


Visual Abstract

Abstract
The peptide hormone ghrelin is produced in cardiomyocytes and acts through the myocardial growth hormone secretagogue receptor (GHSR) to promote cardiomyocyte survival. Administration of ghrelin may have therapeutic effects on post–myocardial infarction (MI) outcomes. Therefore, there is a need to develop molecular imaging probes that can track the dynamics of GHSR in health and disease to better predict the effectiveness of ghrelin-based therapeutics. We designed a high-affinity GHSR ligand labeled with 18F for imaging by PET and characterized its in vivo properties in a canine model of MI. Methods: We rationally designed and radiolabeled with 18F a quinazolinone derivative ([18F]LCE470) with subnanomolar binding affinity to GHSR. We determined the sensitivity and in vivo and ex vivo specificity of [18F]LCE470 in a canine model of surgically induced MI using PET/MRI, which allowed for anatomic localization of tracer uptake and simultaneous determination of global cardiac function. Uptake of [18F]LCE470 was determined by time–activity curve and SUV analysis in 3 regions of the left ventricle—area of infarct, territory served by the left circumflex coronary artery, and remote myocardium—over a period of 1.5 y. Changes in cardiac perfusion were tracked by [13N]NH3 PET. Results: The receptor binding affinity of LCE470 was measured at 0.33 nM, the highest known receptor binding affinity for a radiolabeled GHSR ligand. In vivo blocking studies in healthy hounds and ex vivo blocking studies in myocardial tissue showed the specificity of [18F]LCE470, and sensitivity was demonstrated by a positive correlation between tracer uptake and GHSR abundance. Post-MI changes in [18F]LCE470 uptake occurred independently of perfusion tracer distributions and changes in global cardiac function. We found that the regional distribution of [18F]LCE470 within the left ventricle diverged significantly within 1 d after MI and remained that way throughout the 1.5-y duration of the study. Conclusion: [18F]LCE470 is a high-affinity PET tracer that can detect changes in the regional distribution of myocardial GHSR after MI. In vivo PET molecular imaging of the global dynamics of GHSR may lead to improved GHSR-based therapeutics in the treatment of post-MI remodeling.
Ghrelin is a 28-amino-acid peptide that is a natural ligand of the growth hormone secretagogue receptor (GHSR), a 7-transmembrane, G protein–coupled receptor. Ghrelin is an orexigenic hormone, mediating the appetite and hunger response through hypothalamic GHSR signaling (reviewed by Yanagi et al. (1)). Ghrelin is also produced in cardiomyocytes and acts through myocardial GHSR to promote cardiomyocyte survival and contractility and to inhibit apoptosis, fibrosis, and inflammation (reviewed by Tokudome et al. (2)). These properties of ghrelin have prompted investigations into using ghrelin as a therapy to delay, inhibit, or reverse the post–myocardial infarction (MI) fibrosis and inflammation that may lead to heart failure. For example, in rat models of surgically induced MI, administration of ghrelin improved left ventricular function and inhibited fibrotic remodeling (3), decreased proinflammatory signaling (4), and inhibited angiotensin II–induced cardiomyocyte apoptosis (5). Therefore, modulation of GHSR signaling pathways may translate into beneficial effects on myocardial function and post-MI remodeling in vivo.
If GHSR agonists are to be used as effective therapeutics, there is a need to better understand the dynamics of the receptor to predict the outcomes of such therapies. The distribution of myocardial GHSR can be mapped by microscopy analysis of tissue using fluorescent analogs of GHSR ligands. We have previously generated a far-red fluorescent analog of ghrelin, Cy5-ghrelin(1–19), to image the dynamics of GHSR during induced cardiomyocyte differentiation (6) in a mouse model of diabetic cardiomyopathy (7), in human cardiac tissue before and after transplantation (8), and in heart disease (9). The cellular dynamics of GHSR have also been investigated using a fluorescently labeled N-terminal sequence of another GHSR ligand, liver-expressed antimicrobial peptide 2 (10). Although these studies have provided some information on the spatiotemporal distribution of GHSR, such measures rely on invasive biopsies performed at one or several time points. In vivo molecular imaging that can detect and quantitatively measure the regional dynamics of GHSR will assist in our understanding of the cardiac ghrelin–GHSR system and how it can be more effectively harnessed for therapy after MI to prevent further inflammation and fibrosis that may lead to heart failure.
