


Visual Abstract

Abstract
T lymphocytes are key mediators of the adaptive immune response. Inappropriate or imbalanced T-cell responses are underlying factors in cancer progression, allergy, and other immune disorders. Monitoring the spatiotemporal dynamics of T cells and their functional status has the potential to provide unique biologic insights into health and disease. Noninvasive PET imaging represents an ideal whole-body modality for achieving this goal. With the appropriate PET imaging probes, T-cell dynamics can be monitored in vivo with high specificity and sensitivity. Herein, we provide a comprehensive overview of the applications of this state-of-the-art T-cell PET imaging toolbox and the potential it has to improve the clinical management of cancer immunotherapy and T-cell–driven diseases. We also discuss future directions and prospects for clinical translation.
- T-cell PET imaging
- graft-versus-host disease
- inflammatory bowel disease
- rheumatoid arthritis
- cancer immunotherapy
T lymphocytes play a central role in the adaptive immune response. The interplay between T cells and extracellular factors maintains a careful balance between activation, proliferation, survival, and inhibition. Dysregulation of T-cell responses can contribute to cancer progression, allergy, and immune disorders (1,2). Noninvasive PET imaging represents an ideal approach for visualizing T-cell dynamics in vivo, which could improve our understanding of their role in disease pathogenesis. This has motivated the development and evaluation of numerous T-cell imaging probes in both preclinical and clinical settings (Fig. 1). The T-cell PET imaging toolbox consists of a diverse set of approaches that include direct labeling of cells in vitro, proteins and peptides targeting endogenous T-cell surface and secreted biomarkers, small-molecule metabolic tracers, and engineering cells to express PET reporter genes (3). In direct cell labeling, immune cells are incubated with radiolabels ex vivo before adoptive transfer into a living subject for subsequent imaging. Although this is a