


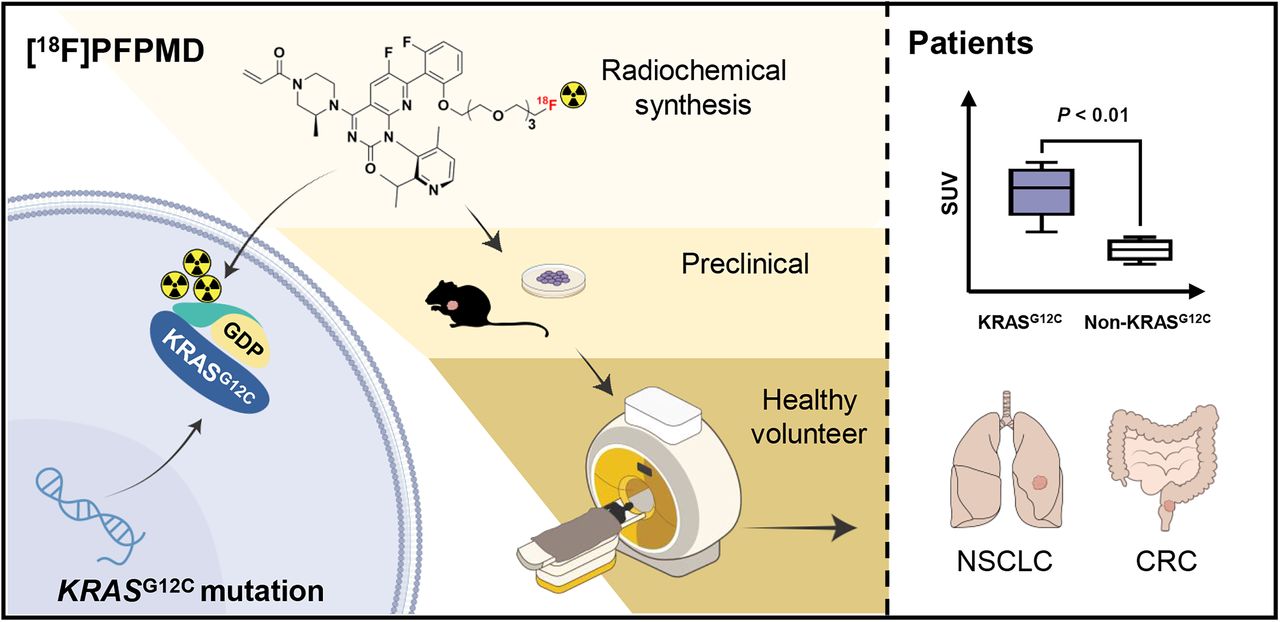
Visual Abstract

Abstract
Kirsten rat sarcoma (KRAS) mutations are an important marker for tumor-targeted therapy. In this study, we sought to develop a KRASG12C oncoprotein–targeted PET tracer and to evaluate its translational potential for noninvasive imaging of the KRASG12C mutation in non–small cell lung cancer (NSCLC) and colorectal cancer (CRC) patients. Methods: [18F]PFPMD was synthesized on the basis of AMG510 (sotorasib) by attaching a polyethylene glycol chain to the quinazolinone structure. The binding selectivity and imaging potential of [18F]PFPMD were verified by cellular uptake, internalization, and blocking (H358: KRASG12C mutation; A549: non-KRASG12C mutation) studies, as well as by a small-animal PET/CT imaging study on tumor-bearing mice. Five healthy volunteers were enrolled to assess the safety, biodistribution, and dosimetry of [18F]PFPMD. Subsequently, 14 NSCLC or CRC patients with or without the KRASG12C mutation underwent [18F]PFPMD and [18F]FDG PET/CT imaging. The SUVmax of tumor uptake of [18F]PFPMD was measured and compared between patients with and without the KRASG12C mutation. Results: [18F]PFPMD was obtained with a high radiochemical yield, radiochemical purity, and stability. The protein-binding assay showed that [18F]PFPMD selectively binds to the KRASG12C protein. [18F]PFPMD uptake was significantly higher in H358 than in A549 and was decreased by pretreatment with AMG510 (H358 vs. A549: 3.22% ± 0.28% vs. 2.50% ± 0.25%, P < 0.05; block: 2.06% ± 0.13%, P < 0.01). Similar results were observed in tumor-bearing mice on PET imaging (H358 vs. A549: 3.93% ± 0.24% vs. 2.47% ± 0.26% injected dose/g, P < 0.01; block: 2.89% ± 0.29% injected dose/g; P < 0.05). [18F]PFPMD was safe in humans and was excreted primarily by the gallbladder and intestines. The whole-body effective dose was comparable to that of [18F]FDG. The accumulation of [18F]PFPMD in KRASG12C mutation tumors was significantly higher than that in non-KRASG12C mutation tumors (SUVmax: 3.73 ± 0.58 vs. 2.39 ± 0.22, P < 0.01) in NSCLC and CRC patients. Conclusion: [18F]PFPMD is a safe and promising PET tracer for noninvasive screening of the KRASG12C mutation status in NSCLC and CRC patients.
