


Abstract
Radiopharmaceutical therapy is an emerging treatment modality that has demonstrated increasing importance as a significant component in the treatment of cancer. Prostate cancer (PCa) remains one of the commonest solid-organ tumors and is associated with significant societal burdens. Despite significant disease heterogeneity, PCa remains an ideal candidate for radiopharmaceutical therapy because of the prolonged disease course, metastatic disease tropism, and sensitivity to radiation therapy. To date, advanced PCa remains one of the most successful arenas for the development and approval of radiopharmaceutical agents. In this review, we aim to summarize the complex processes required to obtain regulatory approval for a novel agent and highlight the limitations and hurdles specific to the approval of radiopharmaceutical agents. In advanced PCa, we outline the importance of a framework for trial design with respect to defining disease state and acceptable outcome measures—as recommended by the Prostate Cancer Clinical Trials Working Group (PCWG). Finally, using the principles mandated by the Food and Drug Administration approval process and the framework provided by the PCWG, we outline experience with the successful approval of the radiopharmaceutical agents 223Ra and 177Lu-PSMA-617.
Broadly speaking, radiopharmaceutical therapy is defined by delivery of radioactive agents to tumor-associated targets (1). Radiopharmaceutical agents comprise both a targeting ligand and a radioactive payload. The targeting ligand facilitates delivery of the therapeutic radionuclide to the tumor microenvironment or directly to the tumor cells. The payload in radiopharmaceuticals is a radioactive nuclide, primarily either α- or β-emitting. Exceptions to this simplistic structural overview of radiopharmaceuticals exist, such as 131I and 223Ra, for which the chemical properties of the compound act to both target and emit therapeutic radiation.
Cellular injury occurs as a result of emitted α- or β-particles via radiation-induced DNA damage (2). β-particle emitters, such as 177Lu, 90Y, and 131I, provide short-range emissions (0.5 to >10 mm) and produce significant cellular injury to nearby cells (3). α-particle emitters, such as 212Bi and 223Ra, produce emissions that travel shorter distances than β-emitters but provide more energy deposition per unit length, resulting in an increased potency and thus frequently causing irreparable DNA damage (4). Unlike traditional radiotherapy approaches, radiopharmaceuticals are typically delivered systemically, which allows the potential for radioactivity exposure to disseminated metastatic disease (1). Further, the ability to target tumor-associated matter allows the radioactivity to be exposed with increasing specificity, preserving physiologic tissue. These tissues targeted by radiopharmaceuticals are typically either malignant cells or cells within the tumor microenvironment, such as osteoclasts and osteoblasts in 223Ra (5). Some radiopharmaceutical agents may accumulate or concentrate in physiologic regions, such as the renal cortex or salivary glands (6); specific morbidity may occur in these organs as a result of radioactivity exposure.
The role of radiopharmaceuticals in oncology is rapidly expanding. A dramatic increase in the number of radiopharmaceuticals approved by the U.S. Food and Drug Administration (FDA) has occurred since 2009, due in part to the release of clear guidelines for FDA approval of radiopharmaceuticals (7). The positive public perception and popularity of radiopharmaceuticals may be attributed to positive trial outcomes that demonstrated improvements in the way patients feel, function, and survive. Additionally, the availability of companion diagnostic imaging with radiopharmaceuticals and visualization and estimation of radiopharmaceutical biodistribution and response provides an attractive feature for patients and clinicians (8). For example, companion imaging for 223Ra-dichloride is 99Tc-bisphosphonate bone scanning, and for 177Lu-PSMA-617, companion imaging is 68Ga-PSMA-11 or 18F-PSMA-DCFPyl PET. From a financial perspective, large pharmaceutical firms are increasingly investing in such technologies (9). Market projections for prostate cancer (PCa) therapies suggest that annual sales of prostate-specific membrane antigen (PSMA)–targeted radiopharmaceuticals could exceed $1.1 billion (10).