relatively straightforward methodology for T-cell tracking, its wider clinical use to date has been limited. Incorporation of radionuclides can cause toxicities, such as radiolysis, and can adversely impact T-cell function. The radiolabel itself becomes diluted as cells divide and proliferate in vivo, reducing the utility of this approach for longitudinal imaging (4). Given the drawbacks of this approach, we will focus our review on the alternative approaches mentioned above, which are summarized in Table 1.
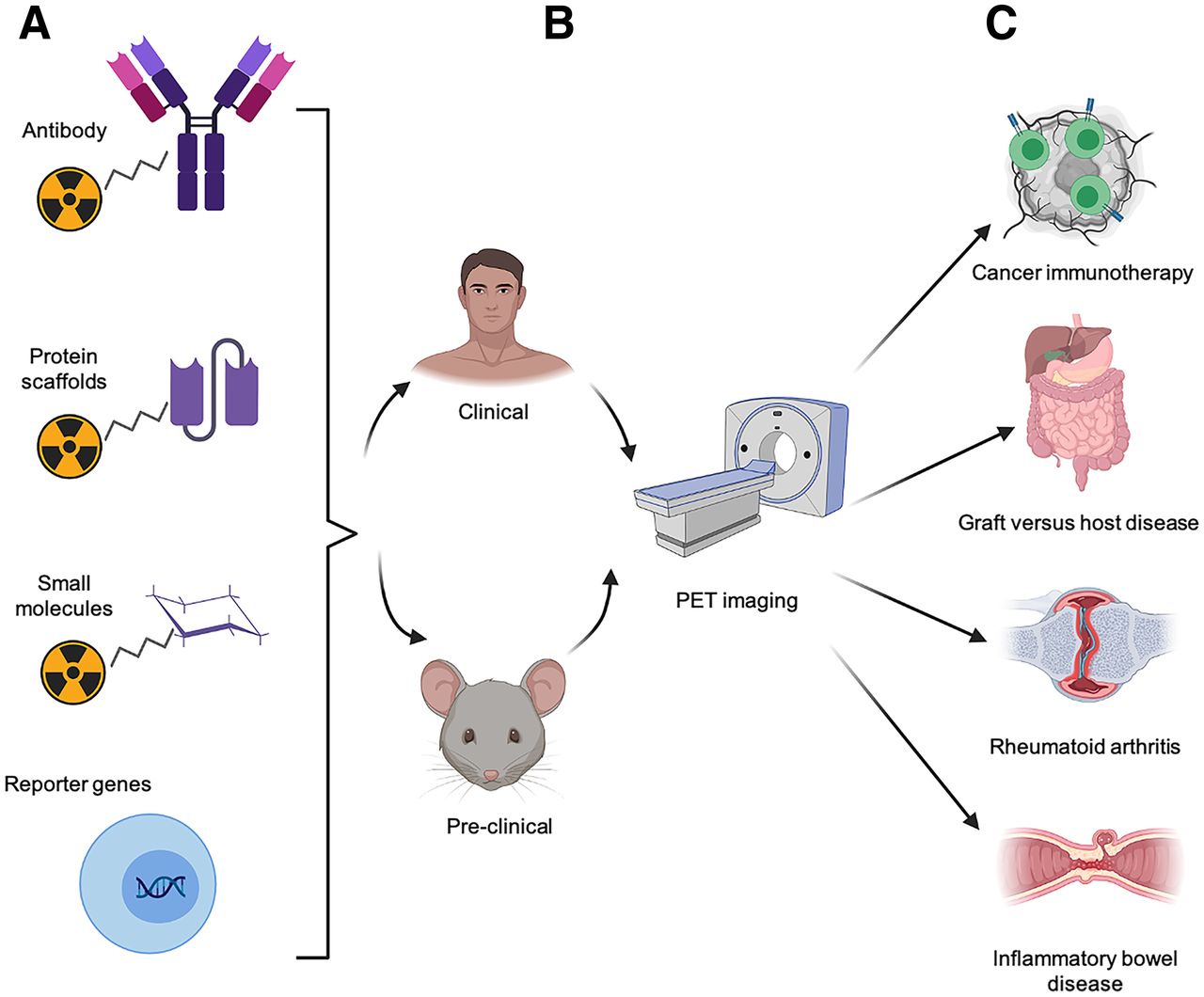
Approaches to PET imaging of T cells. T-cell PET imaging toolbox expanded rapidly over last decade. (A) Numerous T-cell–specific PET tracers have been developed, including radiolabeled antibodies and antibody fragments, protein scaffolds, small molecules, and tracers complementary to reporter genes that can be used to track engineered T cells. (B) These approaches have been evaluated in both preclinical and clinical studies. (C) Noninvasive PET imaging of T cells has potential to be highly useful, allowing preclinical researchers and clinicians to predict or monitor therapeutic response to cancer immunotherapy. This modality also enables early diagnosis of inflammatory diseases, such as GVHD, RA, and inflammatory bowel disease, for timely and effective intervention.
Candidate Biomarkers for PET Imaging of T-Cell Responses
UTILITY OF T-CELL PET IMAGING FOR DIAGNOSIS OF ALLOGENIC AND AUTOIMMUNE DISEASES
Inappropriate T-cell activation and trafficking are seen in a range of pathologies, including acute graft-versus-host disease (GVHD) in the allogenic transplant setting, and autoimmune diseases such as rheumatoid arthritis (RA) and inflammatory bowel disease. It is well documented that early diagnosis and timely therapeutic intervention in these scenarios can lead to better outcomes and minimize organ damage (5,6), illustrating the urgent need to develop more reliable diagnostic tools. T-cell PET imaging represents an ideal approach for achieving noninvasive, early detection of disease, before the onset of clinical symptoms.