Kirsten rat sarcoma (KRAS) is a commonly mutated human oncogene that, in various types of cancer, accounts for 20.4%–67.6% of cases (1). The mutation of KRAS would constitutively activate downstream oncoprotein pathways, including Raf kinase–mitogen-activated protein kinase–extracellular-signal–regulated kinase (ERK) and phosphatidylinositol-3′-kinase/protein kinase B, thereby promoting tumor growth and survival (2). Patients with KRAS mutations usually respond poorly to the standard and epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) therapies (3–5). Therefore, the mutation status of KRAS has been recommended as a standard molecular marker by the National Comprehensive Cancer Network and other leading cancer research centers (6).
Currently, KRAS mutation screening relies on a biopsy combined with amplification-refractory mutation system polymerase chain reaction (PCR) or gene sequencing. Biopsies, however, induce pneumothorax in 20.5% of lung cancer patients, and their use is limited by the quality of the tissue sample and tumor heterogeneity (7,8). Thus, there is an urgent need for accurate yet noninvasive methods of evaluating the KRAS mutation status.
PET is a noninvasive molecular imaging technique that enables accurate detection of specific tumor markers and, consequently, correct diagnosis and choice of targeted therapy (4,9). Nevertheless, noninvasive imaging of KRAS mutations has been limited for a long time by the lack of a specific ligand (10). For 40 y, KRAS mutations were recognized as undruggable, until a series of covalent inhibitors was identified in recent years, such as ARS-1620, MRTX849, and AMG510 (sotorasib) (11,12). These inhibitors could specifically lock the KRASG12C protein, which is the predominant KRAS mutation type in non–small cell lung cancer (NSCLC) patients, binding with guanosine diphosphate (GDP) for an inactive state, eventually suppressing tumor growth (13).
Recently, along with our collaborators, we synthesized several KRASG12C-targeted tracers that were based on ARS-1620. However, their preclinical imaging performance turned out to be insufficient for clinical utility (14). AMG510 (sotorasib), the first Food and Drug Administration–approved (in May 2021) KRASG12C inhibitor for treating NSCLC, has much higher potency than ARS-1620 (half-maximal inhibitory concentration, 0.09 μM vs. 0.831 μM) (11,15). In addition, a recent clinical trial has revealed AMG510 to have benefits in treating colorectal cancers (CRCs) with the KRASG12C mutation (16).
Therefore, in this study we sought to develop a KRASG12C-targeted PET tracer based on AMG510 and to investigate its targeting and imaging capacity in vitro and in animal models. On the basis of the preclinical results, a first-in-humans study was then performed to analyze the safety and biodistribution of the new KRAS mutation tracer in healthy volunteers and to evaluate the feasibility of noninvasive imaging of the KRASG12C mutation in NSCLC and CRC patients.
MATERIALS AND METHODS
Synthesis of [18F]PFPMD
A KRASG12C inhibitor, AMG510, was obtained commercially (MedChemExpress). The detailed synthesis procedure for 4-((S)-4-acryloyl-2-methylpiperazin-1-yl)-6-fluoro-7-(2-fluoro-6-(2-(2-(2-(2-(fluoro-18F)ethoxy)ethoxy)ethoxy)ethoxy)phenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one ([18F]PFPMD) is described in the supplemental methods and summarized in Figure 1A (supplemental materials are available at http://jnm.snmjournals.org). Preparation of the tosylate precursor 2-(2-(2-(2-(2-(4-((S)-4-acryloyl-2-methylpiperazin-1-yl)-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-7-yl)-3-fluorophenoxy)ethoxy)ethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (PPMD-OTs) was based on AMG510, using the substitution reaction (Willamson-type reaction). The [18F]PFPMD was then synthesized on PPMD-OTs through direct bimolecular nucleophilic substitution displacement with [18F]fluoride using an AllinOne (Trasis) universal synthesizer (Supplemental Fig. 1). In addition, the corresponding unlabeled compound PFPMD was synthesized (Supplemental Fig. 2). All nonradioactive targeted compounds were characterized by mass spectrometry (API 3200; SCIEX), high-resolution mass spectrometry (LCT Premier; Waters), and nuclear MR spectroscopy (AvanceCore; Bruker) (Supplemental Figs. 3–5). Analytic radio–high-performance liquid chromatography (HPLC) (1200 series; Agilent Technologies) was used to assess radiochemical purity, and molar activity was analyzed by an ultraviolet standard curve method. The identities of the radiolabeled ligands were established by comparing the retention times with those of the corresponding unlabeled compounds, using coinjection HPLC analysis. The in vitro stability in saline and in mouse serum was evaluated at 6 h by radio-HPLC. The apparent distribution coefficient of [18F]PFPMD was determined by the shake-flask method, as described in the supplemental methods.

Synthesis and characterization of [18F]PFPMD. (A) Synthetic routes of [18F]PFPMD. (B) HPLC coelution profile of [18F]PFPMD and PFPMD. Coinjection of [18F]PFPMD and its corresponding nonradioactive PFPMD HPLC analysis confirmed radioligands. (C) In vitro stability of [18F]PFPMD in saline and in mouse serum at 6 h (n = 3). ACN = acetonitrile; RCP = radiochemical purity; UV = ultraviolet. Kryptofix is a product of Merck.
