (45–100 mCi/m2) of 131I-mAbs (depending on the mAb used) or 555–740 MBq/m2 (15–20 mCi/m2) of 90Y-mAbs have been safely administered (5–10,21,24). With myeloablative RIT, the MTD was found to be 2,725 cGy to the lungs with 131I-anti-CD20 and 131I-anti-CD37 mAbs (18). Third, excellent antitumor responses were demonstrated even with nonmyeloablative RIT, with an ORR and complete remission rate (CRR) of 30%–80% and 10%–50%, respectively, in the various trials using different mAbs labeled with 131I, 90Y, 67Cu, and 186Re (3–10,12–17,19–25). These responses were often observed with tumor doses of <500 cGy regardless of the isotope used, underscoring the exquisite sensitivity of NHL (3–10,12–17,19–25). However, an even higher ORR of 95% was seen with myeloablative RIT, with a CRR of 84% using 131I-anti-CD20 and anti-CD37 mAbs, although the patients treated in the trial with myeloablative RIT were presumably highly selected; only patients with a favorable biodistribution were treated (18). Fourth, with nonmyeloablative RIT, the ORR appeared to be higher in low-grade (indolent) compared with intermediate-grade B-cell NHL (virtually the only form of aggressive NHL treated with RIT). For example, using 131I-B1 mAb, ORR and CRR of 100% and 77%, respectively, were shown in low-grade NHL compared with 43% and 14%, respectively, in intermediate-grade NHL (17). In contrast, similarly high ORRs and CRRs were seen with myeloablative RIT in low-grade and intermediate-grade NHL (18) (Dr. Oliver Press, oral communication, 2000). Fifth, the ORR, and especially the CRR, was higher with even a single nonmyeloablative dose of radiolabeled anti–B-cell lymphoma mAb compared with multiple cycles of a similar unconjugated mAb. For example, in patients with low-grade NHL, the ORR with rituximab, the unconjugated chimeric anti-CD20 mAb, was reported to be about 50%, with a CRR of only 6% despite 4 weekly infusions of 375 mg/m2 per infusion (a total of 1,500 mg/m2) (2). In contrast, the ORR for 90Y-ibritumomab tiuxetan (the murine parent mAb of rituximab) in these patients shown in the initial phase I/II trial was 82%, with a CRR of 27%. Similar ORRs and CRRs were observed using 131I-tositumomab, another similar anti-CD20 mAb (17). This observation, already apparent in these initial studies, was confirmed subsequently in a randomized trial comparing ibritumomab tiuxetan with rituximab (27) (see below).
Phase II and III Trials with Radiolabeled Anti-B-Cell NHL mAbs
The last 3 y or so in the development of RIT of B-cell NHL are characterized primarily by the conduct of definitive phase II or III trials to formally determine the efficacy of radiolabeled mAbs identified as “ideal” candidates for RIT (26–37). To date, these phase II and III trials are available only for 131I-tositumomab, given in nonmyeloablative and myeloablative doses, and for 90Y-ibritumomab tiuxetan, given in nonmyeloablative doses (26–37). A brief summary of the results of these trials follows.
Phase II and III Studies with Nonmyeloablative Doses of 131I-Tositumomab.
Vose et al. (30) have reported the final results of a multicenter phase II study evaluating the efficacy, dosimetry methodology, and safety of 131I-tositumomab in patients with chemotherapy-relapsed/refractory low-grade or transformed NHL. Patients received a dosimetric dose consisting of 450 mg of unlabeled tositumomab followed by 35 mg (185 MBq [5 mCi]) 131I-tositumomab. Serial total-body γ-counts were then obtained to calculate the patient-specific activity (MBq) required to deliver the therapeutic dose. A therapeutic dose of 75-cGy total-body dose (attenuated to 65 cGy in patients with platelet counts of 100,000–149,000 cells per mm3) was given 7–14 d after the dosimetric dose. Forty-five of 47 patients were treated with a single dosimetric and therapeutic dose. Twenty-seven patients (57%) had a response. The ORR was similar in patients with low-grade (57%) or transformed (60%) NHL. The median DR was 9.9 mo. Fifteen patients (32%) achieved a CR, including 5 patients (50%) with transformed NHL. The median duration of CR was 19.9 mo, and 6 patients had an ongoing CR at the time of the report. Treatment was well tolerated, with the principal toxicity being hematologic. The most common nonhematologic toxicities considered to be possibly related to the treatment were mild-to-moderate fatigue (32%), nausea (30%), fever (26%), vomiting (15%), infection (13%), pruritus (13%), and rash (13%). Additionally, 1 patient developed HAMA.
The results of the pivotal trial of 131I-tositumomab in chemotherapy-refractory low-grade or transformed low-grade NHL were reported recently by Kaminski et al. (32). The objective of this trial was to evaluate the efficacy and safety of 131I-tositumomab in these patients and to compare its efficacy to the patients’ last qualifying chemotherapy (LQC) regimens. A total of 60 patients who had been treated with at least 2 protocol-specified qualifying chemotherapy regimens and had not responded or progressed within 6 mo after their LQC were treated with a single course of 131I-tositumomab at the MTD of 75 cGy to the total body. The patients had received a median of 4 prior chemotherapy regimens. Thirty-nine patients (65%) had a CR or PR after 131I-tositumomab compared with only 17 (28%) after their LQC (P < 0.001). Two patients (3%) had a CR after their LQC compared with 12 (20%) after 131I-tositumomab (P < 0.001). The median DR for CR was 6.1 mo after the LQC and had not been reached with follow-up of >47 mo after 131I-tositumomab at the time of the report. An independent review panel verified that 32 of the 43 patients (74%) with nonequivalent DR (>30-d difference) had a longer DR after 131I-tositumomab (P < 0.001). Only 1 patient was hospitalized for neutropenic fever. Five patients (8%) developed HAMA and 1 (2%) developed an elevated thyroid-stimulating hormone (TSH) level after treatment. Myelodysplasia was diagnosed in 4 patients (6.6%) at follow-up.
The interim safety and efficacy results of 131I-tositumomab given in a large multicenter expanded-access trial have been reported recently (33,34). Three hundred fifty-nine patients with low-grade or transformed NHL were enrolled in this trial at 53 community and academic sites between July 1998 and March 2000. The median age of these patients was 58 y (range, 39–87 y). Ninety percent had stage III or IV at entry, 44% had bulky disease (≥5 cm), 60% had an elevated LDH level, and 45% had lymphomatous bone marrow involvement (≤25%). The median number of prior chemotherapeutic regimens was 2 (range, 1–9). Forty-three percent failed rituximab therapy and 21% had prior radiotherapy. Again, an individualized dosing scheme that was similar to that in the phase II studies was used to deliver a total-body dose of 75 cGy. A saturated solution of potassium iodide was used to block the uptake of free 131I by the normal thyroid. As expected, the most common toxicity was reversible myelosuppression. The median ANC nadir was 1,300 cells per mm3, whereas the platelet and hemoglobin nadirs were 68,000 cells per mm3 and 11.2 g/dL, respectively. Fourteen percent of patients developed an ANC nadir of <500 cells per mm3 and 2% developed a platelet nadir of <10,000 cells per mm3 (grade 4 National Cancer Institute toxicity). The most common nonhematologic adverse experiences were grade 1 or grade 2 asthenia (23%), nausea (17%), fever (11%), and pain (10%). Eight percent of patients developed HAMA, 2% exhibited an elevated TSH level after therapy, and 4% percent developed a serious infection requiring hospitalization. Adverse experiences were not more frequent or severe in patients >60 y old compared with patients ≤60 y old. Two hundred seventy-three of the 359 patients could be evaluated for antitumor response at the time of the report. The ORR and CRR were 58% and 27%, respectively. Patients with poor prognostic features achieved significant ORR and CRR rates. The median DR and duration of CR have not been reached with follow-up to 17 mo at the time of the report. The TTP for all patients was about 6.7 mo and has not been reached for responders. Even in patients who failed >4 prior chemotherapeutic regimens, the ORR and CRR were 44% and 17%, respectively. The ORR and CRR were 51% and 17% for patients >60 y of age, 47% and 19% for rituximab failures, 61% and 27% for those with prior radiotherapy, and 40% and 16% for patients with transformed low-grade NHL. The ORR and CRR in patients with bulky tumors (i.e., ≥5 cm) were somewhat lower than those in patients with lower tumor bulk: 47% and 17% versus 66% and 35%, respectively. In contrast, the ORR and CRR in patients with bone marrow involvement were quite similar to those without such involvement: 56% and 25% versus 59% and 28%, respectively.