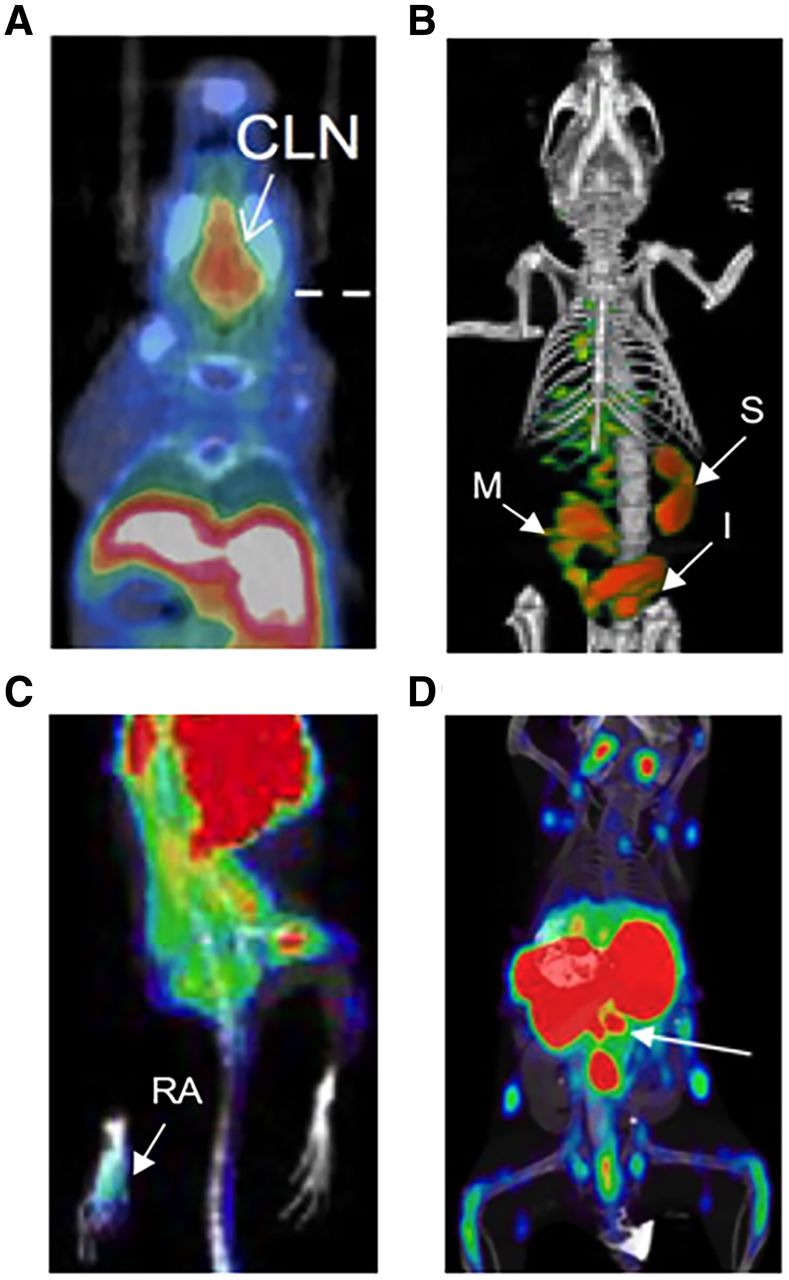
T-cell imaging with 2′-deoxy-2′-18F-fluoro-9-β-d-arabinofuranosylguanine (18F-AraG) has successfully detected activated T cells in secondary lymphoid organs at both early and late stages of disease in a murine acute-GVHD model (Fig. 2A) (7) and is now being evaluated clinically in hematopoietic stem cell transplant recipients (NCT03367962). Imaging of the T-cell surface lineage marker CD3 successfully visualized the total T-cell compartment in vivo in a humanized GVHD model (8). However, downregulation of CD3 expression during T-cell activation poses a limitation to this approach. A 64Cu-labeled OX40 monoclonal antibody demonstrated excellent diagnostic potential in a murine acute-GVHD model, detecting T-cell activation early in disease and before presentation of overt clinical symptoms (Fig. 2B) (9). However, the agonist OX40 monoclonal antibody clone used in this model also accelerated acute GVHD lethality, even when administered at the relatively low mass doses used for imaging. These findings highlight the importance of testing for biologic perturbations and overall safety before clinical translation. Given the utility of OX40 as a biomarker of GVHD, using an OX40 antagonist clone may be a safer imaging approach for GVHD and other indications for which T-cell activation may be deleterious.
