


Abstract
18F-FDG measures glucose consumption and is an integral part of cancer management. Most cancer types upregulate their glucose consumption, yielding elevated 18F-FDG PET accumulation in those cancer cells. The biochemical pathway through which 18F-FDG accumulates in cancer cells is well established. However, beyond well-known regulators such as c-Myc, PI3K/PKB, and HIF1α, the proteins and signaling pathways that cancer cells modulate to activate the facilitated glucose transporters and hexokinase enzymes that drive elevated 18F-FDG accumulation are less well understood. Understanding these signaling pathways could yield additional biologic insights from 18F-FDG PET scans and could suggest new uses of 18F-FDG PET in the management of cancer. Work over the past 5 years, building on studies from years prior, has identified new proteins and signaling pathways that drive glucose consumption in cancer. Here, we review these recent studies and discuss current limitations to our understanding of glucose consumption in cancer.
The glucose analog PET radiotracer 18F-FDG measures glucose consumption and is an important tool for the diagnosis and management of cancer (1,2). 18F-FDG PET imaging has demonstrated value in, for example, the initial staging and the identification of recurrences in lung, colorectal, and esophageal cancers (1). Additionally, preclinical and clinical studies suggest that changes in 18F-FDG accumulation in tumors after certain therapies can be strongly predictive and can function as an early biomarker of therapeutic efficacy (3,4).
However, despite the proven clinical value of 18F-FDG PET in cancer management and a clear understanding of the biochemical pathway that 18F-FDG measures—glucose consumption—a comprehensive understanding of the signaling pathways that regulate glucose consumption and 18F-FDG accumulation in cancer is lacking. Understanding these signaling pathways could provide a better context in which to understand 18F-FDG PET scans in patients, might help to connect mechanistically the level of 18F-FDG accumulation to specific and potentially targetable alterations in a given cancer type, and might aid in identifying additional therapies for which 18F-FDG PET could be leveraged as an early predictor of therapeutic efficacy.
This review describes and categorizes new signaling pathways that regulate glucose consumption in cancer and that have been reported within the last 5 years. Most of these newly described pathways connect to well-established and previously identified regulators of cancer cell glucose consumption such as PI3K, HIF1α, and TXNIP. Work describing these and other previously identified regulators of glucose consumption can be found in prior reviews (5–8). Mechanisms through which therapies block 18F-FDG accumulation and glucose consumption in cancer are important although beyond the scope of this review. Additionally, mechanisms that drive 18F-FDG accumulation in immune cells in the tumor microenvironment and how that information could be used to monitor therapeutic responses to immune checkpoint inhibitors are an important additional topic but also outside the scope of this review and at least partially covered by a recent review (9).
THE BIOCHEMISTRY OF 18F-FDG ACCUMULATION
18F-FDG is transported across the cell membrane by facilitated glucose transporters (GLUTs) and is phosphorylated by hexokinase enzymes to 18F-FDG-6-phosphate (2). The GLUTs are a large family of transporters containing 14 isoforms that transport glucose and related compounds (10). GLUT1 and GLUT3 both transport glucose and 18F-FDG, are highly expressed in a variety of cancers, and are considered the key transporters of 18F-FDG in most cancer cells (10,11). 18F-FDG is not a substrate for sodium-dependent glucose transporters (12), although the exact contribution of these transporters to glucose flux in cancer remains to be determined. Cells express one or more of 4 different hexokinases (13). Hexokinase 1 (HK1) and hexokinase 2 (HK2), alone or in combination, are expressed in most cancer cells and are considered the key enzymes for phosphorylating 18F-FDG to 18F-FDG-6-phosphate in cancer cells (13). 18F-FDG-6-phosphate can be dephosphorylated by glucose-6-phosphatase and transported out of the cell by the GLUTs (14).
Which of these biochemical steps is the major driver of 18F-FDG accumulation is likely context-dependent, and low levels of the GLUTs or hexokinase enzymes or high levels of glucose-6-phosphatase are often found in cancers that accumulate low levels of 18F-FDG. For example, in non–small cell lung cancer, GLUT1 and GLUT3 levels correlate better than HK1 and HK2 levels with 18F-FDG accumulation (15). In pheochromocytomas and paragangliomas, HK2 levels correlate better than GLUT1 or GLUT3 levels with 18F-FDG accumulation (16). 18F-FDG accumulation can be variable in gastric and breast cancers, and in both cases, 18F-FDG accumulation correlates with GLUT1 levels (17,18). Low 18F-FDG accumulation in low-grade hepatocellular carcinoma tumors is associated with low hexokinase activity and high glucose-6-phosphatase activity (19).