Protein-Binding Assay
A protein-binding assay was performed to study the binding interactions between [18F]PFPMD and KRASG12C oncoprotein, as previously described (14). [18F]PFPMD was dissolved in a buffer containing 10% dimethyl sulfoxide, 5 mM MgCl2, and 5 mM GDP or guanosine 5′-[β,γ-imido]triphosphate (GMPPNP) (a guanosine triphosphate analog) with an activity of 37 MBq/mL. Different types of KRAS oncoprotein (G12C, WT, G12D, or G12V) with a final concentration of 2 μM were incubated with 3.7 MBq of a tracer in a buffer containing 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (pH 7.5), 150 mM NaCl, 1 mM MgCl2, and 1 mM DTT for 1 h. For blocking studies, coincubation was performed using different inhibitors (AMG510, ARS-1323, another KRASG12C inhibitor, or gefitinib, an EGFR TKI) and nonradioactive PFPMD at a final concentration of 10 μM. This process was quenched by adding up to 0.2% formic acid. Protein-binding analysis was performed using radio–thin-layer chromatography and autoradiography, respectively.
In Vitro Cellular Uptake Study
In accordance with previous reports, the human NSCLC cell lines H358 and A549 (China Center for Type Culture Collection) were chosen as the KRASG12C mutation and wild-type cell lines, respectively. All cell lines were cultured in RPMI 1640 medium (Thermo Fisher) with 10% fetal bovine serum (Gibco) and 1% penicillin–streptomycin (Thermo Fisher) at 37°C in a moist atmosphere of 5% CO2. To verify the KRASG12C mutation status of each cell line, total DNA was extracted with an Ezup column DNA extraction kit (Sangon Biotech). After being mixed thoroughly with 200 μL of phosphate-buffered saline and 20 μL of proteinase K, the cells were placed briefly in a heated water bath (56°C, 10 min) for lysis. Then, the DNA was extracted according to the manufacturer's instruction. KRAS fragments were then amplified by PCR from the genome with KRAS primers—F 5′-3′ACGTCTGCAGTCAACTGGAATT and R 3′-5′-TCTGTATCAAAGAATGGTCCTGC—and the KRASG12C mutation status was detected using the Sanger sequencing method. Cells (250 μL of 1 × 106 cells/mL) were incubated with [18F]PFPMD (0.5 mL, 110 kBq) at different time points; for the uptake study, the cells were washed with phosphate-buffered saline and digested for radioactivity assessment with a γ-counter (WIZARD2 2470; PerkinElmer). For the internalization study, the cells were washed with phosphate-buffered saline and acid lysate (50 mM glycine and 100 mM NaCl, pH 2.8), respectively, and then for radioactivity assessment with a γ-counter. AMG510, ARS-1323, or nonradioactive PFPMD (10 μM) was used to pretreat H358 for the blocking experiment. In addition, the SW837 CRC cell line (mutated KRASG12C) and gefitinib were used in the cell uptake study to observe the impact of EGFR TKI on tracer uptake. All cell studies were performed in triplicate.
Biodistribution
All animal studies were conducted in accordance with relevant guidelines and under the protocol approved by the Institutional Animal Care and Use Committee of the Fourth Military Medical University. Biodistribution analysis was performed on 4- to 5-wk-old male healthy ICR mice and tumor-bearing BALB/c nude mice. [18F]PFPMD (0.1 mL, about 370 kBq) was injected via the tail vein. At 5, 15, 30, 60 and 120 min after injection, ICR mice were killed while anesthetized (3 at each time point), and major organs were extracted. Tumor uptake of [18F]PFPMD was assessed in H358 or A549 tumor–bearing BALB/c nude mice at 1 h after injection (0.1 mL, about 370 kBq) (5 for each group). In blocking studies, the H358 tumor–bearing BALB/c nude mice were coinjected intravenously with [18F]PFPMD and AMG510 (20, 50, 100 μg) via the tail vein. Ex vivo radioactivity biodistribution was measured with a γ-counter. The results are expressed as percentage injected dose (%ID) per gram of wet tissue.
In Vivo [18F]PFPMD Small-Animal PET/CT Imaging Study
H358 or A549 tumor–bearing BALB/c mice were used for in vivo [18F]PFPMD imaging with a small-animal PET/CT system (Nano PET/CT, Mediso). The mice were anesthetized with isoflurane and injected with 11.1 MBq of [18F]PFPMD via the tail vein. Dynamic [18F]PFPMD small-animal PET/CT scans were acquired within 120 min after injection, with the following parameters (PET: 1–5 coincidence mode, 5-ns coincidence time windows; CT: semicircular scan, 180 projections, 55-kVp x-ray energy, 600-ms exposure time, and maximum zoomed field of view). For the blocking studies, the H358 tumor–bearing BALB/c mice were coinjected intravenously with [18F]PFPMD and PFPMD (20, 50, or 100 μg) via the tail vein. The PET/CT images were iteratively reconstructed as 10, 30, 60, and 120 min using ordered-subset expectation maximization (Tera-Tomo 3D [Mediso]: voxel size, 0.4; 4 iterations; 6 subsets; filter type, cosine; 40 × 40 × 40 mm volume size; 140 × 140 × 140 μm voxel size; 284 × 284 × 284 voxels). The PET/CT images were analyzed using Interview Fusion 1.0 (Mediso) software.