Previous work from our group showed that the structure of the ghrelin peptide can be substantially modified to create molecular imaging probes for PET and SPECT without a significant impact on GHSR binding (11,12). Subsequent studies showed that modified ghrelin peptides, peptidomimetic agents, and small molecules carrying either fluorescent (13,14) or radionuclide-based contrast agents (15,16) could be designed to bind to GHSR at affinities approaching that of the natural ligand. A small-molecule design provides the flexibility to separate the receptor binding domain and the site of labeling and can be further optimized for binding affinities that exceed that of the natural ligand (16–18), thus making them attractive candidates for use as molecular imaging agents for GHSR. A class of small molecules based on quinazolinone derivatives has been developed by pharmaceutical companies to target GHSR as a treatment for obesity (19), and our group has developed strategies for generating libraries of fluorine-bearing azaquinazolinone derivatives for the purpose of developing PET tracers that target GHSR (16).
In this study, our group used a rational design approach in the synthesis of a quinazolinone derivative that could be labeled with 18F, which we termed [18F]LCE470, while maintaining superior binding affinity to GHSR. We measured its sensitivity and in vivo specificity and then tested and validated [18F]LCE470 for preclinical molecular PET imaging in a large-animal model of MI. We demonstrated both acute and chronic changes in the dynamics of [18F]LCE470 distribution within the left ventricle (LV) before and after surgical induction of MI.
MATERIALS AND METHODS
Tracer Design and Synthesis
Details on Figure 2 are described in the supplemental materials (available at http://jnm.snmjournals.org).
Radioligand Binding Assay and Radiosynthesis of [18F]LCE470
Compounds 4a, 4b, 5a, 5b, 7a, and 7b (LCE470) were tested for binding affinity to GHSR as we have done previously (6,20). Production and drying of [18F]fluoride were done as previously described (16). The precursor with tosylate (2.0 mg in 0.5 mL of MeCN) was then added, and the mixture was heated at 100°C for 10 min under sealed conditions. Half a milliliter of water containing 0.1% trifluoroacetic acid was added. The radiolabeled compound [18F]LCE470 was purified by semipreparative high-performance liquid chromatography on an Agilent RP-C18 column (19 × 150 mm, 5-μm pore) using a linear gradient of 40%–80% MeCN in water (0.1% trifluoroacetic acid; flow rate, 4.5 mL/min; 15-min run) followed by a 2-min wash in 95% MeCN. The retention time was 5.7 min. Radiosynthesis details are in the supplemental materials.
Hound Surgery
Four female hounds (19–22 kg) were obtained at 10–11 mo of age (Marshall Bioresource). They were treated in accordance with the ethical guidelines of the Canadian Council on Animal Care. Animal protocol 2017-006 was approved by the Animal Use Subcommittee at Western University. The procedure for MI was an ischemia–reperfusion method previously described (21,22). Details on the surgery and imaging data acquisition are in Supplemental Figure 2. Details on the image reconstruction, time–activity curves, and determination of SUVs and tissue-to-blood ratios (TBRs) are in Supplemental Figure 3. Quantification of 13N-NH3 perfusion imaging as mL/gm/min is in Supplemental Figure 4.
In Vivo Specificity of [18F]LCE470
A blocking study was conducted on 2 female hounds (11 mo of age) that had not undergone surgery or iron chelator therapy. PET/MRI was conducted after injection of a 6.5–7 MBq/kg dose of [18F]LCE470 alone and after injection of a 6.5–7 MBq/kg dose of [18F]LCE470 and a 10 M excess (8.5 μg/kg) of the GHSR ligand hexarelin.
Correlation of [18F]LCE470 and 13N-NH3 Uptake
Details on the correlation of [18F]LCE470 and 13N-NH3 uptake are in Supplemental Figure 5.