Additional enzymes, including phosphoglucose isomerase and phosphoglucomutase, can further metabolize 18F-FDG-6-phosphate (20), but how (if at all) changes in the activity of these enzymes affects 18F-FDG accumulation at the time point usually used for imaging with 18F-FDG remains unclear.
THE SIGNALING PATHWAYS THAT AFFECT GLUCOSE CONSUMPTION AND 18F-FDG ACCUMULATION IN CANCER CELLS
Many well-validated methods exist for quantifying glucose consumption, including measuring 2-DG-6-phosphate levels in cells treated with 2-DG, measuring accumulated radioactivity in cells treated with a radiolabeled glucose analog, or measuring changes in glucose concentrations in cell culture media (21). An often-used but poor measure of glucose consumption is accumulated fluorescence in cells treated with a fluorescent glucose analog such as 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxyglucose or 6-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxyglucose. Various studies have identified multiple systems in which the accumulation of these fluorescent glucose analogs does not measure glucose consumption (22,23). As such, we will focus only on those studies that measure glucose consumption with well-validated methods.
Over the last 5 years, most studies have identified signaling pathways that regulate glucose consumption and 18F-FDG accumulation by regulating the GLUT proteins and hexokinase enzymes at the transcriptional and posttranslational level.
TRANSCRIPTION (FIG. 1A)
Newly identified regulators of GLUT1 and GLUT3 transcription are varied and defy easy categorization. In squamous cell carcinoma cell lines, the transcription factors p63 and SOX2 drive glucose consumption by binding to an enhancer region of the GLUT1 gene and driving GLUT1 messenger RNA (mRNA) and protein expression (24). Our laboratory recently identified over 100 small molecules that block glucose consumption in 1 of 3 different non–small cell lung cancer cell lines using a high-throughput assay. We studied the small molecule milciclib and used it to determine that the cyclin-dependent kinase CDK7 activates GLUT1 transcription, glucose consumption, and 18F-FDG accumulation downstream from mutant PIK3CA (25). In melanoma cells, lysine methyl transferase KMT2D loss-of-function mutants increase glucose consumption by repressing the expression of IGFBP5, a negative regulator of insulin signaling, and increase mRNA expression of GLUT1 and HK1 (26). In pre–B-cell acute lymphoblastic leukemia, B-lymphoid transcription factors Pax5 and IKZF1 function as tumor suppressors and downregulate mRNA levels of GLUT1 and HK2 (27). In breast and colorectal cancer cells, during cell detachment, the protein kinase SGK1 induces the expression of GLUT1 mRNA, leading to increased GLUT1 protein levels and glucose consumption (28). In pancreatic cancer, the transmembrane mucin glycoprotein MUC13 increases glucose consumption by activating nuclear factor κB, leading to increased mRNA expression of GLUT1 (29). Finally, in cervical cancer cells, long noncoding RNA NICI enhances glucose consumption during hypoxia by promoting the recruitment of RNA polymerase 2 to the GLUT3 promoter and increasing GLUT3 mRNA levels (30).
The transcription factor c-Myc continues to be an important regulator of glucose consumption, with attention in the last 5 years focusing on its role in regulating HK2 mRNA levels. In colon cancer cells, interleukin 22 enhances glucose consumption by activating STAT3, which induces c-Myc mRNA and protein levels, leading to increased mRNA and protein expression of HK2 (31). Alternatively, in pancreatic cancer cells, serotonin activates glucose consumption in cells by binding its receptor HTR2B, increasing protein levels of HIF1α and c-Myc, and increasing mRNA expression of HK2 (32). Inhibition of mTOR and c-Myc in lymphoma cell lines decreases glucose consumption, decreases mRNA levels of GLUT1, and increases mRNA levels of glucose-6-phosphatase (33).