Enrollment of Healthy Volunteers and Patients
The clinical validation study was approved by the Ethics Committee Board of Xijing Hospital (KY20212187-F-1), and all participants gave written informed consent. Five healthy volunteers were enrolled to investigate the biodistribution, dosimetry, and safety of [18F]PFPMD (Supplemental Table 1). Biopsy-confirmed NSCLC/CRC patients were recruited for PET/CT imaging. The exclusion criteria were severe or acute systemic disease, pregnancy or lactation, or the inability or refusal to undergo a PET/CT scan. Amplification-refractory mutation system PCR or targeted 425-gene sequencing was used to determine the tumor KRAS mutation status (NovasEquation 6000; Illumina). Amplification-refractory mutation system PCR was performed using a detection kit (human KRAS mutation detection kit; AmoyDx) in the Department of Pathology, Xijing Hospital. The amplification results were evaluated according to the manufacturer’s criterion.
[18F]PFPMD PET/CT and [18F]FDG PET/CT Imaging in Humans
All PET/CT scans were acquired on a hybrid PET/CT system (uMI Panorama; United Imaging). Healthy volunteers were injected intravenously with 229.1 ± 61.22 MBq of [18F]PFPMD diluted with 4 mL of normal saline. A dynamic PET scan was registered within 1 h after the injection, and static PET scans were registered at 2, 4, and 8 h after the injection. The dynamic PET data were reconstructed as 5-, 15-, 30-, 40-, 50-, and 60-min images after the injection. The radiation dose absorbed by organs in each volunteer was calculated using OLINDA/EXM software (version 1.1, Vanderbilt University, 2007), and the radiation dose absorbed by each major organ (Table 1) was estimated as the mean value of the results of the 5 healthy volunteers (17). To analyze the pharmacokinetic profile, blood (2–3 mL) was collected at 10, 30, 60, 120, 240, and 480 min after injection from healthy volunteers and measured with a γ-counter. Vital signs, including blood pressure, heart rate, oxygen saturation, body temperature, and respiratory rate, were monitored after the injection and during PET/CT imaging as a safety assessment. Any potential adverse event was monitored by a phone follow-up within 7 d after PET/CT imaging.
Dosimetry of [18F]PFPMD in Healthy Volunteers
[18F]PFPMD (240.8 ± 43.76 MBq) and [18F]FDG (252.9 ± 61.46 MBq) PET/CT imaging was performed on all NSCLC and CRC patients on 2 separate days within 1 wk. Both types of imaging were performed at 60 min after tracer injection. The patients were required to fast for 6 h and to have a blood glucose level under 180 mg/dL before undergoing [18F]FDG PET/CT imaging.
CT scans were acquired for attenuation correction with the following parameters (100 keV, automatic exposure control, 512 × 512 matrix, 3-mm slice thickness, 0.5-s rotation time, and a pitch index of 0.9). PET scans were subsequently acquired at 2 min/bed position and 5 positions in total, with a 192 × 192 matrix. The data were transferred to a workstation (uWS-MI R002 4.0; United Imaging). Regions of interests were drawn on the PET images, and SUVmax was used to measure tracer accumulation in organs. All images were analyzed by 2 experienced nuclear medicine physicians unaware of the KRAS mutation results or the groups. Any discrepancies were resolved by consensus.
Statistical Analysis
Statistical analysis was performed on GraphPad Prism 9. Continuous variables are presented as mean ± SD. One-way ANOVA was used to compare cell uptake or PET SUVmax among groups. Differences with a P value of less than 0.05 were considered statistically significant.
RESULTS
Synthesis and Characterization of [18F]PFPMD
[18F]PFPMD was synthesized as shown by the routes in Figure 1A. The overall synthesis time for [18F]PFPMD was around 90 min. After purification, [18F]PFPMD was obtained in a 21.5% ± 1.5% radiochemical yield (n = 3, corrected for decay). The radiochemical purity was more than 99%, and molar activity was 18.20 ± 5.43 GBq/μmol (Supplemental Fig. 6). The identity of the radioligands was confirmed by coinjection HPLC analysis (Fig. 1B). The log D (pH 7.4) value was 2.17 ± 0.08, indicating the lipophilicity of the tracer is good for entering the cell. The in vitro stability of [18F]PFPMD was more than 99% (Fig. 1C).
Protein-Binding, Molecular-Docking, Cell Uptake, Internalization, and Blocking Studies on [18F]PFPMD
The protein-binding assay showed that [18F]PFPMD bound to KRASG12C oncoprotein under the coexistence of GDP but not GMPPNP. Nevertheless, it failed to bind to KRASWT, KRASG12D, or KRASG12V under the GDP condition. Moreover, after preblocking with AMG510, PFPMD, or ARS-1323, the binding of [18F]PFPMD and KRASG12C/GDP dissolved. However, gefitinib did not block the binding (Fig. 2A; Supplemental Fig. 7). In addition, molecular docking estimated the complementarity of PFPMD to the binding site of KRASG12C protein. The binding energy of PFPMD and KRASG12C oncoprotein was −5.49 kcal/mol, which was similar to the −5.11 kcal/mol between AMG510 and KRASG12C oncoprotein (Supplemental Fig. 8). All these results collectively support the thesis that [18F]PFPMD selectively binds to KRASG12C oncoprotein in the GDP-bound state and that polyethylene glycol modification and 18F labeling do not preclude selective binding.