MRI Data Acquisition and Analysis
Details on the MRI data acquisition and analysis are in Supplemental Figure 6.
Tissue Harvesting, Fluorescence Image Acquisition, and Analysis
Details on fluorescence imaging of GHSR are in Supplemental Figure 7. Regional fibrosis and tissue morphology was determined as described previously (Supplemental Fig. 8) (8,9).
Correlation of [18F]LCE470 Uptake and to Tissue GHSR and Plasma Ghrelin
To determine the sensitivity of [18F]LCE470, the TBRs at 18 mo were correlated with fluorescence intensities of Cy5-cyclo-ghrelin(1–20) (14). Values from all tissue regions were pooled to obtain a robust sample size. Correlation to plasma ghrelin is in Supplemental Figure 8.
Statistical Analyses
Statistical analysis was performed with Prism version 8.2.0 (GraphPad Software). For TBR analyses, a repeated-measures ANOVA for both [18F]LCE470 and 13N-NH3 was used to determine regional differences at any given time point before and after MI. Corrections for multiple comparisons were performed using the Tukey post hoc test. MR data were analyzed with ANOVA and the Tukey post hoc test for multiple comparisons. Unpaired Student t tests and 1-way ANOVA were used to compare overall Cy5 fluorescence intensities. Correlations of tissue and PET values were performed using linear regression analysis. Significance was set at a P value of less than 0.05.
RESULTS
Tracer Design and Synthesis
Previously, we have performed extensive modifications to quinazolinone derivatives (16), resulting in the identification of 1 fluorine-bearing compound with picomolar binding affinity (20 pM), representing the highest—to our knowledge—binding affinity for GHSR reported to date (Fig. 1). However, efforts to radiolabel this compound were not successful (16). Molecular docking identified a small cavity in the opposite direction of the fluorine substituent (Fig. 1). We therefore synthesized quinazolinone derivatives to incorporate a small fluorine-containing substituent in the R4 position (compounds 4a–7a and 4b–7b in Fig. 2A).

Tracer design. (A) Picomolar binding affinity ligand identified from our previous modification. (B) Small cavity identified in opposite direction of fluorine substituent. IC50 = half-maximum inhibitory concentration.

(A) Synthesis of quinazolinone derivatives 4a–7a and 4a–7b. (B) Radiolabeling of compound 7b ([18F]LCE470). Details are in supplemental materials.
Receptor Binding Affinity and Radiochemistry
Six compounds were evaluated for GHSR-binding affinity (Table 1). The ligand with the highest binding affinity, 7b (LCE470), was selected for radiolabeling with 18F. The radiolabeling was performed using compound 6b as the precursor, resulting in a decay-corrected radiochemical yield of 8% ± 4%, molar activity of 87 ± 64 GBq/μmol, and a radiopurity of more than 99% (n = 11) (supplemental methods).
Structure–Activity Relationships of Quinazolinone Derivatives
Regional Analysis of [18F]LCE470 Tracer Uptake in LV
Time–activity curves were generated for 3 regions of interest (ROIs) (infarct, remote myocardium, and left circumflex coronary artery [LCX]) and for the blood pool before and at all time points after MI (Figs. 3A–3E). The pattern of uptake of [18F]LCE470 in all tissue ROIs was similar before surgery (Fig. 3A) and distinct from that of the blood pool, indicating retention in target tissues. In contrast, at all time periods after MI, the patterns of uptake in the infarct diverged from the remote myocardium and LCX (Figs. 3B–3D), and by month 11, there appeared to be a washout pattern in the infarct, similar to the blood pool (Fig. 3E).

Time–activity curves showing regional uptake of [18F]LCE470 in infarct, remote myocardium, LCX, and blood pool: baseline (A), day 3 (B), day 21 (C), week 16 (D), and month 11 (E). Uptake decreased slightly in infarct after MI from baseline, whereas both remote and LCX tissues increased after MI.