Bringing any drug to market requires time, strategy, financial resources, data, and adherence to regulatory guidance. It is an intrinsically complex process, and most drugs in clinical trials will not achieve this goal. In this review, we highlight PCa as a case study in bringing radiopharmaceuticals to FDA approval. PCa is among the common malignancies in the developed work and is responsible for considerable public burden in contemporary clinical practice. PCa has a widely heterogeneous disease course, with a proportion of patients progressing to advanced, disseminated disease. Patients are generally elderly, with diverse comorbidities and medical risks. The disease is bone-tropic, and anticancer responses are therefore difficult to assess as endpoints in early clinical trials. Despite these challenges, significant advances in therapeutic strategies that leverage the biology of the disease, and clinically qualifying interim endpoints in clinical trials, have resulted in new tools exerting long-term disease control even in the metastatic state. These treatments for metastatic disease include chemotherapy (11,12) and therapies that target the androgen receptor (13) and DNA repair (14). These principles, in addition to the radiosensitivity of PCa, make PCa an ideal disease to assess radiopharmaceutical therapy. Recently 2 radiopharmaceuticals have been approved for PCa, making it one of the most successful arenas for developing radiopharmaceuticals.
Given that the role of these agents is projected to expand, it is imperative that clinicians and investigators, particularly those involved in nuclear medicine, are aware of the pathways and hurdles required to obtain FDA approval for novel radiopharmaceuticals. The current review aims to provide an educational overview of the process for acquiring FDA approval for a novel radiopharmaceutical for PCa. We highlight the potential barriers and pitfalls to this process and outline several cases of successful approval of agents for advanced PCa.
OVERVIEW OF THE FDA APPROVAL PROCESS
In the United States, regulation of the development, production, and sale of novel pharmaceuticals and devices, including radiopharmaceuticals, is governed by the FDA. After the federal Food, Drug, and Cosmetics Act of 1938 (15), all drugs in the United States required approval for safety by the FDA. Multiple subsequent iterations in 1962 and 1976 broadened the regulation to include devices; further, approval required demonstration of efficacy in addition to safety (16). Over the past decades, the approval process has become increasingly complex; presently, the FDA approval process is among the most comprehensive and stringent regulatory processes worldwide (Fig. 1). Although such complex processes ensure public safety, the current timeline from initial conception of molecular agents to regulatory approval by the FDA may take up to 10–15 y and cost up to billions of dollars (17).

Overview of FDA process for new drug approval (19).
IND Application
Although the conceptualization and preclinical phases of pharmaceutical agents are not covered in the scope of this review, these stages are fundamental in the progression to clinical-stage trials and subsequent FDA approval. When the intended agent is ready to progress to human clinical trials, initial contact with the FDA should be sought. At this time, the drug’s sponsor applies for a commercial investigational-new-drug (IND) application with the FDA. IND applications to the FDA require disclosure of available preclinical data and human data, manufacturing information, and the protocols of the intended studies (18). The preclinical and human data relevant for the IND application may include pharmacologic profile, toxicity, and efficacy data for the respective disease process. Manufacturing information may include composition profile, manufacturer methodology and controls, and compound stability.
Clinical Trials
As part of IND approval, ideally an early-stage trial design is submitted, with the objective of achieving the specific goals required for FDA approval—demonstration of safety and efficacy. Defining efficacy is imperative, as FDA applications will be rejected because of “a lack of substantial evidence that the drug will have the effect it purports or is represented to have” (15). Broadly speaking, efficacy may be established if the drug or device provides a positive and clinically meaningful effect on how an individual feels, functions, or survives (19). Feel and function may be measured by patient-reported outcomes, clinically reported outcomes, observer-reported outcomes, performance outcomes, or biomarker data. Data relevant to these questions are acquired by methodically designed and conducted phase I, II, and III trials. After each trial is completed, data are submitted to the FDA before subsequent trial phases proceed.
Phase I trials are typically low-volume studies on healthy individuals, aimed to evaluate treatment safety, acceptable dosing strategies, and adverse effects (20). However, completion of phase I trials in the realm of radiopharmaceuticals is not mandatory, and a phase II dose might be advanced despite the absence of a formal phase I study (21). For example, 177Lu-PSMA-617 did not complete formal phase I trials before progressing to phase II trials. This was possible because of the publication of safety and tolerability in the form of retrospective series from international centers where regulations for novel radiopharmaceuticals are less restrictive (22).