In vitro binding study and preclinical imaging study with [18F]PFPMD. (A) Protein-binding results for [18F]PFPMD based on radio–thin-layer chromatography results. (B) Uptake of [18F]PFPMD in 2 NSCLC cell lines at indicated time points (n = 3). (C) Cell uptake and blocking study at 60 min (n = 3). (D) Internalization study in 2 NSCLC cell lines. (E) Representative [18F]PFPMD small-animal PET/CT images at 1 h in tumor-bearing mouse models and before treatment with AMG510 (n = 3). (F) Quantitative analysis of tumor and muscle uptake of [18F]PFPMD in H358/A549 tumor–bearing mice at 1 h after injection (5 for each group). *P < 0.01. ARS-1323 = a KRASG12C inhibitor; GMPPNP = guanosine 5′-[β,γ-imido]triphosphate; GTP = guanosine triphosphate; ns = not statistically significant; WT = wild type.
The H358 cell line exhibited a 76.9% KRASG12C mutation rate, which was not observed in the A549 cell line, as revealed by Sanger sequencing results (Supplemental Fig. 9). H358 cells displayed significantly higher uptake of [18F]PFPMD than did A549 cells at different time points (Fig. 2B). The internalization study showed that approximately 50% and 60% of radioactivity was retained at 1 h in the H358 and A549 cell lines, respectively (Fig. 2C). Moreover, pretreatment with AMG510, PFPMD, or ARS-1323 significantly decreased the [18F]PFPMD uptake in H358 cells (AMG510: 3.22% ± 0.28% vs. 2.06% ± 0.13%, P < 0.01; PFPMD: 3.22% ± 0.28% vs. 2.48% ± 0.17%, P < 0.01; ARS-1323: 3.22% ± 0.28% vs. 2.14% ± 0.09%, P < 0.01; Fig. 2D). In addition, we assessed the possible drug effect of PFPMD on the H358 cell line and found that it significantly inhibited the downstream phospho-ERK expression (Supplemental Fig. 10A). To explore the impact of the combination therapy on tracer uptake, we performed a cell uptake study on the SW837 cell line, and this study revealed that coincubation with gefitinib did not impact tracer uptake (3.28% ± 0.23% vs. 3.21% ± 0.14%, P > 0.05, Supplemental Fig. 10B).
Biodistribution and [18F]PFPMD Small-Animal PET Imaging in Mice
As the biodistribution and imaging study showed, the 2 major organs with the highest uptake of [18F]PFPMD were the gallbladder and intestines (Supplemental Table 2; Supplemental Fig. 11A), indicating that [18F]PFPMD cleared primarily via the digestive system.
[18F]PFPMD was further tested on a tumor-bearing mouse model, as the small-animal PET/CT imaging showed rapid accumulation of [18F]PFPMD in H358 tumors (KRASG12C mutation, Supplemental Figs. 11B–11E). Biodistribution revealed that tumor uptake was significantly higher in H358 than in A549 (wild type) (3.93 ± 0.24 vs. 2.47 ± 0.26 %ID/g, +59.1%, P < 0.01, Figs. 2E and 2F). Pretreatment with AMG510 significantly decreased H358 tumor uptake (3.93 ± 0.24 vs. 2.48 ± 0.26 %ID/g, P < 0.01, Figs. 2E and 2F), and no significant difference was observed among different AMG510 blocking dosages (Supplemental Fig. 11F) These results not only jointly confirmed the feasibility of [18F]PFPMD for PET/CT imaging but also substantiated the tracer targeting specificity in vivo.
Biodistribution, Dosimetry, and Safety of [18F]PFPMD in Healthy Volunteers
In humans, [18F]PFPMD accumulated predominantly in the gallbladder and intestines. The tracer rapidly accumulated in the intrahepatic bile ducts and renal pelvis after the injection and then gradually concentrated in the gallbladder and bladder from 15 to 60 min, as detailed in Figures 3A and 3B. [18F]PFPMD uptake was clearly observed in the small intestine at 1 h after injection, but after 2 h, when secretion of the tracer to the colon began, uptake in the small intestine decreased. Colon uptake remained high and stable even at 8 h after injection. However, with time, uptake decreased in most major organs, including the brain, heart, liver, spleen, and lungs. Radioanalysis of the blood revealed blood clearance of [18F]PFPMD—that is, from 0.0042 ± 0.0012 %ID/mL at 10 min after injection to 0.0019 ± 0.0008 %ID/mL at 120 min after injection (Fig. 3C).

Biodistribution of [18F]PFPMD in healthy volunteers. (A) Representative PET images of [18F]PFPMD in healthy man at 5 min, 30 min, 1 h, 2 h, 4 h, and 8 h after injection. (B) Biodistribution of [18F]PFPMD in major organs at 5, 15, 30, 40, 50, 60, 120, 240, and 480 min after injection (n = 5). (C) Radioanalysis of [18F]PFPMD biodistribution in blood samples at 10, 30, 60, 120, 240, and 480 min after injection (n = 3).
The radiation dosimetry in major organs was estimated (Table 1). The gallbladder showed the highest absorbed dose (65.3 ± 44.8 μGy/MBq). The effective dose was 14.3 ± 6.1 μSv/MBq. No significant changes in vital signs were recorded during the imaging period. Moreover, there were no abnormal results on laboratory examinations, and no adverse events were reported during the course of the study, indicating that the [18F]PFPMD tracer is safe for human PET imaging.