Additional regulators of HK2 transcription include the transcription factor BACH1, the cytokine Gremlin-1, and the micro RNA miR-34c-3p. In lung cancer cells, BACH1, which is stabilized in the presence of antioxidants, binds to the HK2 promoter and induces HK2 mRNA levels, leading to increased glucose consumption (34). In breast cancer cells, Gremlin-1 induces glucose consumption by activating the reactive oxygen species–PKB (Akt)–STAT3 pathway, which leads to increased HK2 mRNA (35). Finally, in hepatocellular carcinoma cell lines, miR-34c-3p drives glucose consumption by downregulating guanylate kinase MAGI3. MAGI3 inhibits β-catenin transcriptional activity, and decreased MAGI3 levels leads to increased HK2 mRNA levels and glucose consumption (36).
PROTEIN EXPRESSION (FIG. 1B)
A few studies only looked at protein levels of GLUT1 and HK2. In osteosarcoma cells, the long noncoding RNA MALAT1 sponges up miR-485-3p, leading to higher levels of c-MET and Akt3, whose mRNA miR-485-3p targets. Higher c-MET and Akt3 protein levels increase mTOR signaling, GLUT1 and HK2 protein levels, and glucose consumption (37). In cervical cancer cells, programmed-death ligand 1 activates glucose consumption through the induction of SNAI1, which represses SIRT3 expression, leading to higher protein levels of HK2 and GLUT1 (38).
POSTTRANSLATIONAL REGULATION AND LOCALIZATION (FIG. 1C)
Recent work has focused on the posttranslational modifications of HK2 (39,40). SUMOylation and ubiquitination represent the conjugation of specific polypeptides to lysine residues on proteins. HK2 SUMOylation at lysine 315 and lysine 492 is added by the conjugating enzyme UBC2 and removed by the protease SENP1. In prostate cancer cells, HK2 SUMOylation weakens the interaction between HK2 and the anion channel VDAC on the outer mitochondrial membrane, leading to lower glucose consumption (39). In hepatocellular carcinoma, the COP9 signalosome subunit CSN5 is overexpressed and deubiquitinates HK2. HK2 ubiquitination is associated with its degradation, so CSN5 overexpression leads to increased HK2 protein levels and glucose consumption (40).
The α-arrestin family member TXNIP is a well-studied protein whose levels are induced by glucose, which binds to GLUT1, leading to GLUT1 internalization, and which decreases GLUT1 mRNA levels (41). In this capacity, TXNIP is a key effector of a negative-feedback loop that limits excess glucose consumption and a key node that is dysregulated in cancer cells to enhance glucose consumption (41). In recent work, the role of TXNIP in regulating glucose consumption continued to be affirmed, with studies identifying additional regulators of TXNIP levels (42–45).
Lower TXNIP levels lead to increased GLUT1 at the plasma membrane and increased glucose consumption (42). PI3K/Akt signaling in non–small cell lung cancer cell lines decreases TXNIP expression and increases glucose consumption. In a hepatocellular carcinoma cell line, growth factors induce rapid glucose uptake by activating PI3K/Akt signaling, leading to the phosphorylation of TXNIP and yielding a weaker interaction between TXNIP and GLUT1 (43,44). Across a variety of cancer cell lines, reducing hyaluronan levels activates receptor tyrosine kinase signaling, leading to induction of the zinc finger protein ZNF36, which degrades TXNIP mRNA (42). ZNF36 also binds to and contributes to the degradation of HK2 mRNA (46), suggesting that ZNF36 may regulate glucose consumption through multiple mechanisms. In metastatic pancreatic cancer cells, elevated activity of the metabolic enzyme phosphogluconate dehydrogenase increases glucose consumption by depleting levels of its substrate, 6-phosphogluconate, leading to retention of the transcription factor MondoA in the cytoplasm and diminished expression of the Mondo target gene TXNIP. Lower TXNIP levels lead to increased GLUT1 surface expression (45). In pancreatic cancer cells, the cell-surface glycoprotein MUC13 also directly interacts with and stabilizes GLUT1 at the cell surface, potentially synergizing with decreased levels of TXNIP (29).
The GTPase KRAS is highly mutated and activated in many different types of cancer and is well known to activate glucose consumption (47). However, mechanisms for how KRAS activates glucose consumption are not fully understood. KRAS is expressed as 2 splice variants, KRAS4A and KRAS4B. Recent work has shown that in multiple cancer cell lines, KRAS4A directly interacts with HK1 at the outer mitochondrial membrane, where it relieves glucose-6-phosphate–dependent product inhibition of HK1, leading to increased glucose consumption (48).