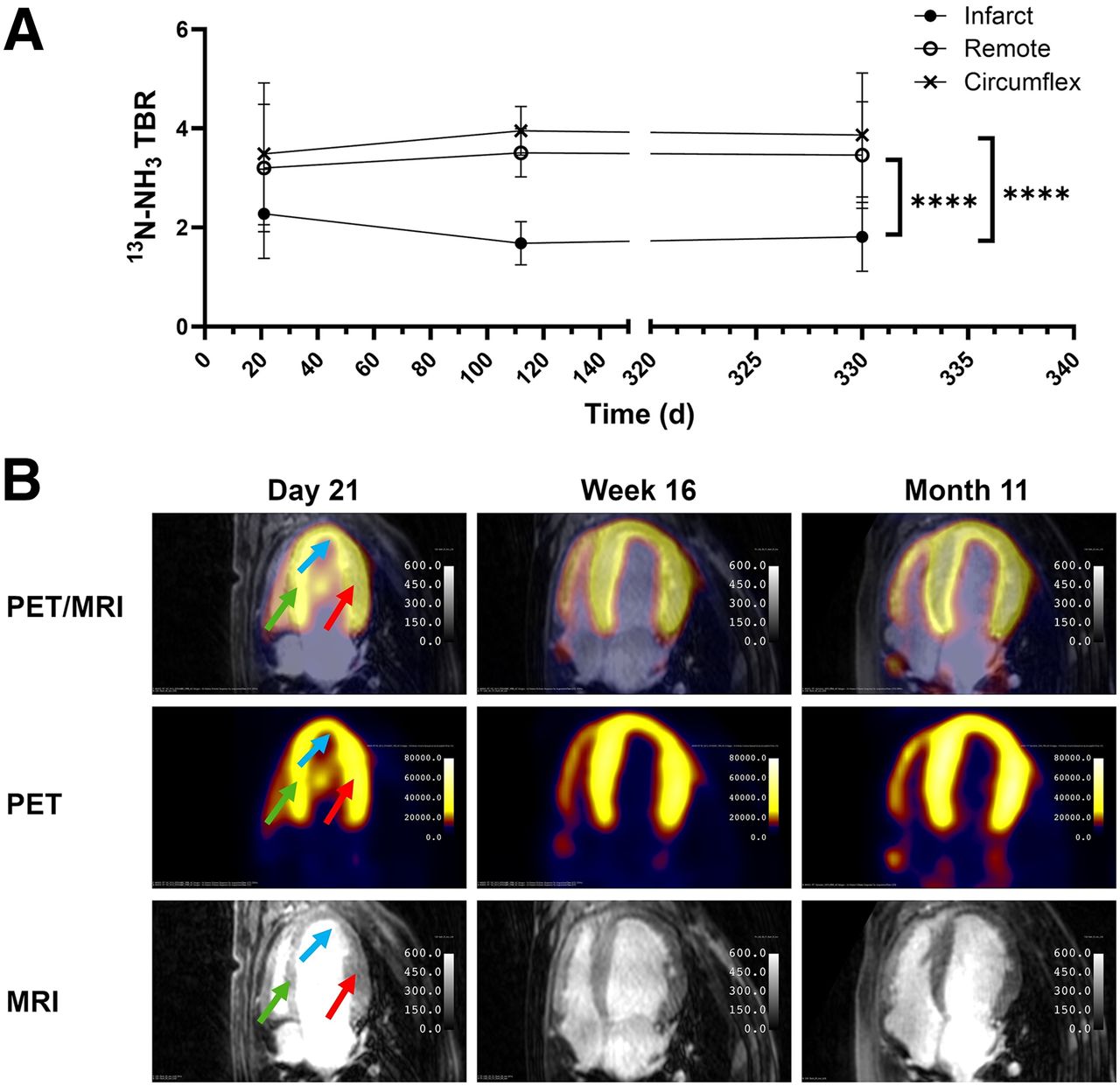
To calculate uptake of [18F]LCE470 in the 3 ROIs over the time course of the study, the TBRs of [18F]LCE470 were determined from the SUVs for each ROI before surgery and at specified time points after MI (Fig. 4A). Before surgery, [18F]LCE470 uptake was evenly distributed among all 3 regions (time 0, Fig. 4A). Twenty-four hours after MI, [18F]LCE470 uptake decreased more sharply in the infarct area than in the remote and circumflex regions. Uptake of [18F]LCE470 remained low in the infarct area throughout the time course of the study and was significantly lower than uptake in either the remote (P < 0.05) or the LCX (P < 0.001) regions. In the remote region, [18F]LCE470 uptake after MI remained lower than that in the LCX region throughout the time course of the study (P < 0.05). In contrast, uptake of [18F]LCE470 in the LCX region remained relatively unchanged. Representative PET/T1-weighted MR images of global [18F]LCE470 uptake from 1 hound are shown (Fig. 4B).