[18F]PFPMD PET Imaging of NSCLC and CRC Patients
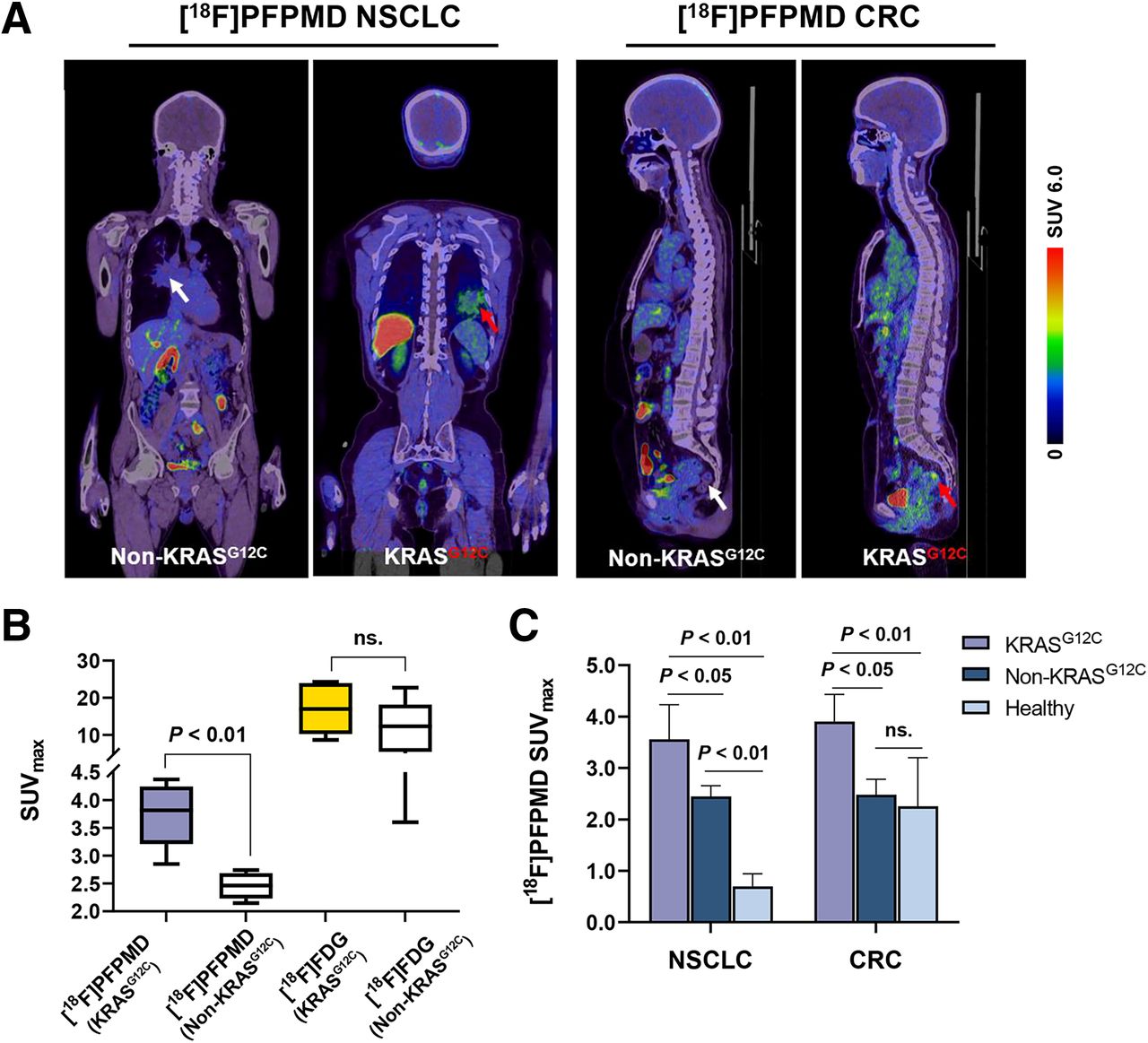
Eight NSCLC patients and 6 CRC patients underwent [18F]PFPMD and [18F]FDG PET/CT imaging (Fig. 4). The baseline characteristics of the patients are summarized in Table 2. The accumulation of [18F]PFPMD was significantly higher in KRASG12C mutation tumors than in non-KRASG12C mutation tumors (SUVmax: 3.73 ± 0.58 vs. 2.39 ± 0.22, +56.1%, P < 0.01, Figs. 5A and 5B). In contrast, although tumor uptake was higher for [18F]FDG than for [18F]PFPMD in all patients, no significant difference was found between the 2 groups (17.07 ± 6.97 vs. 11.70 ± 7.21, P = 0.25, Fig. 5B).

Study design for [18F]PFPMD PET/CT imaging of cancer patients. (A) Schematic diagram of patient enrollment. (B) Summary of finally involved patients for [18F]PFPMD PET/CT scan. ARMS = amplification-refractory mutation.