Whereas all of the trials with 131I-tositumomab summarized above have been conducted in patients who were treated previously with chemo- or immunotherapy, Wahl et al. (35) have conducted a phase II trial with this mAb in previously untreated (i.e., chemotherapy-naive) patients with advanced-stage low-grade or transformed NHL. This trial was in fact the first to evaluate the toxicity and therapeutic efficacy of RIT in previously untreated NHL patients of any histologic type. Seventy-six patients were studied, of whom 54 (71%) had follicular small-cleaved and 22 (29%) had follicular mixed cell histology. All had stage III or IV disease, 65% had lymphomatous bone marrow involvement of ≤25%, and 29% had high tumor burdens (>500 g). These patients were also treated using the dosimetry-based approach at an MTD of 75 cGy to the total body. Seventy-four of 76 patients (97%) showed a PR or CR, with 63% achieving a CR. The median DR has not been reached with follow-up of up to 38 mo and a median follow-up of 16.2 mo at the time of the report. Molecular CRs by polymerase chain reaction were common. Grade 4 neutropenia occurred in only 5% of patients, with no grade 4 thrombocytopenia noted. No patient required hematologic supportive care. However, HAMA was seen in 49 of 76 patients (64%) to this murine mAb, and about two thirds of these patients had flu-like symptoms lasting <1 wk within the first 2 wk after therapy.
In summary, the reported phase II and III trials with 131I-tositumomab show that this mAb, when given at the MTD of 75 cGy to the total body, is quite safe and effective in patients with low-grade and transformed NHL at the various stages of their disease. Moreover, the results of the phase II study conducted in patients with previously untreated low-grade or transformed NHL may provide, in principle, strong justification for considering this relatively new modality as front-line therapy for such patients either alone or in combination with chemotherapy. Unfortunately, a potential drawback related to the use of the murine tositumomab in this setting is the quite high incidence of HAMA response. This could potentially jeopardize the possibility of front-line multicycle therapy (in hopes of obtaining more frequent molecular CRs or sustained responses) or retreatment at the time of relapse. The use of humanized chimeric or hyperchimeric mAbs, mAb regimens containing smaller amounts of murine mAb (e.g., murine ibritumomab tiuxetan in combination with rituximab), and combining RIT with immunosuppressive cytotoxic chemotherapy may, at least in part, circumvent this problem (3,4,19,24–29,39–41).
Phase II and III Studies with Nonmyeloablative Doses of 90Y-Ibritumomab Tiuxetan.
The results of the formal phase II trial with 90Y-ibritumomab tiuxetan were reported by Witzig et al. (3) and Wiseman et al. (4) in conjunction with the findings of the phase I study and were described above.
Wiseman et al. (26) have reported subsequently the results of another phase II trial, in which the investigators focused exclusively on mildly thrombocytopenic patients (100,000–149,000 platelets per mm3) with relapsed/refractory low-grade or transformed B-cell NHL to evaluate the safety and efficacy of 90Y-Zevalin at the determined MTD dose of 11.1 MBq/kg (0.3 mCi/kg) in this group (a reduced dose compared with the 14.8-MBq/kg [0.4 mCi/kg] dose in patients with ≥150,000 platelets per mm3). Thirty patients were treated at this dose using the same treatment schedule as that used in the initial phase I/II study with this mAb (Fig. 2), except that, in this case, patients received the determined MTD rather than escalating doses of 90Y-ibritumomab tiuxetan. Patients (median age, 61 y; 90% stage III/IV at study entry; 83% follicular lymphoma; 67% with bone marrow involvement) had a median of 2 prior regimens (range, 1–9). An ORR of 83% (37% CR, 6.7% unconfirmed CR, and 40% PR) was found, with an estimated TTP of 9.4 mo (range, 1.7–24.6 mo). Toxicity was primarily hematologic, transient, and reversible. The incidence of grade 4 neutropenia, thrombocytopenia, and anemia was 33%, 13%, and 3%, respectively.
Witzig et al. (27) have reported the final results of a prospective randomized phase III trial of 90Y-ibritumomab tiuxetan and rituximab immunotherapy in relapsed/refractory low-grade, follicular, or transformed NHL. This was the first prospective randomized trial comparing the efficacy of nonmyeloablative RIT with unconjugated mAb in the same patient population. As stated earlier, 90Y-ibritumomab tiuxetan is simply the murine parent mAb of the chimeric unconjugated rituximab. Patients randomized into the 90Y-ibritumomab tiuxetan arm were given a tracer dose of 185 MBq (5 mCi) 111In-ibritumomab tiuxetan on day 0, evaluated with dosimetry, and then administered a therapeutic dose of 14.8 MBq/kg (0.4 mCi/kg) 90Y-ibritumomab tiuxetan on day 7. Both ibritumomab tiuxetan doses were preceded by an infusion of 250 mg/m2 of rituximab. Patients randomized into the rituximab arm received a standard course of rituximab immunotherapy (375 mg/m2 weekly × 4). The final efficacy analysis performed on 143 patients enrolled into this prospective phase III trial showed an ORR of 80% for 90Y-ibritumomab tiuxetan (n = 73) versus 56% for rituximab (n = 70) (P = 0.002). The CRR was 30% for 90Y-ibritumomab tiuxetan versus only 16% for rituximab (P = 0.04). The Kaplan-Meier estimated median DR was 14.2 mo for 90Y-ibritumomab tiuxetan versus 12.1 mo for rituximab (P = not significant), and the TTP was 11.2 versus 10.1 mo (P = not significant). The response to 90Y-ibritumomab tiuxetan was statistically superior to that of rituximab within prognostic subsets, including age, sex, bone marrow involvement, extranodal disease, bulk of disease, number of prior regimens, splenomegaly, and IPI risk groups. A secondary objective of this trial was to determine if radiation dosimetry before 90Y-ibritumomab tiuxetan administration is required for safe treatment in this patient population. It was found that 90Y-ibritumomab tiuxetan administered to NHL patients at its nonmyeloablative MTD delivers acceptable radiation doses (defined as ≤300 cGy to red marrow and 2,000 cGy to normal organs) to uninvolved critical organs (red marrow, lungs, liver, and kidneys). This finding clearly established the safety of administering 90Y-ibritumomab tiuxetan in this defined patient population without pretreatment 111In-based radiation dosimetry. On the basis of this and similar findings in >250 patients, the FDA has approved the administration of 90Y-ibritumomab tiuxetan without pretreatment radiation dosimetry, although an 111In-ibritumomab tiuxetan study is still required to ascertain the expected biodistribution of this mAb before administering therapy (91,109).