LIMITATIONS TO OUR CURRENT UNDERSTANDING OF GLUCOSE CONSUMPTION IN CANCER CELLS
Most proteins identified to regulate cancer cell glucose consumption have been shown to do so only in established cancer cell lines and in cell cultures despite the fact that metabolism can be very different between cell cultures and in vivo. Small-animal 18F-FDG PET is an ideal and well-established assay for conducting such studies but is rarely used. Additional studies to evaluate regulators of glucose consumption in vivo are necessary. Ideally, these studies would be done using patient-derived xenografts implanted in orthotopic locations or genetically engineered mouse models.
Most proteins that have been shown to alter glucose consumption have been studied in one or a limited number of cell lines from the same cancer type. The activity of these proteins is often not linked to any known genetic alteration. Studies evaluating which signaling pathways are cancer-type–specific or are shared across different cancer types will be important for identifying the scope of any pathway identified. Studies determining the genetic or epigenetic alteration that activates a certain pathway to drive glucose consumption will serve the same purpose.
The data so far suggest that glucose consumption is regulated at multiple levels by various mechanisms. Largely understudied is the relative importance of each regulator in affecting glucose consumption. Proteins or pathways that drive a large proportion of the glucose consumption would likely have greater relevance for human 18F-FDG PET.
An emerging but understudied theme is that blocking glucose consumption by affecting the identified pathways limits cell growth. For example, we have shown that the cyclin-dependent kinase inhibitor milciclib blocks cell growth by inhibiting glucose consumption (25). If this proves to be true across many different therapies and cancer types, it would provide a causal basis for correlative clinical studies showing that early changes in tumor 18F-FDG accumulation in response to therapy can be predictive of therapeutic efficacy.
To identify additional drugs for which 18F-FDG could function as a pharmacodynamic biomarker, it will be important to understand the kinetic time course over which glucose consumption is affected after a drug treatment. For example, because EGFR inhibitors block glucose consumption in glioblastoma cells within 4 h and through 24 h (4), a patient could be imaged with 18F-FDG PET shortly after or up through a day after administration of an EGFR inhibitor. Milciclib affects glucose consumption only after 24 h (25); a patient would therefore need to be imaged with 18F-FDG PET 24 h after milciclib treatment. Earlier imaging of a patient treated with milciclib could lead to the false impression that milciclib was not working in the tumor.
Tumors contain a complex mixture of cells, including cancer cells, immune cells, and stromal cells. The amount of glucose consumption in these various cell types and how that relates to the overall accumulation of 18F-FDG in the tumor across different cancer types and locations remains to be fully determined. However, a recent study suggested that at least in certain tumor models, myeloid cells and T cells consume glucose at a higher rate than tumor cells (49). Further studies will be necessary to understand how much 18F-FDG accumulation in tumors represents 18F-FDG accumulation in cancer cells versus other cell types.
CONCLUSION
18F-FDG PET is an established imaging approach for the management of many different types of cancer, but the full repertoire of signaling pathways that drive 18F-FDG consumption in cancer are not well understood. A better understanding of these signaling pathways could yield new insights into 18F-FDG PET imaging and will be important for developing additional uses of 18F-FDG PET in cancer.
Critical and important work has been accomplished over the last 5 years to further elucidate the detailed mechanisms that cancer cells use to elevate their glucose consumption and that lead to increased 18F-FDG accumulation in cancer cells. These studies continue to support the role of well-studied primary regulators of glucose consumption, including PI3K/Akt, mTOR, and HIF1ɑ, while detailing the different ways in which these primary regulators control glucose consumption at the transcriptional and posttranslational level. Studies in the next 5 years should focus on how and whether the regulators of glucose consumption often identified in limited cell lines and in cell cultures translate in vivo to mice and potentially humans and across various types and subtypes of cancer.