General Characteristics of Patients Who Underwent [18F]PFPMD and [18F]FDG PET/CT Imaging

[18F]PFPMD PET/CT imaging of NSCLC and CRC patients. (A) Representative [18F]PFPMD PET/CT images of non-KRASG12C and KRASG12C tumors in NSCLC and CRC patients, respectively. (B) SUVmax of [18F]PFPMD and [18F]FDG in non-KRASG12C and KRASG12C tumors, respectively (KRASG12C, n = 6; non-KRASG12C, n = 8). (C) [18F]PFPMD SUVmax of non-KRASG12C tumors, KRASG12C tumors, and healthy tissue in NSCLC patients, CRC patients, and healthy volunteers, respectively (NSCLC: KRASG12C, n = 3; non-KRASG12C, n = 5; healthy, n = 5) (CRC: KRASG12C, n = 3; non-KRASG12C, n = 3; healthy, n = 5). ns = not statistically significant.
Further subgroup analysis revealed that in NSCLC patients, [18F]PFPMD uptake in KRASG12C mutation tumors not only was significantly higher than in non-KRASG12C mutation tumors (3.56 ± 0.67 vs. 2.45 ± 0.21, P < 0.05, Fig. 5C) but also was significantly higher than in healthy lung tissue (3.56 ± 0.67 vs. 0.69 ± 0.25, P < 0.01, Fig. 5C). Similarly, in CRC patients, uptake was higher in KRASG12C mutation tumors than in non-KRASG12C mutation tumors (3.90 ± 0.53 vs. 2.48 ± 0.29, P < 0.05, Fig. 5C), but there was no significant difference between the non-KRASG12C group and the healthy-intestine group (2.48 ± 0.29 vs. 2.25 ± 0.95, P > 0.05, Fig. 5C). Collectively, these data support the potential of [18F]PFPMD PET imaging to distinguish KRASG12C mutations from non-KRAS mutations in NSCLC and CRC patients.
DISCUSSION
To the best of our knowledge, this was the first-in-humans study of noninvasive imaging of the KRAS mutation using a molecularly specific imaging tracer. Three principal findings were made. First, we found a method to synthesize a novel KRASG12C oncoprotein–targeted PET imaging tracer [18F]PFPMD and proved its targeting selectivity in vitro and in vivo. Second, we found that [18F]PFPMD is safe for human PET/CT imaging and is cleared primarily by the hepatobiliary system and the intestines. Third, we found that [18F]PFPMD showed a significantly higher accumulation in KRASG12C mutation tumors than in non-KRASG12C mutation tumors in NSCLC and CRC patients.
The potential of the SPECT tracer 131I-ARS-1620 inspired us to design another PET tracer based on a third-generation KRASG12C inhibitor, AMG510, which was recently reported to have much higher potency than ARS-1620 (1). To this end, by modifying the quinazolinone structure with a polyethylene glycol chain, we enabled 18F radiolabeling of AMG510. The modification and 18F labeling did not preclude penetration of and selective binding to the KRASG12C oncoprotein, as confirmed by the protein-binding study and the uptake, internalization, and blocking studies using cells and the mouse model. Previous attempts at KRAS-targeted tracer development were based on a messenger RNA antisense imaging method, which allowed for preclinical imaging of KRAS messenger RNA (the G12D mutation) in pancreas cancer xenografts using 64Cu- or 99mTc-radiolabeled DO3A-KRAS-peptide nucleic acid nanoparticles (18,19). However, these nanoparticles required that the tumor cell itself highly express insulinlike growth factor 1 receptor in order to be internalized and hybridized with KRAS messenger RNA, limiting the clinical relevance and translation of the findings. [18F]PFPMD is advantageous over peptide nucleic acid–based nanoparticles since it directly targets KRAS oncoprotein. Moreover, in contrast to previous nanoparticle tracers, the small and simple chemical structure of [18F]PFPMD makes it easier and safer for clinical translation.
Dynamic PET imaging of healthy volunteers revealed stable and low [18F]PFPMD uptake in the lungs, brain, and muscle, which may favor a higher contrast for distinguishing NSCLC tumors. Moreover, PET imaging also indicated that [18F]PFPMD was cleared predominantly by the hepatobiliary system and the intestines, as is consistent with a recent study on the pharmacokinetics of sotorasib. In that study, a 14C-isotopic labeling method was used, and 74.4% of the drug was detected in human fecal specimens (20). Although a radioisotope technique was used, individual properties of 14C rendered it unsuitable for in vivo PET imaging. Interestingly, lingering signal on the injected arm was observed in 2 individuals at 1 h after injection, possibly indicating dysfunction of a venous valve, since the distribution of the signal appeared to closely match the location of a venous valve. A similar phenomenon was noted in patients who had undergone [18F]FDG PET/CT imaging at our center (Supplemental Fig. 12).
No adverse events were observed during or after [18F]PFPMD PET imaging. The effective dose of [18F]PFPMD was 14.3 μSv/MBq, which was comparable to that of [18F]FDG (15.3 μSv/MBq) (21). After a single scan, with an average 240 MBq in an injected dose of [18F]PFPMD, the subject would be exposed to a radiation dose of about 3.44 mSv, which was far lower than the Food and Drug Administration criterion of 500 mSv/y. On the other hand, though [18F]PFPMD inhibits the downstream marker phospho-ERK, suggesting that the drug has an effect, the mass of the tracer (∼0.00975 mg) was considerably lower than the 960-mg treatment dose (22). Therefore, multiple rounds of scanning may not induce drug resistance. Overall, [18F]PFPMD can safely be used as a tracer in clinical PET imaging.