More recently, Witzig et al. (28) have reported the results of a nonrandomized phase III open-label clinical trial to evaluate the efficacy and safety of 90Y-ibritumomab tiuxetan in follicular NHL patients refractory to rituximab, defined as those who did not achieve a response or had a TTP of <6 mo with the most recent course of rituximab. A total of 57 patients (54 follicular, 2 small lymphocytic, and 1 transformed large cell) were treated. Toxicity was primarily hematologic, transient, and reversible. The incidence of grade 4 neutropenia, thrombocytopenia, and anemia was 35%, 9%, and 4%, respectively. The ORR was 74% with a CRR of 15%, was significantly better than the ORR to prior rituximab (ORR of 32%; P = 0.002), and was similar to that of the last chemotherapy (67%). The Kaplan-Meier estimated TTP was 6.8 mo (range, 1.12 to ≥25.9 mo) for all patients and 8.7 mo for responders.
Witzig et al. (29) recently reported the “integrated” safety data of the various clinical RIT trials with 90Y-ibritumomab tiuxetan, consisting of the above-described trials in addition to an open-label expanded-access trial and comprising a total of 349 patients with relapsed or refractory low-grade, transformed, or intermediate-grade B-cell NHL. All patients had ≤25% bone marrow involvement, circulating lymphocytes of <5,000 per mm3, ANC of ≥1,500 per mm3, platelets of ≥100,000 per mm3, adequate renal and hepatic function, and no prior stem-cell therapy. These patients (median age, 60 y; range, 24–85 y) represented a refractory population with advanced disease. Ten percent had splenomegaly; 42% had bone marrow involvement; 15% were in the IPI intermediate/high- or high-risk group; and 31% had 4 or more prior therapies. As expected, ibritumomab tiuxetan therapy consisted of day 1 rituximab (250 mg/m2) and 111In-ibritumomab tiuxetan (185 MBq [5 mCi]) and day 7, 8, or 9 rituximab (250 mg/m2) followed by a 10-min intravenous injection of 90Y-ibritumomab tiuxetan for therapy (14.8 MBq/kg [0.4 mCi/kg], reduced to 11.1 MBq/kg [0.3 mCi/kg] for patients with 100,000–149,000 platelets per mm3; maximum dose, 1.18 GBq [32 mCi]). Toxicity was primarily hematologic. Median nadirs were an ANC of 800 per mm3, a platelet count of 40,000 per mm3, and a hemoglobin level of 10.3 g/dL. Grade 3 neutropenia (ANC of ≥500 but <1,000 per mm3) and thrombocytopenia (≥10,000 but <50,000 platelets per mm3) occurred in 28% and 52% of patients, respectively, whereas grade 4 neutropenia and thrombocytopenia occurred in 30% and 10% of patients, respectively. The median duration of grade 4 neutropenia (i.e., time from grade 4 to grade 3) was 8 d, whereas that of grade 4 thrombocytopenia was 10 d. Medication and transfusion data were collected in 4 of the trials and revealed that 22% and 20% of patients received platelet and red blood cell transfusions, respectively. Also, 12.8% and 8.1% of patients received filgrastim and erythropoietin, respectively. The median duration of grade 3 or grade 4 neutropenia was 8 d, shorter in those patients who received filgrastim than in those who did not. The presence of bone marrow involvement at baseline was associated with a significantly greater incidence of grade 4 neutropenia (P = 0.001) and thrombocytopenia (P = 0.013), with an increasing incidence with greater bone marrow involvement at baseline. The chemotherapy history also was predictive of hematologic toxicity. Only 6.6% of patients were hospitalized with infections. Bleeding events occurred in 18% of patients and were grade 3 or grade 4 in only 1.7%. Two patients died of traumatic intracranial bleeding while thrombocytopenic; one of these patients was also taking oral anticoagulant and ibuprofen. Myelodysplasia or acute myeloblastic leukemia was reported in 5 patients (1.4%) from 8 to 34 mo after 90Y-ibritumomab tiuxetan treatment, which corresponds to an estimated annualized incidence of 0.6% from the date of lymphoma diagnosis and 1.1% from the date of the first infusion, based on the Kaplan-Meier estimate.
In summary, the reported phase II and III trials with 90Y-ibritumomab tiuxetan also demonstrate that this mAb, when given at its recommended dose, is safe, although a significant percentage of patients do develop transient grade 4 hematologic toxicity, particularly neutropenia. However, this toxicity appears to be easily manageable in most patients. 90Y-ibritumomab tiuxetan also demonstrates remarkable antitumor activity in patients with low-grade or transformed B-cell NHL, is clearly superior to rituximab therapy, and is effective even in patients refractory to rituximab. Considering the 95% confidence interval for the ORR, CRR, and DR expected in the various phase II and III efficacy studies with 90Y-ibritumomab tiuxetan and 131I-tositumomab, it appears that these parameters are quite similar for both mAbs, when given at their respective MTDs. Overall, the reported studies clearly demonstrate that both mAbs are quite effective in the treatment of relapsed/refractory low-grade and transformed NHL with the ORR and CRR probably ranging from 60% to 80% and 20% to 30%, respectively, with a median DR of about 1 y. Only limited data are available for aggressive NHL, predominantly from the phase I/II studies with these 2 mAbs (3,17,38). These studies suggest that the ORR and CRR may be about 30%–40% and 10%–30%, respectively. It is important to note, however, that no formal phase II efficacy trial using either mAb has been reported in patients with aggressive B-cell NHL.
Phase II Study with Myeloablative Doses of 131I-Tositumomab.
As stated earlier, Press et al. (18) have determined the myeloablative single-dose MTD for the 131I-anti-CD20 (1F5 and B1) and anti-CD37 (MB-1) mAbs. However, because only the B1 131I-anti-CD20 mAb (i.e., 131I-tositumomab) was selected for further development, definitive phase II studies are currently available only for this mAb (36,37). Press et al. (36) have reported the results of their phase II trial in 25 patients with relapsed B-cell NHL given the myeloablative MTD of 131I-tositumomab combined with autologous HSCT. The patients were first evaluated with trace-labeled doses (2.5 mg/kg; 185–370 MBq [5–10 mCi]) of this mAb to determine if they achieved the desired favorable biodistribution before treatment was administered. Twenty-two patients (88%) achieved favorable biodistribution and, of those, 21 were treated subsequently with radioactive amounts of 131I-tositumomab designed to deliver 2,700 cGy to the critical organs, again found to be the lungs in most patients. The treatment was then followed by HSCT. Eighteen of the 21 treated patients (86%) had an OR, with 16 (76%) achieving a CR. One patient died of progressive lymphoma and 1 patient died of sepsis. The investigators have further analyzed the efficacy results of their phase I and II trials with myeloablative doses of 131I-anti-B-cell lymphoma mAbs involving a total of 37 patients, of whom 29 received 131I-tositumomab. Their analysis revealed an ORR of 89%, CRR of 79%, progression-free survival (PFS) of 62%, and overall survival (OS) of 93% with a median follow-up of 2 y at the time of their report. Liu et al. (37) updated these results and reported on the long-term toxicity after a median follow-up period of >3 y. The more recently reported median DR was in excess of 38 mo. Of particular interest was the finding that myeloablative therapy resulted in a very high ORR and CRR even in patients with intermediate-grade aggressive NHL. Unlike with nonmyeloablative RIT, where the ORR and CRR are only about 30%-40% and 10%–30%, respectively, ORR and CRR similar to those seen with low-grade NHL were demonstrated in patients with intermediate-grade NHL, who made up about one third of the total number of patients entered into the phase I and II trials. However, the DR was substantially shorter in these patients compared with that of patients with low-grade NHL (a median of 24 mo compared with ≥38 mo for low-grade NHL). Late toxicities have been uncommon except for elevated TSH levels found in about 60% of patients; 2 patients developed second malignancies, but none have developed myelodysplasia.