Phase II trials are moderate-sized trials on individuals with the target disease and aim to determine efficacy and provide additional safety data. Phase III trials are large trials to discern treatment efficacy, monitor adverse effects, and compare with current treatment regimens. Historically, 2 adequate and well-controlled clinical studies that are designed “to distinguish the effect of a drug from the other influences, such as spontaneous change, placebo effect or biased observation” are required for FDA approval (23). However, the FDA Modernization Act in 1997 provided flexibility so that based on relevant science, data from a single well-controlled clinical investigation and confirmatory evidence are sufficient to establish efficacy.
New-Drug Application (NDA)
After completion of the phase III trials, an NDA may be submitted to the FDA. The NDA application requires all data pertaining to the drug, including manufacturing, quality control, chemical properties (formula, pharmacokinetics), indications, and data obtained from the phase I, II, and III clinical trials. Expedited reviews may be requested at an early point in the regulatory process and include “fast track,” “breakthrough therapy,” or “accelerated approval” (24).
ISSUES WITH DEVELOPMENT OF RADIOPHARMACEUTICALS
Despite the well-defined FDA approval process via IND and NDA applications, several specific challenges exist in fostering the progression of novel radiopharmaceuticals (25). Logistically, development of radiopharmaceuticals is complex because of the broad, multidisciplinary approach required to recruit and treat patients. Specifically, the target population of patients intended for treatment with radiopharmaceuticals is those with disseminated disease, often managed by medical oncologists. However, radiopharmaceuticals and other radioactive therapies are typically administered and managed by clinicians in the nuclear medicine and radiology fields.
Radiopharmaceutical use can be more complex than systemic therapies such as immunotherapy or cytotoxic chemotherapeutic agents. Within the United States, the Nuclear Regulatory Commission maintains stringent training and education requirements for physicians to be authorized users of radiopharmaceuticals for medical purposes. The formulation location of the radiopharmaceutical compound must be considered, keeping in mind that many agents will have a limited half-life that requires timely administration. Such formulation, dispensing, and disposal of radiopharmaceuticals require specific expertise by radiochemists or radiopharmacists, of whom there is a recognized shortage (26). Radiation safety before, during, and after treatment must be considered, as well as the availability of qualified personnel to ensure staff, patient, and caregiver safety. Associated set-up costs for radiopharmaceuticals and associated imaging are considerable (27).
Finally, as with all novel treatments, production of scientific data is critical in development and eventual clinical application. Development of such agents requires motivated investigators because of the regulatory and logistic barriers and the significant periods from benchtop preclinical research to clinical trials. In some regions of the world, there may be limited regulatory oversight of drugs, and the resulting ease of clinical access for new treatments provides no impetus to perform clinical trials and contribute data to the medical literature.
ISSUES WITH DRUG APPROVALS IN PCa
PCa treatment options have expanded, with increasing complexity in various stages of PCa. Given the variety of therapeutic approaches and lengths of treatment, the risk a patient faces at a certain time point varies widely. For example, in metastatic PCa, patients are typically treated with a combination of androgen deprivation therapy, androgen receptor inhibitors, or cytotoxic chemotherapy (docetaxel) (11). Accordingly, a patient’s risk may vary on the basis of prior therapies or non–disease-related factors, such as risk of death from competing causes. For example, after extended periods of androgen deprivation therapy with or without androgen receptor inhibitors or chemotherapy, PCa progression in the setting of continued castration denotes castration-resistant PCa (CRPC) (28). Contextualizing the various disease settings of metastatic PCa is critical in exploring the role of various novel radiopharmaceuticals under investigation or development.