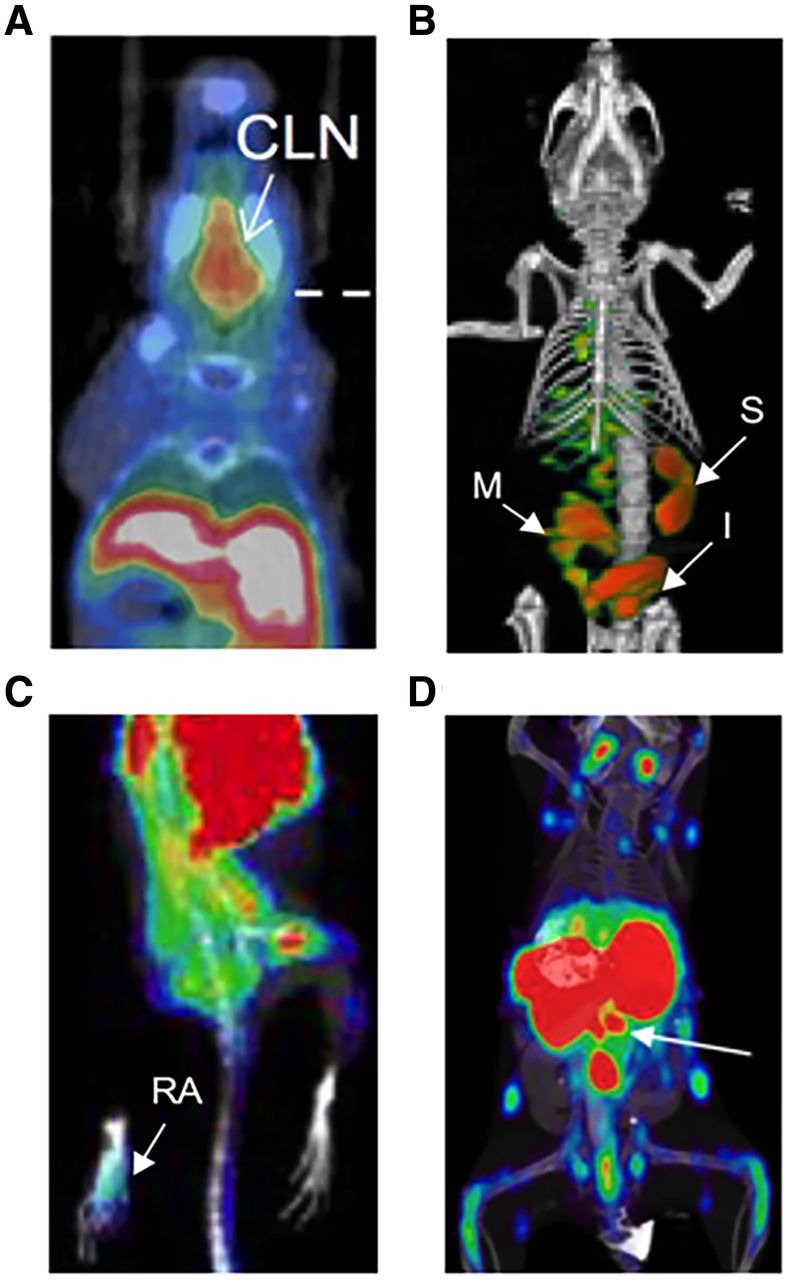
PET imaging of T cells for early detection of inflammatory diseases. (A) 18F-AraG enables detection of T-cell activation in cervical lymph nodes during acute GVHD (7). (B) OX40 immuno-PET allows early diagnosis of acute GVHD, before overt clinical symptoms (9). (C) 18F-AraG imaging detects RA in mouse model of adjuvant-induced arthritis (11). (D) 89Zr-labeled CD4-targeting cys-diabody allows detection of inflammatory bowel disease (white arrow indicating mesenteric lymph nodes) (13). CLN = cervical lymph nodes; I = intestine; M = mesenteric lymph node, S = spleen.