In summary, despite the still-limited experience with myeloablative RIT in B-cell NHL and the nonrandomized nature of the studies conducted, the study by Press et al. (36,37) strongly suggests that this treatment approach results in a higher CRR and longer DR compared with that of nonmyeloablative RIT using the same radiolabeled mAb (i.e., 131I-tositumomab) in relapsed/refractory low-grade B-cell NHL patients. In this context, it is noteworthy that, unlike what was apparent in the phase I study with myeloablative RIT reported by the same investigators (18), there was no significant patient selection in the phase II study using 131I-tositumomab: 88% of patients initially entered into the study achieved favorable biodistribution and all, except one, were treated subsequently. However, it is important to point out that not only the efficacy but also the toxicity with the potential for fatal complications need to be taken into consideration when assessing the relative merits of the nonmyeloablative and myeloablative treatment approaches in these patients, particularly considering the often indolent course of disease. The situation may be different, however, in patients with aggressive intermediate-grade NHL. In this case, the substantially higher ORR and CRR and, presumably also the DR, may need to be strongly considered when choosing the optimal treatment approach in these patients. However, it is important to note that no prospective randomized trials have been conducted to date comparing the nonmyeloablative with the myeloablative RIT approaches in patients with low-grade or intermediate-grade NHL. It is also noteworthy that a study with myeloablative doses of the single agent 90Y-ibritumomab tiuxetan has not been reported to date. However, Juweid et al. (39) have initiated a phase I/II study with myeloablative doses of 90Y-hLL2 mAb (epratuzumab) in patients with predominantly aggressive NHL (see below).
Other Approaches and Developments Using RIT as a Single Modality
In addition to the conduct of the phase II and III trials described above, important advances have been made with respect to the use of RIT as a single modality in patients with relapsed/refractory NHL. Perhaps most notable among these advances are the initiation of phase I/II studies using nonmyeloablative and myeloablative RIT in patients who failed prior high-dose chemotherapy [HDC] with HSCT (38,39) and the development of new approaches for RIT delivery using the streptavidin-biotin method (40).
B-cell NHL patients with prior HSCT have been entered into some of the pilot or phase I/II RIT trials along with patients without prior HSCT (17,20,24). It was observed that such patients exhibited greater hematologic toxicity compared with those without prior HSCT and that it might be more prudent to determine the MTD separately in this group (24). Using 131I-tositumomab, Kaminski et al. (38) have conducted separate dose escalations of total-body dose in B-cell NHL patients who had or had not undergone prior HDC with HSCT until a nonmyeloablative maximally tolerated total-body dose was established. They found that the MTD in the group of patients with prior HSCT was only 45 cGy to the total body compared with 75 cGy in patients without this prior experience. Overall, a total of 14 patients with prior HSCT were treated in the phase I/II trial, of whom 7 (50%) responded, with 5 (36%) achieving a CR. The median PFS of responders was not reached after 4.7 y and 4 patients remained disease free >3 y after therapy at the time of the report (38). Juweid et al. (39) have used a similar approach to determine the nonmyeloablative MTD of 90Y-hLL2 (epratuzumab) in patients with prior HSCT. In this trial, escalating doses of 90Y-hLL2 starting at a dose of 185 MBq/m2 (5 mCi/m2), with increments of 185 MBq/m2 (5 mCi/m2), were given to patients with prior HSCT compared with a starting dose of 370 MBq/m2 (10 mCi/m2) with a similar dose increment of 185 MBq/m2 (5 mCi/m2) to patients who did not have prior HSCT. To date, 10 patients with prior HSCT (8 aggressive and 2 indolent NHL) were treated at dose levels of 185 MBq/m2 (5 mCi/m2) (n = 7) and 370 MBq/m2 (10 mCi/m2) (n = 3), whereas the other 13 patients without prior HSCT (6 aggressive and 7 indolent) were treated at dose levels of 370 MBq/m2 (10 mCi/m2) (n = 7), 555 MBq/m2 (15 mCi/m2) (n = 3), and 740 MBq/m2 (20 mCi/m2) (n = 3). Interestingly, 1 of the 6 assessable patients with prior HSCT treated at the first dose level and another treated at the second dose level developed hematologic DLT. In contrast, none of the 13 patients without prior HSCT developed DLT despite the higher administered dose of up to 740 MBq/m2 (20 mCi/m2). Overall, 3 of the 10 patients with prior HSCT and 6 of the 13 patients without HSCT had an OR. Thus, the initial results available from the nonmyeloablative trial with 90Y-hLL2 show a trend toward higher hematologic toxicity in patients with prior HSCT compared with those without prior HSCT, and it appears likely that the nonmyeloablative MTD of this mAb will also be lower in such patients.
Juweid et al. (39) have initiated the first myeloablative RIT trial in patients with prior HSCT. Here again, separate dose escalations were undertaken in patients who had or had not undergone prior HSCT. Escalating doses of 90Y-hLL2 starting at a dose of 555 MBq/m2 (15 mCi/m2), with increments of 370 MBq/m2 (10 mCi/m2), were given to patients who had compared with a starting dose of 740 MBq/m2 (20 mCi/m2), with a similar dose increment of 370 MBq/m2 (10 mCi/m2), in patients who had not undergone prior HSCT. Only 9 patients (all with aggressive NHL) were treated to date. Seven patients with prior HSCT were treated, 4 at a dose of 555 MBq/m2 (15 mCi/m2) and 3 at 925 MBq/m2 (25 mCi/m2). No significant (i.e., >grade 1) nonhematologic toxicity was observed in these patients and all but 1 patient had grade 2 or grade 3 hematologic toxicity. One patient, treated at 925 MBq/m2 (25 mCi/m2), had a grade 4 hematologic toxicity lasting 5 wk, which was considered DLT in this trial. However, this patient had 2 prior HSCTs (tandem transplant) and, because of this experience, such patients were excluded subsequently from the trial. The 2 patients treated who had no prior HSCT also had grade 2 or grade 3 hematologic toxicity with grade 1 nonhematologic toxicity observed in one. Overall, 7 of the 9 patients had an OR, 5 of the 7 with and 2 of the 2 without prior HSCT. This initial experience suggests acceptable toxicity and encouraging antitumor responses with this treatment in patients with prior HSCT, who usually have a very poor prognosis.