In vivo [18F]LCE470 in canine model of MI (n = 4). (A) TBRs for [18F]LCE470 in LV from baseline to 330 d after MI. Values were calculated from static scans of delineated ROIs. There was significant difference between infarct and remote regions (*P = 0.0365), remote and circumflex regions (*P = 0.0348), and infarct and circumflex regions (***P = 0.0003). (B) Representative images of axial view of LV from 1 dog depicting PET, MR, and PET/MR images for all time points shown in A. [18F]LCE470 uptake on day 3 after MI is indicated by arrows in 3 ROIs (infarct [blue], remote myocardium [green], and LCX [red]).
Specificity of [18F]LCE470 Binding in Cardiac Tissue
Two hounds that had not undergone surgery were coadministered [18F]LCE470 and a 10 M excess of hexarelin to determine binding specificity (20). In hound 1, there was a 32% reduction in global myocardial [18F]LCE470 uptake on coadministration of hexarelin and a 29% reduction in the LAD, a 26% reduction in the LCX, a 30% reduction in the apex, and a 40% reduction in the right coronary artery. There were slight reductions (between 11% and 16%) in [18F]LCE470 uptake on blocking in hound 2 (Fig. 5).

In-vivo blocking study for [18F]LCE470 shows TBRs for global tracer uptake, left anterior descending artery (LAD), LCX, right coronary artery (RCA), and apex for dog 1 (black) and dog 2 (gray) injected with tracer alone (solid bars) and hexarelin plus [18F]LCE470 (striped bars).
A metabolic stability study of [18F]LCE470 in the 2 control dogs did not yield a complete dataset; more than 70% of the tracer remained at 10, 25, 40, and 55 min after injection in at least 1 dog per time point). A more complete metabolic study is needed to more precisely quantify tracer stability.
Sensitivity of [18F]LCE470 Binding in Cardiac Tissue
Tissue quantification of GHSR was determined with quantitative fluorescence microscopy using the fluorescent ghrelin analog Cy5-cyclo-ghrelin(1–20) (14). In all hounds, Cy5 fluorescence in the LCX was significantly increased compared with that in the infarct (P < 0.0001), and in 2 hounds, Cy5 fluorescence in the LCX was significantly increased compared with that in the remote region (P < 0.001) (Supplemental Figs. 7A and 7B). Specificity of Cy5-cyclo-ghrelin(1–20) was demonstrated through a blocking study (Supplemental Fig. 7C) in which fluorescence decreased in the presence of excess hexarelin.
To determine the sensitivity of binding of [18F]LCE470, we conducted a linear regression analysis between [18F]LCE470 TBRs (in vivo tracer uptake) and fluorescence intensities (GHSR abundance). Figure 6 shows a significant (P = 0.007) positive correlation, indicating that tracer uptake increased with increasing GHSR expression in cardiac tissue.

Correlation of GHSR tissue analysis to [18F]LCE470 TBR.
Relationship Between [18F]LCE470 Distribution and Perfusion
The TBRs of 13NH3 showed significantly (P < 0.0001) decreased tracer uptake in the infarct compared with both the circumflex region and the remote region at all time points (Fig. 7A). In contrast to [18F]LCE470, there were no significant differences in 13NH3 uptake between the remote and circumflex regions. Representative 13NH3 PET/MR images from 1 hound indicate that the 13NH3 PET signal in the LV was localized to the LV wall at all time points after MI, as expected, with some distribution in the right ventricle (Fig. 7B).