Imaging activated T cells also represents a promising strategy for early diagnosis of RA. Although 18F-FDG was tested in preclinical and clinical RA studies, the glucose metabolism pathway is shared by multiple tissues, which results in a high rate of false-positives, especially when the region of interest is adjacent to metabolically active tissues (10). Evaluation of 18F-AraG in a preclinical adjuvant-induced arthritis model (Fig. 2C) (11) showed significantly higher accumulation of 18F-AraG in RA-affected paws in both the acute and the chronic phases of disease, demonstrating its potential utility for RA diagnosis. Because of the enormous challenge of developing small-molecule binders, antibody-based PET tracers known as immuno-PET probes are favored as a reliable tool for immune cell imaging given their robust and facile radiolabeling protocols. Immuno-PET tracers targeting T-cell surface or secreted biomarkers should also warrant evaluation for RA detection in future studies (12).
For inflammatory bowel disease detection, an anti-CD4 monoclonal antibody labeled with 111In was previously evaluated for SPECT imaging of CD4-positive T cells. More recently, an 89Zr-labeled CD4-specific GK1.5 cys-diabody was developed and evaluated in a mouse model of dextran sulfate sodium–induced colitis (Fig. 2D) (13). Increased accumulation of 89Zr-maleimide-deferoxamine–GK1.5 cDb was detected in the distal colon of colitis mice and further corroborated by increased CD4-positive immunohistochemistry staining, demonstrating the sensitivity of this probe for CD4-positive T cells in vivo. Whether PET quantification correlated with disease severity was not discussed in this study; further evaluation is therefore required to explore the feasibility of this approach for determining disease severity. Additionally, naïve CD4-positive T cells residing in the gut may raise the background signal in CD4-targeted imaging. Since activated T cells are the true mediators of inflammatory bowel disease pathogenesis, PET tracers specific to activated T cells may be preferred.
T-CELL PET IMAGING FOR PREDICTING AND MONITORING IMMUNE RESPONSE TO CANCER IMMUNOTHERAPIES
Immunooncology has rapidly evolved over the last decade (14), with numerous clinical trials demonstrating the unprecedented success of cancer immunotherapies in treating late-stage and recurrent malignancies, including relapsed or refractory B-cell malignancies, melanoma, bladder cancer, and non–small cell lung carcinoma (15–18). Despite the meteoric rise of these innovative therapies, only a small fraction of patients exhibits durable responses, highlighting the urgent need for reliable tools to monitor and predict therapeutic response (19). Anatomic imaging often lacks sensitivity and does not collect direct molecular or functional information about the T-cell compartment. Biopsies are invasive, risk iatrogenic complications, fail to capture whole-body information, and do not assess tumor heterogeneity (20,21). Longitudinal PET imaging and quantitative analysis of T-cell distribution in tumors or secondary lymphoid organs before and after treatment may correlate with therapeutic response, helping clinicians to identify patients most likely to benefit from treatment (22). With this motivation, a wide range of T-cell–specific PET tracers has been developed and evaluated in preclinical studies and clinical trials.
Endogenous T-cell biomarkers include those specifically expressed on the cell surface, such as T-cell lineage markers (CD4, CD8); those secreted by certain T-cell subsets, such as granzyme B; and activation markers, such as CD278 and CD134 (Figs. 3A and 3B). A major advantage of using immuno-PET probes is their high specificity and affinity toward their homologous target, generating high signal-to-noise ratios and high-contrast images. Neutral binders without agonistic and antagonistic functions have the potential to be widely applicable without concern about perturbative effects. However, because of their large size, intact antibodies (150 kDa) can suffer from poor penetration into target tissues and slow clearance. Additionally, the crystallizable fragment regions of whole antibodies can bind nonspecifically to crystallizable fragment receptors on other cells, such as macrophages and natural killer cells (23). To overcome these challenges, antibody fragments such as the minibody, diabody, and single-domain antibody formats and other engineered protein scaffolds have been evaluated as radiotracers (24,25). These smaller vectors are likely to exhibit better tissue penetration and faster clearance and are more suited to radiolabeling with PET isotopes with shorter half-lives, making same-day imaging more feasible (26).