Weiden et al. (40) have evaluated the feasibility of pretargeted RIT (PRIT) in patients with B-cell NHL. The PRIT approach used in their study consisted of a multistep delivery system in which rituximab was used to target streptavidin to the CD20 antigen on B-cell NHL (through a rituximab-streptavidin conjugate), with biotin used subsequently to target 90Y to the tumor-localized streptavidin (via 90Y-DOTA-biotin). The rituximab-streptavidin conjugate was first administered to the patients, followed 34 h later by a clearing agent, synthetic biotin-N-acteyl-galactosamine, to remove nonlocalized conjugate from the circulation. A DOTA-biotin ligand, labeled with 111In for imaging or 90Y for therapy, was then administered. Ten patients with relapsed/refractory B-cell NHL were studied. Seven patients received 1.11 GBq/m2 (30 mCi/m2) or 1.85 GBq/m2 (50 mCi/m2) 90Y-DOTA-biotin. Radiolabeled biotin localized well to the tumor and unbound biotin was removed rapidly from the whole body and normal organs. The mean tumor dose calculated was 0.78 ± 0.62 cGy/MBq (29 ± 23 cGy/mCi) compared with a whole-body dose of only 0.02 ± 0.008 cGy/MBq (0.76 ± 0.3 cGy/mCi). Only grade 1 or grade 2 nonhematologic toxicity was observed, with 5 patients experiencing transient grade 3 hematologic toxicity. None had grade 4 toxicity. Although 6 of 10 patients developed humoral immune responses to the streptavidin, these were delayed and transient. Six of the 7 patients treated had an antitumor response (3 CR, 1 PR, and 2 minor response). Thus, the initial findings of this study suggest that PRIT in B-cell NHL is feasible and results in encouraging antitumor responses with only moderate toxicity. Most notable was the very high average tumor:total-body radiation dose ratio achieved (38:1), which is about 1.5-fold higher than the 25:1 ratio determined for the conventionally administered 90Y-anti-CD20 mAbs (e.g., 90Y-ibritumomab tiuxetan) and is also higher than the respective ratios reported for other radiolabeled anti-B-cell NHL mAbs (3–43,108). The only moderate toxicity observed suggests that further dose escalation may be possible, thereby further enhancing the efficacy of this treatment approach.
COMBINED RIT WITH CHEMOTHERAPY
An increasingly important development in the field of RIT of NHL is the attempt to combine this modality with chemotherapy with both treatments given either sequentially or concurrently. Two approaches of combined RIT and chemotherapy have been used: (a) a nonmyeloablative approach, in which low-dose RIT is combined with standard or low-dose chemotherapy; and (b) a myeloablative approach, in which high-dose RIT is given with either standard-dose or HDC—hence, requiring HSCT to control myelotoxicity. A brief review of the results with both approaches follows.
Nonmyeloablative Approaches
Leonard et al. (41) have recently reported the findings of the first study on the feasibility of sequential standard-dose chemotherapy and low-dose RIT using fludarabine and 131I-tositumomab, respectively, in patients with untreated low-grade NHL. Because fludarabine is an active agent alone or in combination for treatment of NHL and in vitro data have shown that 131I-tositumomab and fludarabine have a markedly supraadditive effect on tumor cell killing, this study was conducted with the aim of evaluating the safety and efficacy of a sequential regimen of 3 cycles of fludarabine (25 mg/m2 × 5 d every 5 wk) followed 6–8 wk later by 131I-tositumomab for patients with previously untreated low-grade, transformed, or follicular NHL. Thirty-eight patients were enrolled, 14 of whom were evaluable for response at least 3 mo after treatment with 131I-tositumomab. The radioactive dose of 131I given was that required to deliver 75 cGy to the whole body for patients with ≥150,000 platelets per mm3 or 65 cGy for patients with 100,000–149,000 platelets per mm3. The toxicity after fludarabine was mainly hematologic. The principal toxicity after 131I-tositumomab was also hematologic: An ANC of <500 was seen in 5 patients (36%), and <10,000 platelets were seen in 1 patient (7%). Nonhematologic toxicity was typically mild to moderate and the most frequent events were nausea, asthenia, headache, and rhinitis. No patient developed HAMA. An investigator-assessed response was seen in 13 of 14 patients (93%) (2 CR, 11 PR) after fludarabine. After treatment with 131I-tositumomab, 13 of 14 patients (93%) had a response (6 CR, 7 PR), with conversion of 4 of the 11 PRs after fludarabine into CR. Thus, this study has demonstrated the feasibility of sequential standard-dose chemotherapy and RIT in patients with untreated low-grade NHL with acceptable toxicity and increased efficacy compared with fludarabine alone. Moreover, it appears that the prior administration of fludarabine suppresses the HAMA response to the murine mAb. However, it will be interesting to investigate if RIT can be given concurrently with multiple cycles of standard-dose chemotherapy without additive toxicity and if this approach can prove even more effective than the design used by Leonard et al. The concurrent administration of both modalities may in fact be ideal to exploit the potential radiosensitizing effect of chemotherapy and, hence, the potential of synergistic rather than simply additive antitumor effects.
Myeloablative Approaches
The first trial of combined myeloablative RIT with chemotherapy (in this case, HDC) in relapsed/refractory NHL was reported by Press et al. (42). In this study, HDC using etoposide and cyclophosphamide, was given approximately 10–14 d after RIT with escalating myeloablative doses of 131I-B1 (131I-tositumomab), when the whole-body radiation exposure was ≤5.2 × 10−7 C/kg/h (≤2 mR/h), thereby allowing the administration of chemotherapy without substantial radiation hazard to treating personnel. As mentioned above, these investigators have shown in a phase I/II study that high doses of single-agent 131I-B1 combined with HSCT resulted in very high ORR and CRR (36,37). However, although 39% of the patients have remained alive and free of any recurrences for 5–10 y without any further therapy, more than one half of the patients eventually relapsed with this single-modality therapy (37). With combined therapy consisting of up to 2,500 cGy of 131I-anti-B1 (2.5 mg/kg), etoposide (60 mg/kg), and cyclophosphamide (100 mg/kg), these investigators showed an OS rate of 85% and a PFS rate of 73% after a median follow-up of 2 y. These figures were statistically superior in a multivariable analysis to the OS and PFS (50% [P = 0.0l] and 38% [P = 0.006], respectively) of a nonrandomized control group of patients treated at the investigators’ institution with the same doses of etoposide and cyclophosphamide but who received total-body irradiation (TBI) rather than 131I-B1. Of the 52 patients treated, 4 died of opportunistic infections.
Winter et al. (43) used a similar approach of combined HDC with RIT, this time, however, using 90Y-ibritumomab tiuxetan. Interestingly, a dosimetry-based approach was also used in this trial; the investigators’ objective was to determine the MTD of absorbed irradiation to critical organs delivered by 90Y-ibritumomab tiuxetan in combination with standard high-dose BEAM (carmustine-etoposide-cytarabine-melphalan) chemotherapy and autologous HSCT. To date, 12 patients with relapsed or refractory CD20+ NHL were treated and doses up to 500 cGy to the liver were delivered in this phase I study. The toxicity and efficacy results remain preliminary at this point.
In summary, the study by Press et al. (42) has demonstrated the feasibility of sequential high-dose RIT and HDC, and it showed that it was possible to deliver the full dose of HDC with about 90% of the MTD of single-agent high-dose RIT combined with HSCT (i.e., critical organ doses, in this case to the lung, of 2,500 cGy could be administered with HDC compared with 2,700 Gy with single-agent high-dose RIT). Although the approach used is quite aggressive, the initial therapeutic results are very promising, and it is remarkable that the PFS and OS rates with this therapy were statistically superior to the combination of HDC and TBI with presumably similar or lower toxicity. In addition, the investigators’ data also suggest that this may be more effective than high-dose RIT alone. The mortality rate of about 8%, which was due to opportunistic infections and not nonhematologic toxicities, is comparable to what has been reported for the combination of HDC with TBI. Assuming a median tumor-to-lung radiation absorbed dose ratio of about 1.5, reported in a previous study with this mAb (37), it is expected that a median tumor dose of about 38 Gy was delivered to treated tumors compared with only 12 Gy expected to be delivered using TBI, which probably explains the better antitumor effects seen.