Analysis of perfusion imaging tracer in dogs (n = 4). (A) TBRs for 13N-NH3 over time in LV from days 7 to 330 after MI. Values were calculated from static scans of 3 delineated regions. There were significant differences between infarct and remote regions (****P < 0.0001) and between infarct and circumflex regions (****P < 0.0001) but no significant difference between remote and circumflex regions. (B) Representative images of axial view of LV from 1 dog depicting PET, MR, and fused PET/MR images for all time points shown in A. Distribution of 13NH3 at day 21 is indicated by arrows for infarct (blue), remote myocardium (green), and LCX (red). To assess whether regional distribution of [18F]LCE470 could be accounted for by perfusion alone, linear regression analysis was done for TBRs of [18F]LCE470 and 13N-NH3 (Supplemental Fig. 5). There were no significant correlations at any selected time points.
Fibrotic Deposition and Circulating Ghrelin
There was significant fibrosis in the infarct compared with the remote region and the LCX. Hematoxylin and eosin showed changes in tissue morphology between the infarct and both remote myocardium and LCX (Supplemental Fig. 8A).
To determine whether [18F]LCE470 administration affected circulating ghrelin levels, serum ghrelin concentrations were measured at all time points. Linear regression analysis showed no significant correlations of plasma ghrelin concentrations with [18F]LCE470 TBRs in any of the ROIs (Supplemental Fig. 8B).
Heart Function Before and After MI
Left ventricular ejection fraction and stroke volume were determined from baseline to 11 mo after MI (Supplemental Fig. 6). There were no significant differences at any time point.
DISCUSSION
Molecular imaging offers a unique opportunity to visualize the cardiac metabolic and cellular landscape in vivo. This imaging strategy can yield information on physiologic and pathophysiologic changes over time and within different cardiac regions. Advances in PET tracer development have enabled specific detection of clinically relevant markers of inflammation, metabolism, immunity, and fibrosis that may yield insights into the development and evolution of heart disease and heart failure.
One potential marker of myocardial function and disease is the GHSR and its ligand ghrelin. Ghrelin and GHSR are both synthesized in cardiomyocytes (23), and myocardial GHSR signaling is associated with cardioprotection through antiapoptosis (5,24,25), antiinflammation (4,26), and regulation of contractility (27–29). Myocardial tissue expression of GHSR in patients is elevated in end-stage heart failure (30) and cardiac transplantation (8). In patients with valvular disease, there was a decrease in myocardial ghrelin expression (9). In rat models of MI, global expression of GHSR increased over time (31). Therefore, GHSR may be an imaging target for detecting molecular changes associated with the evolution of heart disease and possibly heart failure.
In this study, we designed and synthesized an 18F-labeled quinazolinone derivative, [18F]LCE470, that demonstrated picomolar binding affinity to GHSR. This tracer was then tested and validated in a canine model of moderately extensive anterior MI. Using simultaneous multitracer PET/MRI, we determined the relationships between GHSR, perfusion, and heart function before and after MI. Our 18F-tracer was found to bind in a sensitive and specific manner to GHSR in the LV independently of perfusion tracer distributions and changes in global cardiac function. We found that the regional distribution of [18F]LCE470 within the LV diverged significantly within 1 d after MI and remained that way throughout the 1.5-y duration of the study. These results demonstrate how PET molecular imaging with a high-affinity radioligand can track spatiotemporal changes in myocardial GHSR.
Radioligands based on the peptide structure of ghrelin labeled with 64Cu or 68Ga have been developed to target GHSR (32,33). More recently,18F-labeled peptidomimetic and small-molecule GHSR agonists have been developed as clinically applicable tracers. Our group has previously used a peptidomimetic, [1-NaI4, Lys5(4-[18F]-FB)]G-7309, for in vivo imaging of cardiac GHSR in healthy mice (34,35). However, this tracer was neither sensitive nor specific, either because of the animal model or because of its unfavorable pharmacokinetics of high lipophilicity and moderate binding affinity (69 nM). Our present study characterizing [18F]LCE470 as a GHSR radioligand represents an improvement since it has a much higher binding affinity (0.33 nM), and we tested it in an animal model that was clinically relevant for cardiac imaging.