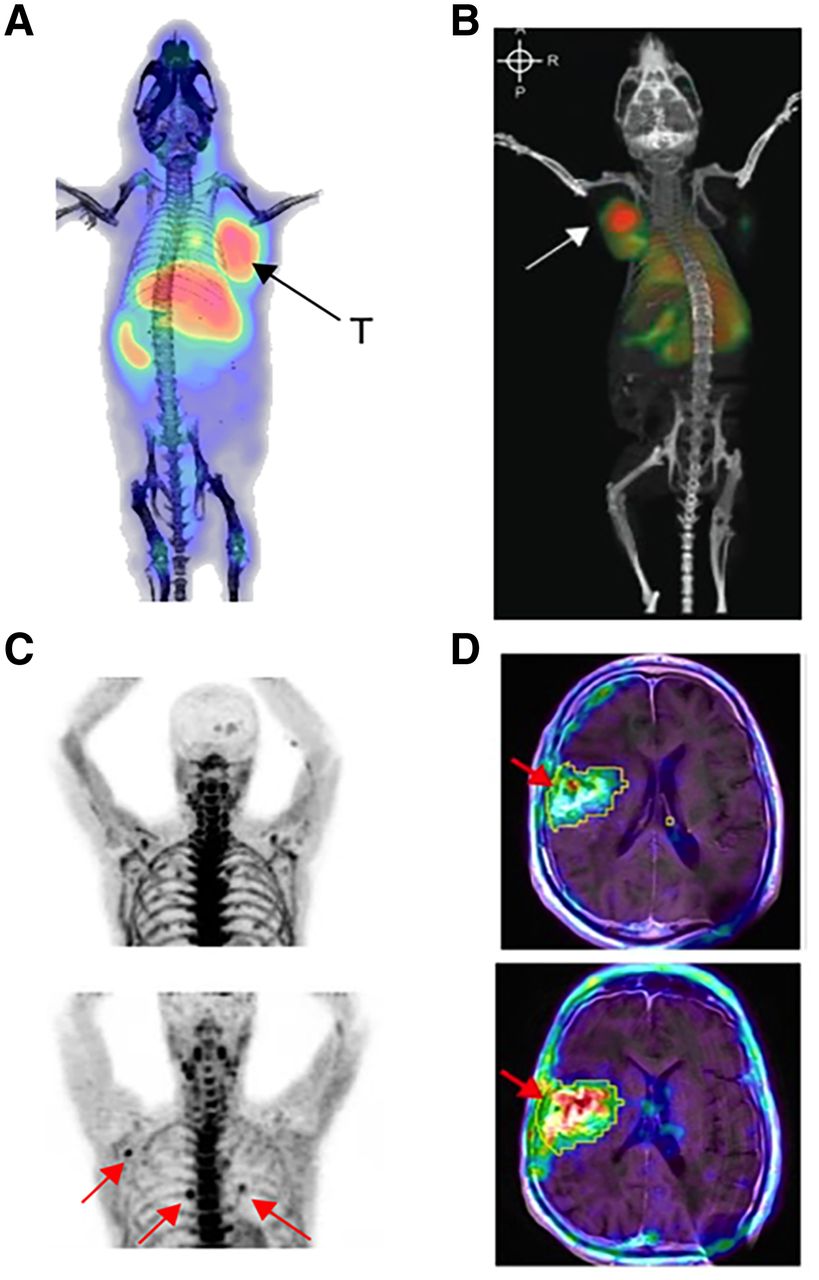
PET imaging of T cells enables monitoring of treatment response in cancer immunotherapy. (A) ICOS immuno-PET with 89Zr-DFO-ICOS monoclonal antibody visualizes and predicts therapeutic response in mouse model of Lewis lung cancer treated intratumorally with STING agonist and PD-1 blockade (37). (B) OX40 immuno-PET imaging enables visualization of activated T cells in A20 tumor–bearing mouse treated intratumorally with CpG (arrow represents CpG-treated tumor) (44). (C) Elevated uptake of 2-chloro-2′-deoxy-2′-18F-fluoro-9-b-d-arabinofuranosyl-adenine was observed in several lymph nodes (arrows) of recurrent glioblastoma patient after treatment with tumor lysate–pulsed DCVax and PD-1 blockade (bottom), compared with before treatment (top) (29). (D) 18F-FHBG enables visualization of HSV-TK1 reporter gene–modified interleukin-13 CAR-T cells (arrows) in recurrent glioblastoma patients (top, before CAR-T infusion; bottom, after CAR-T infusion) (32). T = treated tumor.