ANTI-ANTIBODY RESPONSE
Patients receiving murine mAbs can develop HAMA, which frequently results in altering the mAb pharmacokinetics and markedly reduced tumor targeting. However, in contrast to what is seen in RIT of solid tumors, HAMA has not been a major problem in previously treated NHL patients who have received even multiple and relatively high doses of murine anti-B-cell lymphoma mAbs (5–22,30–34,36–38). This is presumably due to the significant immunosuppression caused by the previous, often multiple, chemotherapy regimens and could also be related to immunosuppression caused by the disease itself. For example, the incidence of HAMA in the large multicenter expanded- access trial with the murine 131I-tositumomab mAb involving a total of 359 patients who received a cumulative mAb dose of about 970 mg over a 1-wk period was only 8% (33). An even lower incidence of HAMA of only 1.4% was seen in the multiple trials using 111In/90Y-ibritumomab tiuxetan (349 patients), in which a much smaller cumulative dose of murine mAb (<10 mg) is used to radiolabel ibritumomab tiuxetan with 111In and 90Y (29). The chimeric rituximab used as preinfusion before ibritumomab tiuxetan has a much smaller amount of mouse protein and is therefore presumably much less immunogenic (the incidence of HACA was only 0.5%) (29). Thus, anti-antibody responses (HAMA or HACA) do not appear to be a major problem in B-cell NHL patients who were treated previously. However, as the study of Wahl et al. (35) showed, the situation may be quite different in previously untreated (i.e., chemotherapy-naive) patients. Here, the incidence of HAMA was 64% with similar mAb protein doses, which could potentially jeopardize repeated treatments in such patients. It is quite possible that the use of mAb regimens consisting either entirely of chimeric or hyperchimeric mAbs (e.g., epratuzumab) or containing only a small amount of murine mAb (e.g., ibritumomab tiuxetan) may be preferable in this situation (3,4,19,23,24–29). As shown by Leonard et al. (41), combining RIT with immunosuppressive cytotoxic chemotherapy could also obviate this problem.
ORGAN AND TUMOR DOSIMETRY
Even if pretreatment radiation dosimetry is not required before the administration of the now commercially available ibritumomab tiuxetan mAb, the nuclear medicine physician should be aware of at least the range of organ and tumor radiation doses delivered with nonmyeloablative and myeloablative RIT. In general, the radiation doses to the various normal organs from nonmyeloablative RIT, even when given at the MTD, do not exceed 2,000 cGy and are usually in the range of 50–1,000 cGy (2,3,17,22,23,27,108).
Wiseman et al. (108) reported recently on the organ and tumor radiation dose estimates from the MTD of 14.8 MBq/kg (0.4 mCi/kg) 90Y-ibritumomab tiuxetan calculated for 72 patients with low-grade or transformed B-cell NHL treated in their phase III randomized trial. They found that the median radiation doses to the total body, red marrow, lungs, liver, and kidneys were about 60 cGy (range, 23–79 cGy), 71 cGy (range, 18–221 cGy), 216 cGy (range, 94–457 cGy), 532 cGy (range, 234–1,856 cGy), and 15 cGy (range, 0.3–76 cGy), respectively. The median radiation doses to bone surfaces, spleen, and urinary bladder were 60 cGy (range, 20–157 cGy), 848 cGy (range, 76–1,902 cGy), and 95 cGy (range, 44–270 cGy), respectively, whereas the median dose to the other organs and tissues, such as the brain, gastrointestinal tract, and so forth was only about 44 cGy (range, 13–59 cGy). In contrast, the median radiation dose for 39 tumors in 29 patients, for which reasonable dose estimates could be obtained, was 1,484 cGy (range, 61–24,274 cGy), about 24.7-, 20.9-, 6.9-, 2.8-, and 98.9-fold higher than the median radiation doses to the total body, red marrow, lungs, liver, and kidneys, respectively. Other investigators, using various 131I-and 90Y-labeled mAbs given in nonmyeloablative doses have found similar ranges of dose estimates to normal organs, although the median radiation doses to the liver and spleen at the MTD of an 90Y-labeled mAb are almost always higher than those at the MTD of the same or similar mAb labeled with 131I, whereas those to the kidneys and urinary bladder are higher for the 131I-labeled mAb (5–10,12,14,15–17,19–22,24). The latter is presumably related to the renal clearance of free 131I, in contrast to negligible renal clearance of 90Y.
Although the knowledge of the range of organ and tumor radiation doses with 90Y-ibritumomab tiuxetan is important, more precise patient-specific estimates of the radiation doses to certain organs may sometimes be necessary when consulting radiation oncologists who often require at least an approximate estimate of the radiation dose delivered to certain normal organs from prior RIT before contemplating XRT in a particular patient. The intent is then not to exceed the maximal threshold doses that can be delivered safely to these organs over time from both XRT and RIT. In this case, presumably reasonable patient-specific radiation dose estimates may be obtained retrospectively because, per current clinical protocol, whole-body imaging will be performed on at least 2 time points, with an optional third time point, after 111In-ibritumomab tiuxetan. Obviously, more frequent imaging (i.e., on at least 3 or 4 time points after injection) would provide more accurate dose estimates, but this may not always be clinically feasible or desired. Imaging on as few as 4 time points on only 3 separate days (e.g., at 0, 2–4, 48–72, and 90–120 h) would seem to be a reasonable and practical imaging approach that takes into account the recommendations in the ibritumomab tiuxetan (Zevalin) package insert (109) and yet provides quite accurate dosimetric data in clinical 111In/90Y-ibritumomab tiuxetan studies. This is because these time points capture both the uptake and the clearance phases of the 111In/90Y-ibritumomab tiuxetan in normal organs (110).
As expected, much higher radiation doses are delivered to both normal organs and tumors with myeloablative RIT compared with nonmyeloablative RIT. In fact, per definition, the myeloablative MTD with 131I-tositumomab is a “critical organ” (lungs, liver, and kidneys) radiation dose of 2,700 cGy. Although, in the studies by Press et al. (36) and Liu et al. (37) the lungs represented the organ with the highest radiation absorbed dose in the most patients, thus receiving 2,700 cGy, the kidneys and liver usually received doses of >1,500 cGy. These doses either exceed or are only slightly below threshold doses allowed with XRT. A more precise knowledge of the delivered radiation doses is therefore critical in this case.
Similar to the findings of Wiseman et al. (108) using 90Y-ibritumomab tiuxetan, most investigators have found that the tumor radiation doses are, on average, higher than those to the total body or normal organs, with mean tumor-to-total body, red marrow, and critical normal organ (i.e., lungs, liver, and kidneys) dose ratios usually in the range of 10–25, 10–20, and 1.5–10, respectively (5–10,12,14,15–18,19–22,24,36,37). Several other observations are noteworthy. First, the radiation dose (on a cGy/MBq basis) to relatively small tumors (i.e., <2–3 cm in diameter) is considerably higher (up to 10-fold) than the radiation dose to larger tumors, primarily because of their substantially better vascularization compared with the larger ones (24). Second, the ORRs and CRRs of 60%–80% and 20%–30%, respectively, observed in the phase II and III trials with the 131I- and 90Y-labeled anti-CD20 mAbs in patients with low-grade and transformed lymphoma were achieved with median tumor doses of only about 1,000–1,500 cGy. In contrast, median tumor doses of >1,500 cGy were delivered to patients with solid malignancies with very few ORs seen (111,112), emphasizing the exquisite radiosensitivity of B-cell NHL. Finally, although there is generally a rather weak correlation between the tumor dose and response and, hence, it is still not possible to predict if a patient will respond to RIT on basis of the estimated tumor radiation dose, preliminary evidence suggests a considerably higher probability of response with the higher tumor doses compared with the lower tumor doses (36,113). This is the most likely explanation for the higher ORR and CRR observed with myeloablative RIT compared with nonmyeloablative RIT using the same radiolabeled mAb in similar patient populations (30–32,34,36,37).