We previously designed and synthesized a fluorine-containing quinazolinone derivative with even greater picomolar binding affinity (0.02 nM) for GHSR (16). However, this compound could not be successfully labeled with 18F. Our rational design approach using molecular docking, based more on machine learning–based scoring than physics-based scoring (36), predicted a potential space within the quinazolinone-bound GHSR binding pocket for a small fluorine-containing substituent that would not sterically hinder binding of the radioligand. On the basis of this prediction, we successfully radiolabeled an 18F-labeled quinazolinone derivative with picomolar binding affinity (0.33 nM) for PET imaging of GHSR. Another study that generated PET radioligands for GHSR using quinazolinone derivatives reported a compound with a half-maximum inhibitory concentration of 2.2 nM (37), almost 1 order of magnitude lower than our compound. Therefore, we have generated a PET tracer with the highest—to our knowledge—known receptor binding affinity for a radiolabeled GHSR ligand, making it an optimal tracer for PET imaging of GHSR.
We chose to evaluate our compound in a canine preclinical model of MI instead of rodent models. The primary reasons are, first, that this a well-established model of MI developed at our institute that has resulted in clinical translation of MRI of myocardial viability (38) and PET/MRI of myocardial inflammation (39); second, that we could use the protocols for tracer injection and imaging on our clinical PET/MRI scanner; and third, that the study could be performed over 18 mo without significant growth or aging of the hounds, unlike pigs or rodents (40).
The kinetics of [18F]LCE470 distribution before MI showed a washout curve in the blood pool and retention in the LV regions (infarct, remote myocardium, and LCX). In the absence of cell uptake data, this experiment is evidence of tissue binding and was corroborated by the TBRs of the calculated SUVs and by the PET/MR images showing a defined pattern of tracer uptake in the LV wall. This pattern of uptake is also exhibited by 18F-FDG, the gold standard PET measure of myocardial viability, as well as by other 18F-labeled tracers for myocardial perfusion and neurohumoral activation (41). Further evidence of specific binding to myocardial GHSR was provided by an in vivo competitive blocking study on 2 healthy hounds. Although a perceived limitation of this experiment is the low number of animals, it was done in a large animal in multiple regions within an endogenous tissue before and after a disease condition, as opposed to the implanted, and often genetically engineered, cells in rodent models (12,33,42). The apparent differences in the extent of blocking (from 26% to 40%) in 1 dog may reveal some heterogeneity in the distribution of GHSR in the healthy LV. We used a coinjection protocol since hexarelin is a competitive agonist, not an antagonist, and would therefore exhibit the same displaceable binding kinetics regardless of the injection protocol. Hexarelin has a greater binding affinity for GHSR than does LCE470 (18 vs. 330 pM, respectively) and has high serum stability. Therefore, coadministration of hexarelin in molar excess would ensure effective displacement of [18F]-LCE470 from available binding sites. Because of institutional ethics requirements, only 2 animals were approved for this study, and the blocking study had to be a terminal experiment. Further investigation into the distribution of myocardial GHSR in healthy LV may lead to a greater understanding of the significance of changes after MI.
After surgical induction of MI, there were significant heterogeneities in the distribution of [18F]LCE470 in the 3 regions of the LV, especially between the LCX and the infarct. This difference persisted throughout the course of the study, suggesting that permanent alterations in this area have occurred as a result of acute MI. Since [18F]LCE470 is a high-affinity ligand for GHSR, the attenuated tracer uptake may be due to a decrease in GHSR number or density. This hypothesis is supported by fluorescence microscopy analysis using a fluorescent ghrelin analog that binds with high affinity to GHSR (14), showing similar changes in the distribution of GHSR in all dogs. The highly significant positive correlation between [18F]LCE470 uptake and tissue GHSR provides strong evidence that [18F]LCE470 distribution is a sensitive measure of myocardial GHSR. Therefore, [18F]LCE470 may be used to discern fine changes in the regional distribution of GHSR after acute MI, which in turn may provide additional information on the biology of subsequent LV remodeling.