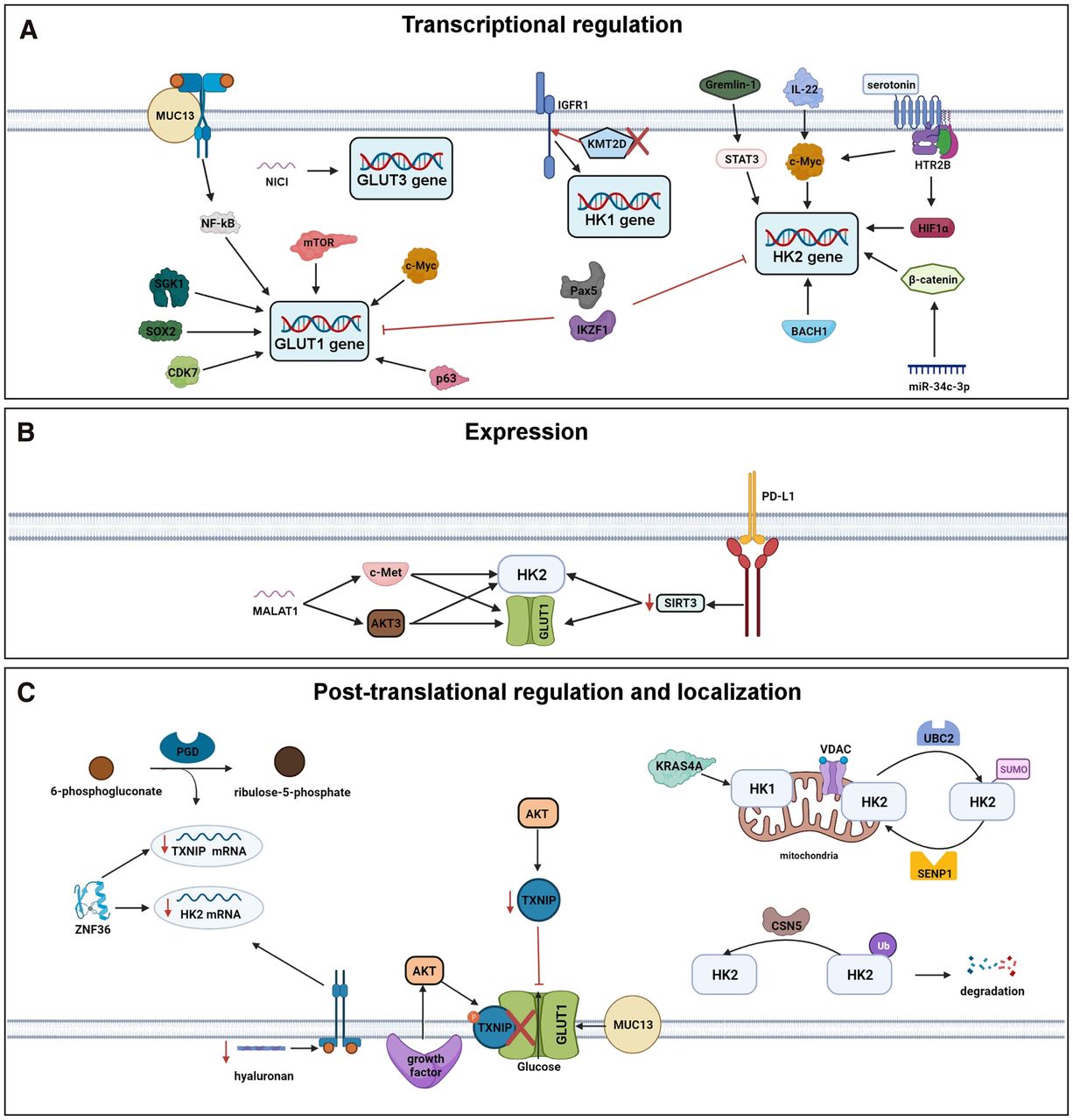
Recently described regulators of GLUT and hexokinase transcription (A), expression (B), and posttranslational regulation and localization (C).
DISCLOSURE
No potential conflict of interest relevant to this article was reported.
Footnotes
Guest Editor: Carolyn Anderson, University of Missouri
Published online Mar. 3, 2022.
- © 2022 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication December 21, 2021.
- Revision received February 22, 2022.
In this issue





{kind=link}
Jump to section
- Article
- Abstract
- THE BIOCHEMISTRY OF 18F-FDG ACCUMULATION
- THE SIGNALING PATHWAYS THAT AFFECT GLUCOSE CONSUMPTION AND 18F-FDG ACCUMULATION IN CANCER CELLS
- TRANSCRIPTION (FIG. 1A)
- PROTEIN EXPRESSION (FIG. 1B)
- POSTTRANSLATIONAL REGULATION AND LOCALIZATION (FIG. 1C)
- LIMITATIONS TO OUR CURRENT UNDERSTANDING OF GLUCOSE CONSUMPTION IN CANCER CELLS
- CONCLUSION
- DISCLOSURE
- Footnotes
- REFERENCES
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.