Development of novel radiopharmaceuticals in PCa poses specific issues relevant to the FDA approval process. For several reasons, there are barriers for novel agents proposing to improve survival outcomes in PCa. PCa is a heterogeneous disease that may compromise survival in some patients but demonstrate a more indolent course in others. Further, PCa patients are an older population, typically with comorbidities that may compete with PCa. Additionally, the propensity for bone metastatic deposits is problematic, given the difficulty in determining treatment response based on RECIST or other standard response criteria. PCa also lacks a biomarker that is accepted by the FDA as an indicator of clinical benefit. Neither prostate-specific membrane antigen (PSMA) nor any other serum biomarker has been shown to be a surrogate for clinical benefit; neither of these is recognized by regulatory agencies as being clinically qualified as an endpoint for drug approval (29,30). Finally, long PCa survival times and the lack of an accepted intermediate endpoint lead to trial designs requiring prolonged periods of follow-up (29).
To assist in providing consensus and meeting these clinical trial design challenges, the Prostate Cancer Clinical Trials Working Group (PCWG) initially issued recommendations to standardize outcomes in metastatic PCa trials. The PCWG provides a framework to organize clinical trial design by prior treatments, disease distribution and extent, risk of death from PCa or competing causes, and primary outcome measures. The first iteration of these recommendations, PCWG1, was published in 1999 and outlined recommendations for phase II clinical trials on CRPC (31). Two subsequent iterations have been published to broaden the recommendations for clinical trials in PCa: PCWG2 in 2008 (29) and PCWG3 in 2016 (32). PCWG2 and PCWG3 furnished a framework for regulatory drug approval that outlined the clinical course of PCa in a series of clinical states. They also provided recommendations on standardized eligibility criteria, assessment intervals, and endpoints for clinical trials.
Given the prolonged nature of overall survival and the lack of measurable disease in bone, PCWG2 and PCWG3 noted the need for a intermediate-progression endpoint, rather than a treatment-response surrogate endpoint. Accordingly, radiographic progression-free survival (rPFS) was identified in PCWG2 and PCWG3 as a potential surrogate for overall survival. Clear, objective definitions for rPFS were proposed in PCWG2, denoted by the “2 + 2” rule: at least 2 new lesions on the first posttreatment scan with at least 2 additional lesions on the next scan. After the flare period, progression was defined as 2 new lesions relative to the first posttreatment scan, confirmed on a subsequent scan. Use of the 2 + 2 rule is beneficial as it does not require specialized software, it is generalizable, it is not time-consuming, and only 2 lesions are required to be counted. Additionally, this definition compensates for posttreatment flare that may be observed in bone scintigraphy (33). rPFS was subsequently credentialed in several trials, including the COUGAR-302 and PREVAIL trials (13). rPFS was shown to be closely associated with overall survival, with an estimated correlation coefficient of 0.72 (13,34).
PATHWAYS TO APPROVAL FOR 223RA-DICHLORIDE AND 177LU-PSMA-617
Despite the barriers to FDA approval of radiopharmaceuticals, there have been several recent success stories. Early radiopharmaceutical approval was successfully obtained for palliation of painful bone metastases via 89Sr-chloride (35) and 153Sm-ethylenediamine tetra(methylene phosphonic acid) (36). Unlike these older bone-seeking radiopharmaceuticals that were tested in studies designed to demonstrate relief of pain, the more recent radiopharmaceuticals 223Ra and 177Lu-PSMA617 achieved FDA approval in advanced PCa on the basis of prolonging overall survival. As outlined below, approval of these agents was achieved by adherence to the principles outlined in the PCWG framework and compliance with FDA regulatory requirements.
223Ra
223Ra is an α-emitting radionuclide, and the radiopharmaceutical 223Ra-dichloride is used to treat skeletal metastases and is in commercial use under the trade name Xofigo (Bayer). PCa bone metastases are characterized by dysregulated bone metabolism, are mediated by tumor-associated growth factors, and result in an abundance of new disorganized bone formation (37). 223Ra is an earth alkali metal that is substituted for calcium in hydroxyapatite within areas of osteoblast-mediated new bone formation (38). Accordingly, for PCa, 223Ra targets the bone microenvironment rather than metastatic tumor cells themselves. The resulting local α-emission causes DNA double-strand breaks in adjacent tumor cells and in osteoblasts and osteoclasts (39). This mode of targeting and cell injury is advantageous because the treatment effect is directed to a metastatic disease compartment rather than cell-by-cell.