RIT OF B-CELL NHL IN CLINICAL PRACTICE
The approval of 90Y-ibritumomab tiuxetan establishes RIT in the management of patients with B-cell NHL. Because this mAb has been approved for RIT of patients with relapsed/refractory low-grade or transformed NHL, this should be the most important group of patients that receive RIT clinically. However, considering the fact that about 40% of patients with intermediate-grade NHL are not cured with conventional therapy and the encouraging initial experience with 90Y-ibritumomab tiuxetan in similar patients entered into the phase I/II trials with this mAb (3,4), 90Y-ibritumomab tiuxetan will also be used for RIT of intermediate-grade NHL and, presumably, other forms of aggressive NHL. Definitive phase II and III trials in well-defined patient populations with aggressive NHL are needed to determine the true potential of 90Y-ibritumomab tiuxetan RIT in this setting. A phase II study evaluating the efficacy of ibritumomab tiuxetan in patients with diffuse large B-cell lymphoma will soon be initiated at the University of Iowa and New York Presbyterian Hospital. 90Y-Ibritumomab tiuxetan will also be used clinically in patients who failed prior HDC with HSCT. However, because there is no documented experience with this mAb in such patients, but prior results with other radiolabeled mAbs indicate that these are at increased risk of myelotoxicity, the MTD of 90Y-ibritumomab tiuxetan in this setting may need to be determined or “dose-reduction” strategies established before it is clinically used in such patients. A phase I dose-escalation trial with the aim of determining the MTD of 90Y-ibritumomab tiuxetan in patients with prior HSCT is currently underway at the University of Nebraska and the University of Iowa. On the other hand, 90Y-ibritumomab tiuxetan may also be considered for treatment of patients not treated previously with chemotherapy and radiation. In this case, it is likely that a lower toxicity will be found and that attempts will be made to investigate whether a higher dose or more frequent doses of this mAb can be tolerated in untreated patients with advanced low-grade B-cell NHL. Various dose schedules of 90Y-ibritumomab tiuxetan and Rituxan administered sequentially or concurrently are also likely to be used. However, it is expected that these schedules will be first investigated in phase I/II clinical trials before certain regimens or dose schedules are recommended in the clinical setting. More critical are any attempts to administer 90Y-ibritumomab tiuxetan with chemotherapy, even when using the nonmyeloablative approach. In this case, it appears prudent to conduct well-controlled clinical trials with the aim of establishing the appropriate doses and schedules (order of administration, interval between doses, and so forth) of both modalities given in combination. The necessity for conducting these trials should by no means be considered a limitation, because these trials will eventually allow the safe and smooth integration of this modality into the other currently more-established treatments and will provide a more solid basis for any proposed combination that is based on sound clinical research. Conduction of these trials would now be greatly facilitated because of fewer regulatory constraints and presumably more rapid patient accrual, possibly through conduction of multicenter trials. In summary, the recent approval of 90Y-ibritumomab tiuxetan will expand the use of RIT into both the routine clinical setting and the arena of clinical research. The scope of these studies is likely to expand even further as other radiolabeled anti-B-cell NHL mAbs become commercially available.
CONCLUSION
RIT has emerged as an important treatment modality for B-cell-NHL, particularly low-grade or transformed forms of this disease. Well-controlled phase II and III trials involving hundreds of patients have established its efficacy in these patients, ranging from the previously untreated to those refractory to chemo- and immunotherapy. Its efficacy in other tumor types and settings, such as in aggressive NHL and in posttransplant relapses will likely continue to be tested in upcoming clinical trials. Attempts at combining RIT with other treatment modalities using various doses and dose schedules will increase. It is likely that comparisons with other treatment modalities will be made, a process that will expand as additional radiolabeled mAbs or other novel treatments become available for NHL patients. Although prior phase II and III trials have led to the introduction of RIT for B-cell NHL into clinical practice, new clinical trials will be needed to establish the role for this treatment modality in the standard management of B-cell NHL patients.
Acknowledgments
The author is extremely grateful to his first mentor in nuclear medicine, Peter Georgi, MD, Professor Emeritus, at the University of Heidelberg School of Medicine, Heidelberg, Germany. The author thanks Drs. Michael M. Graham, Michael W. Vannier, David Bushnell, Yusuf Menda, and Brian Link from the Departments of Radiology and Internal Medicine at the University of Iowa for thoughtful comments on the manuscript. In addition, the author is grateful to Dr. Gregory A. Wiseman from the Mayo Clinic Foundation for helpful input during preparation of the manuscript. This research was supported in part by the lymphoma Specialized Programs of Research Excellence (SPORE) grant from the National Cancer Institute (CA972784) at the University of Iowa and the Mayo Clinic.
Footnotes
Received Apr. 25, 2002; revision accepted Jul. 9, 2002.
For correspondence or reprints contact: Malik E. Juweid, MD, Department of Radiology, University of Iowa College of Medicine, 200 Hawkins Dr., Iowa City, IA 52242.