Radiolabeled small molecules are usually designed to target specific intracellular metabolic pathways, such as carbohydrate metabolism and DNA synthesis. The most widely used clinical PET tracer for cancer diagnosis and staging, 18F-FDG, has also been explored for monitoring immune responses (27). Since the glycolytic pathway is shared by both activated immune cells and cancer cells, its specificity for the interrogation of T-cell responses is low. Rapidly proliferating T cells rely heavily on the nucleoside salvage pathway for DNA synthesis. Deoxycytidine kinase and deoxyguanosine kinase are key enzymes that regulate this pathway, motivating the development of radiolabeled small molecules targeting each. The first deoxycytidine kinase-targeting PET tracer, 1-2′-deoxy-2′-18F-fluoroarabinofuranosyl cytosine, was able to distinguish proliferating CD8-positive T cells from naïve T cells (28), but rapid catabolism in vivo impeded its clinical translation. Another deoxycytidine kinase tracer with improved metabolic stability, 2-chloro-2′-deoxy-2′-18F-fluoro-9-b-d-arabinofuranosyl-adenine, was subsequently developed and evaluated in glioblastoma patients treated with PD-1 blockade. Increased accumulation of this tracer was observed in secondary lymphoid organs and tumor tissue, and the PET signal in the latter strongly correlated with the concentration of tumor-infiltrating lymphocytes (Fig. 3C) (29). 18F-AraG, a guanosine analog with high specificity for deoxyguanosine kinase, was successfully used to detect activated T cells induced by anti-PD-1 therapy in mouse models, enabling early prediction of therapeutic response (30). Given its favorable imaging characteristics, several clinical trials are currently evaluating the feasibility of monitoring T-cell responses with 18F-AraG (NCT04186988, NCT04726215).
Engineering cells to express reporter genes encoding proteins that can be detected with complementary PET tracers is a promising approach suited to tracking adoptively transferred T cells (31). The most extensively evaluated PET reporter gene is the herpes simplex virus type 1 thymidine kinase (HSV1-tk) and its mutant version HSV1-sr39tk. In a pilot clinical study, CD8-positive cytotoxic T lymphocytes were engineered to express both HSV1-tk and interleukin-13 zetakine chimeric antigen receptor (CAR) and then adoptively transferred into recurrent high-grade glioblastoma patients. 18F-FHBG was subsequently administered to monitor the trafficking and proliferation of the engineered cytotoxic T lymphocytes in the brain (Fig. 3D) (32). This landmark study demonstrated the feasibility of using the HSV1-tk system to clinically track engineered immune cells. The HSV1-sr39tk system can also act as a suicide gene on treatment with the prodrug ganciclovir, enabling ablation of CAR-T cells for safe control of potential toxicities (33). Human PET reporter genes including sodium iodide symporter, norepinephrine transporter, and somatostatin receptor 2 have also been developed to overcome the potential immunogenicity of HSV1-tk and evaluated for CAR-T-cell imaging. However, because of their endogenous expression, as well as the internalization of hNET and somatostatin receptor 2, their application has been limited (34). Recently, a highly promising prostate-specific membrane antigen/18F-DCFPyL reporter system was evaluated for CD19-positive CAR-T-cell imaging (35). To specifically prevent the internalization of prostate-specific membrane antigen, human CD19-targeted CAR-T cells were transduced with an N-terminally modified variant, tPSMA(N9del). 18F-DCFPyL, a prostate-specific membrane antigen–specific PET tracer, subsequently allowed detection of these cells with high sensitivity and specificity in vivo. Importantly, quantification of the 18F-DCFPyL PET signal from CD19-positive CAR-T cells in these tumors led to more accurate assessment of therapeutic response than that from measurement of CAR-T cells in peripheral blood.
CONCLUDING REMARKS
Here, we have reviewed state-of-the-art PET imaging approaches for in vivo T-cell visualization and their applications in T-cell–mediated diseases and cancer immunotherapy. Noninvasive PET imaging has the potential to provide comprehensive information about the distribution and abundance of immune cell subsets in real time (36,37). With these imaging approaches, physicians and research scientists can be better informed about the adaptive immune response and T-cell behaviors in different settings, which would greatly facilitate personalized medicine.