[18F]PFPMD PET imaging showed varied uptake in KRASG12C mutation tumors, and the SUVmax was significantly higher than in non-KRASG12C mutation tumors, according to our preliminary patient study. The uptake increase (KRASG12C vs. non-KRASG12C) was generally consistent between the patient (+56.1%) and animal (+59.1%) studies, indicating that [18F]PFPMD is highly selective toward the targeted KRASG12C mutation status. Consequently, the results of this study seem to indicate that [18F]PFPMD PET is a new molecular imaging tool of significant clinical relevance. In the next step, assessment of the SUVmax threshold based on a larger number of patients would be useful. Inclusion of a larger number of patients would allow assessment of the KRASG12C mutation status at diagnosis and during treatment. How the SUVmax might change after KRASG12C inhibitor therapy, and whether this change might serve as an accurate and instant guideline detailing more effective treatment, are other interesting questions that warrant further investigation. Moreover, PET tracers can also quantify the inter- and intraheterogeneity of therapeutic targets, such as PD-L1, EFGR, and αvβ3 (9,23,24). Thus, the heterogeneity of KRASG12C between primary and metastatic lesions may greatly increase the overall clinical relevance of the tracer in question. In addition, acquired drug resistance, along with its complex mechanism (i.e., secondary mutation, high-level amplification of KRASG12C, and reactivation of KRAS), is a common limitation of monotherapies (1,25). A recent clinical trial indicated that combination therapy with adagrasib (a KRASG12C inhibitor) and cetuximab (an EGFR-targeted monoclonal antibody) had a higher response rate, longer median response duration, and longer median progression-free survival than adagrasib monotherapy in CRC patients with mutant KRASG12C(26). As we observed in the in vitro studies, cell uptake was not blocked by gefitinib in the CRC cell line with the KRASG12C mutation, indicating that a combination therapy might not affect the PET signal.
Encouraged by the promising results of a phase 2 clinical trial with AMG510 to treat KRASG12C-mutation CRC patients, we look forward to seeing the performance of the tracer in CRC patients (16). The biodistribution of the tracer in healthy volunteers suggests that it needs a relatively long time to reach the rectum. The clearance mechanism of the tracer leaves a time window to observe rectal uptake. However, performing PET imaging on CRC patients other than those with rectal cancer—for example, patients with cancer of the transverse colon—may be difficult. Previous studies reported a significantly higher [18F]FDG uptake in KRAS-mutated CRC patients than in patients with wild-type CRC (27,28). This higher uptake would indicate that the KRAS mutation may be related to glucose metabolism due to the KRAS mutation–upregulated glucose transporter 1 and hypoxia-inducible factor 1α expression (29). In this study, we observed that [18F]FDG uptake was generally higher than [18F]PFPMD uptake in tumors, but no correlation was found between the two. Moreover, [18F]FDG uptake was higher in the KRASG12C group than in the non-KRASG12C group, but this difference was without significance. This discrepancy between previous reports and our finding was possibly due to the different groupings.
Chemical modification based on the existing structure of therapeutic drugs is a common way of developing tracers—fibroblast-activation-protein inhibitors and EGFR, for example. However, not every drug can easily be modified to a best imaging agent, although the use of [18F]PFPMD enabled tumor uptake and contrast similar to those of a previously distinguished EGFR mutation–targeted PET tracer, [18F]MPG, in NSCLC patients (24). A relatively high unspecific uptake was also observed in this study. This uptake could have been caused by the lipophilicity of the tracer and its binding to the cell membrane, as revealed by the cell internalization study. Moreover, since blood clearance of the tracer is relatively slow, the high radioactivity from blood may also be responsible for the unspecific signal in tumor. This can still be perceived as a limitation of the tracer and warrant further optimization to improve the imaging contrast. Moreover, although the results are encouraging, the sample size is not large enough. Thus, the performance of [18F]PFPMD PET in screening of the KRASG12C mutation status and heterogeneity, monitoring of therapeutic response, and determination of drug resistance did not yet provide solid scientific evidence and warrants further investigation.
CONCLUSION
[18F]PFPMD is a safe and promising PET tracer for clinically noninvasive imaging of the KRASG12C mutation status in NSCLC and CRC patients.
DISCLOSURE
This study was supported by National Natural Science Foundation of China grants 91959208, 92259304, 82122033, 81971646, 82202208, and 81871379. No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Can the KRASG12C mutation status be noninvasively imaged in NSCLC and CRC patients?
PERTINENT FINDINGS: We synthesized a PET imaging tracer, [18F]PFPMD, which is highly target-specific to the KRASG12C oncoprotein and suitable for in vivo PET imaging. First-in-humans PET imaging studies on healthy volunteers and patients revealed that [18F]PFPMD is safe in humans and can detect KRASG12C mutations in NSCLC and CRC patients.
IMPLICATIONS FOR PATIENT CARE: [18F]PFPMD is a safe and promising PET tracer for noninvasive screening of the KRASG12C mutation status in NSCLC and CRC patients.
Footnotes
↵* Contributed equally to this work.
Published online Oct. 12, 2023.
- © 2023 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication March 12, 2023.
- Revision received September 7, 2023.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}