The area of the infarct showed the most dramatic sustained decrease in [18F]LCE470 uptake, which was accompanied by attenuated perfusion and significant fibrosis. We have previously shown that GHSR is not detectable fibrotic tissue (8), which may partially explain the decrease in tracer uptake. The sustained decrease in perfusion may result in either reduced delivery of the tracer or reduced receptor density or number. Although we cannot rule out reduced tracer delivery, microscopy analysis showed a decrease in GHSR in tissue obtained at the end of the study. Therefore, LV remodeling after acute MI may include a loss of GHSR in areas of reduced perfusion and increased fibrosis.
LV remodeling also involves upregulation of adaptive processes in the area around the edge of the infarct, termed border myocardium, represented by the LCX region in our study. As measured by both [18F]LCE470 uptake and fluorescence microscopy, GHSR was upregulated in this region in both the acute and the chronic phases after MI. GHSR signaling is coupled to the Gqa/phospholipase C β pathway, which plays a role in cardioprotection through regulation of Ca2+ homeostasis to promote contractility (29) and also the Akt pathway to promote cardiomyocyte survival (24,43), leading some to propose that GHSR may be a target to combat post-MI remodeling (43). In later stages of LV remodeling, it is known that the border myocardium modulates scar formation to preserve LV function. This study is limited by a lack of late-gadolinium-enhancement MRI, and future studies investigating the temporal relationship between scar formation and GHSR with [18F]LCE470 PET and late-gadolinium-enhancement MRI may reveal a direct role for GHSR signaling in the reduction of myocardial scarring in vivo.
In this study, we did not determine the agonist properties of [18F]LCE470. We also did not conduct a whole-body biodistribution study or a detailed tracer kinetic analysis of binding potential. Quinazolinone derivatives have been developed as possible therapies for obesity and diabetes and are GHSR antagonists (19,44), and 1 other study of fluorinated derivatives reported compounds with weak antagonist activity (37). Therefore, it is likely that [18F]LCE470 may have antagonist properties. In our study, serum ghrelin concentrations were not affected by the degree of tracer uptake, so it is unlikely that there is any biologic effect of the tracer itself. Since GHSR is distributed somewhat ubiquitously (1), a whole-body biodistribution would not have yielded clear results. Finally, quantitative analysis of binding potential would confirm changes in GHSR density or number. We are currently deriving an appropriate modeling approach to determine the kinetics of [18F]LCE470.
CONCLUSION
[18F]LCE470 is a high-affinity PET tracer that can detect changes in the regional distribution of myocardial GHSR after MI. Such in vivo studies investigating the global dynamics of GHSR may lead to improved GHSR-based therapeutics in the treatment of post-MI remodeling.
DISCLOSURE
This work was supported by an operating grant from the Natural Sciences and Engineering Research Council of Canada and the Canadian Institutes for Health Research to Savita Dhanvantari, Leonard Luyt, Gerald Wisenberg, and Frank Prato. Rebecca Sullivan was supported by a Canada Graduate Studentship-Doctoral from the Natural Sciences and Engineering Research Council of Canada. No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Can a high-affinity 18F-labeled compound that can track the dynamics of cardiac GHSR in health and disease be developed?
PERTINENT FINDINGS: [18F]LCE470 had a receptor binding affinity of 0.33 nM, the highest known for a radiolabeled GHSR ligand. In vivo and ex vivo blocking studies showed the specificity of [18F]LCE470, and sensitivity was demonstrated by a positive correlation between tracer uptake and GHSR abundance. Post-MI changes in [18F]LCE470 uptake occurred independently of changes in perfusion and global cardiac function. The distribution of [18F]LCE470 within the LV diverged significantly within 1 d after MI and remained that way throughout the 1.5-y duration of the study.
IMPLICATIONS FOR PATIENT CARE: Tracking the regional dynamics of cardiac GHSR after MI may help to better predict the effectiveness of ghrelin-based therapeutics.
Footnotes
Published online Sep. 12, 2024.
- © 2024 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication February 22, 2024.
- Accepted for publication August 13, 2024.
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.