For novel T-cell PET tracers to translate from bench to bedside, lessons learned in the clinic should be brought back to the bench to facilitate probe optimization. Although many T-cell–targeted PET tracers have yielded promising preclinical results, only a few candidates are currently under clinical investigation, and they have yet to be approved for routine clinical use. This imbalance may be attributed to redundant preclinical studies. To streamline PET tracer development, unsupervised analysis of prospective biomarkers using both preclinical and clinical “omic” datasets would help identify the optimal target for a given application. In a recent study, we used RNAseq to compare several candidate T-cell activation markers expressed on activated CD19-positive human CAR-T cells, and inducible costimulator was identified as a promising imaging biomarker that was specifically and consistently upregulated; subsequent PET imaging was focused accordingly (38). In the future, advanced techniques such as Slide-seq and CODEX (CO-Detection by indEXing) should be incorporated to identify the most relevant biomarker for T-cell imaging in a given scenario (39,40). Another reason for the imbalance is the slow adoption of novel molecular imaging techniques in the clinic. In conventional clinical workflow, anatomic imaging remains the gold standard for therapeutic response evaluation and monitoring; novel immunotherapy adjuvants are also approved by the Food and Drug Administration under certain criteria. Future clinical studies need to demonstrate that these PET approaches provide actionable insights for improved patient management. In a recent landmark study, PD-L1 imaging in cancer patients indicated that noninvasive immuno-PET outperformed RNA and immunohistochemistry-based biomarker measurements from tissue biopsies in the selection of patients most likely to benefit from PD-L1 blockade (41). These are compelling reasons to accelerate the clinical evaluation of new probes and the selection of the most promising candidates for further consideration.
To optimize the T-cell PET imaging toolbox, we should carefully compare different biomarkers, pathways, and probe formats. For example, intact antibodies exhibit higher binding affinity than small molecules but lower tissue penetration. Although the reporter gene strategy has the advantage of low background, immunogenicity usually limits its wider applications. Thus, among all the potential candidates, we should consider their affinity, specificity, sensitivity, immunogenicity, tissue penetration, and clearance and then select the best candidate for further investigation. Moreover, with current imaging probes, although we can visualize the in vivo distribution of distinct T-cell populations, we are unable to delineate their antigen specificity. A recent published study reported a novel engineered PET imaging probe—64Cu-labeled synTac (synapse for T-cell activation)—which was able to distinguish antigen-specific CD8-positive T cells from bystander CD8-positive T cells (42). This powerful approach could allow the detection of cytotoxic CD8-positive T cells specific for invading pathogens or tumor cells, which would provide more precise assessment of cancer immunotherapy efficacy or disease severity. Finally, the groundbreaking total-body PET will substantially improve sensitivity, enabling enhanced detection of T cells while also lowering the radioactive dose typically required to acquire high-resolution images, allowing for safer repeat imaging (43).
In summary, the T-cell PET imaging toolbox has great potential for improving clinical management of cancer immunotherapy and diagnosis of T-cell–driven immunopathology. Further optimization of these approaches is still needed to overcome limitations in their specificity, sensitivity, and safety. We hope that our discussion will highlight the utility of the T-cell PET imaging toolbox to both researchers and physicians and encourage the translation and wider adoption of these tools in clinical practice.
DISCLOSURE
This work was supported by Heilongjiang Postdoctoral funding (LBH-Z18197). Israt Alam was supported by NIH/NCI (R01 CA201719-05). No other potential conflict of interest relevant to this article was reported.
ACKNOWLEDGMENTS
We thank our colleagues in the fields of molecular imaging, immunology, and oncology, among others, for their contributions toward the development of T-cell PET imaging strategies and probes discussed in this review.
Footnotes
Published online December 09, 2021.
- © 2022 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication July 16, 2021.
- Revision received December 6, 2021.





{kind=link}
{kind=link}
{kind=link}
{kind=link}