E-mail: malik-juweid{at}uiowa.edu
REFERENCES
In this issue




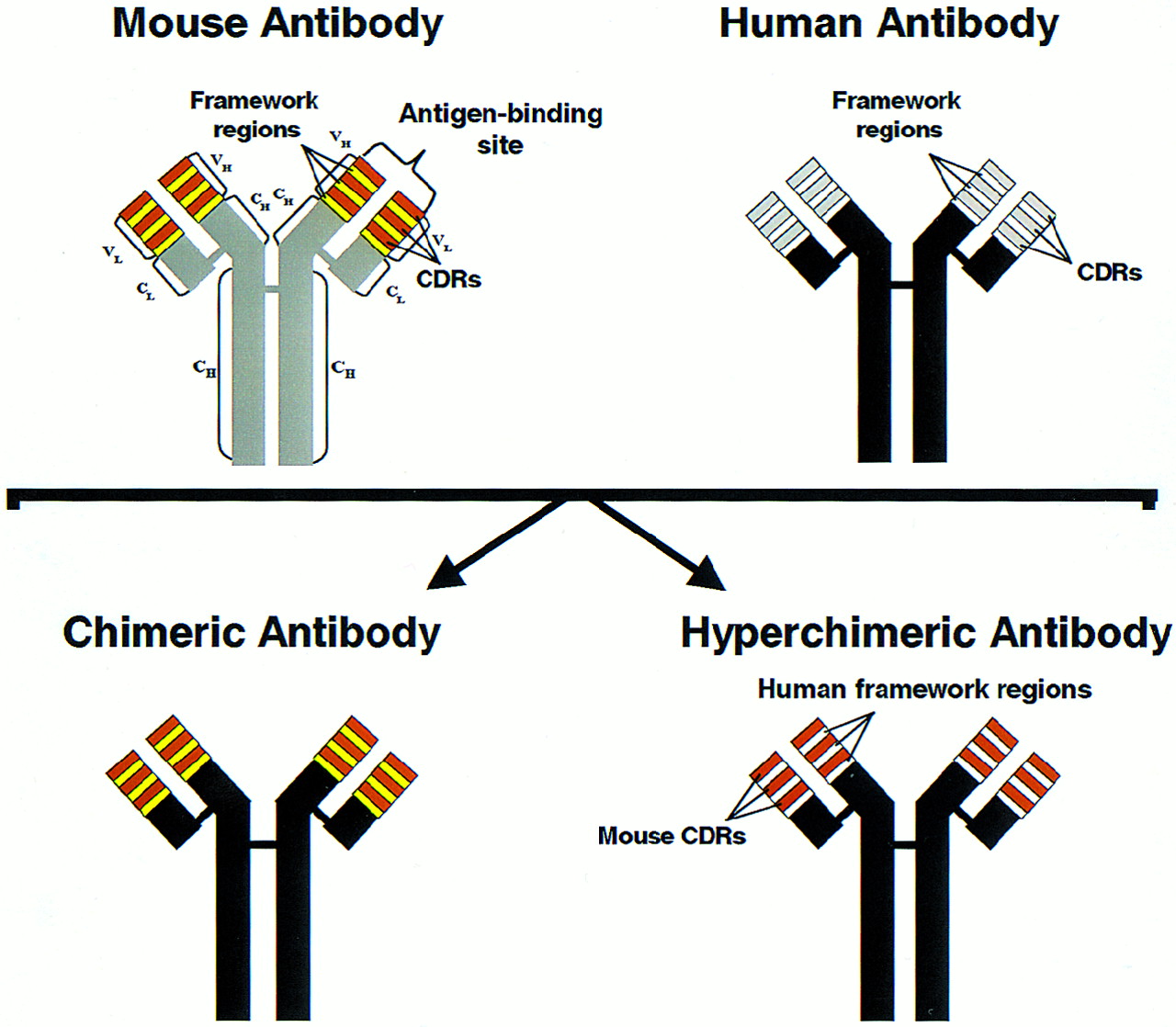{kind=link}
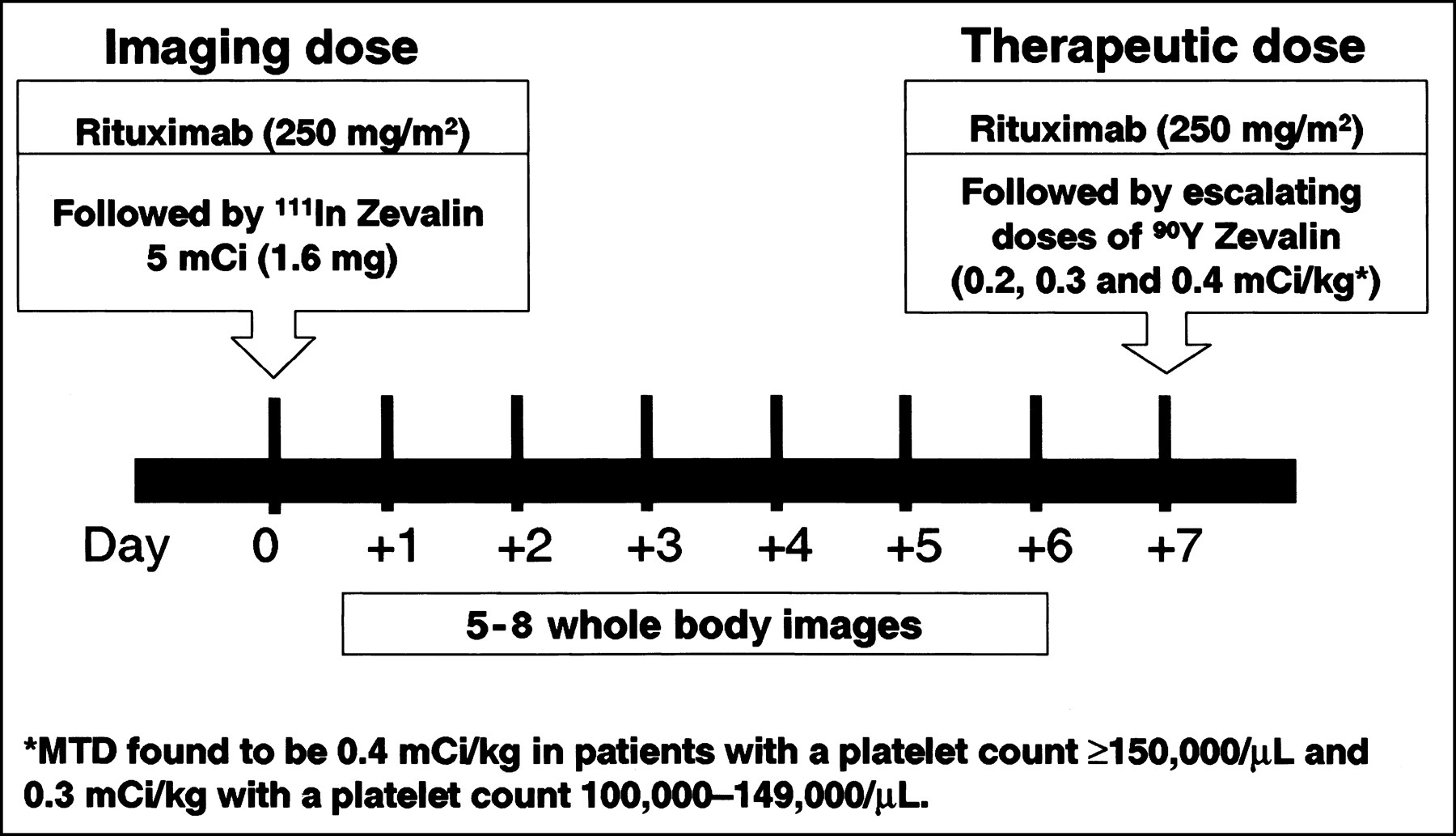{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Ofatumumab monotherapy in rituximab-refractory follicular lymphoma: results from a multicenter study
- Comparative Toxicity and Efficacy of Combined Radioimmunotherapy and Antiangiogenic Therapy in Carcinoembryonic Antigen-Expressing Medullary Thyroid Cancer Xenograft
- Selective High-Affinity Ligand Antibody Mimics for Cancer Diagnosis and Therapy: Initial Application to Lymphoma/Leukemia
- Comparison of Medium- and High-Energy Collimators for 131I-Tositumomab Dosimetry
- Therapeutic Potential of 90Y- and 131I-Labeled Anti-CD20 Monoclonal Antibody in Treating Non-Hodgkin's Lymphoma with Pulmonary Involvement: A Monte Carlo-Based Dosimetric Analysis
- Collimator Selection, Acquisition Speed, and Visual Assessment of 131I-Tositumomab Biodistribution in a Phantom Model
- Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody Drug Conjugate
- Chemotherapy With or Without Radiotherapy in Limited-Stage Diffuse Aggressive Non-Hodgkin's Lymphoma: Eastern Cooperative Oncology Group Study 1484
- 186Rhenium-labeled anti-CD20 antibody radioimmunotherapy followed by autologous peripheral blood stem cell transplantation in patients with relapsed or refractory non-Hodgkin lymphoma
- Impact of Nodal Regression on Radiation Dose for Lymphoma Patients After Radioimmunotherapy