223Ra-dichloride was among the first radiopharmaceuticals achieving FDA approval in May 2013 for advanced PCa on the basis of prolonging overall survival (40). Data used to support the IND and NDA applications originated from early phase I and II trials. Phase I trials assessed 15 patients with PCa and bone metastases who received increasing doses of 223Ra-dichloride, starting at 46 kBq/kg and then increasing to 93, 163, 213, and 250 kBq/kg (41). With this, safety and tolerability were demonstrated, although dose-limiting toxicity and maximal tolerated dose were not formally identified. Encouragingly, most patients reported pain palliation by 8 wk; on the basis of these promising results, subsequent trials proceeded. Phase II trials on patients with CRPC and bone metastases demonstrated acceptable safety and tolerability (42). Nilsson et al. (42) enrolled 64 patients to receive either 223Ra-dichloride (50 kBq/kg every 4 wk) or placebo, with a primary endpoint of total alkaline phosphatase and time to skeleton-related events. This trial reported a significant response to alkaline phosphatase (−65.6% vs. 9.3%, P < 0.0001) and delayed skeleton-related events (hazard ratio [HR], 1.75; 95% CI, 0.96–3.19; P = 0.065) in the 223Ra-dichloride group. A difference in overall survival was also reported in the 223Ra-dichloride group (HR, 2.12; 95% CI, 1.13–3.98; P = 0.020).
Efficacy was further demonstrated in the subsequent phase III ALSYMPCA trial (43), which recruited 922 metastatic PCa patients with 2 or more bone metastases on skeletal scintigraphy and no evidence of visceral metastases. Patients were randomized 2:1 to receive the standard of care either with or without 223Ra-dichloride (at a dose of 50 kBq/kg of body weight). The primary endpoint was overall survival, powered to detect an HR of 0.76 for the risk of death in the 223Ra group. At a preplanned interim analysis, the trial reached its primary endpoint, with patients who received 223Ra-dichloride demonstrating an improved overall survival (14.0 vs. 11.2 mo in the placebo arm; HR, 0.699; P = 0.002). Secondary outcomes were observed, including a reduction in alkaline phosphatase (P < 0.001), a delayed time to an increase in alkaline phosphatase (6.4 vs. 3.8 mo; HR, 0.17; P < 0.001), and a delayed time to an increase in prostate-specific antigen (PSA) (3.6 vs. 3.4 mo; HR. 0.64; P < 0.001) in the 223Ra-dichloride group. Patients in the 223Ra-dichloride group also demonstrated a prolonged time to the first symptomatic skeletal event (15.6 vs. 9.8 mo; HR, 0.66; P < 0.001). However, no imaging-based outcome measures were used in the ALSYMPCA design. After the interim analysis, the trial was stopped early and FDA approval was granted after a priority review due to unmet medical need and successful achievement of the primary endpoints (44). Despite the success of the ALSYPMPCA trial, trial design limitations existed. The absence of radiologic outcomes or monitoring during the trial resulted in a paucity of data highlighting metastatic bone disease during treatment with 223Ra. Moreover, the trial design did not allow for collation of data on asymptomatic pathologic fractures.
A postapproval modification to the use of 223Ra-dichloride was released by the European Medicines Agency, based on the results of the ERA 223 trial (45). ERA 223 randomized 806 patients to abiraterone acetate (1,000 mg daily) plus prednisolone with or without 223Ra-dichloride (55 kBq/kg once every 4 wk) versus placebo. Eligible men had chemotherapy-naïve CRPC with bone metastases, and the primary endpoint was symptomatic skeletal event–free survival. The study was unmasked prematurely after more fractures and deaths were noted in the 223Ra group; patients died on average 2.6 mo earlier than in the placebo group and demonstrated a higher rate of factures (29% vs. 11%). Accordingly, the European Medicines Agency recommended that 223Ra be restricted to patients who had 2 previous treatments for PCa and that 223Ra not be used with abiraterone acetate plus prednisolone (46). A comparable trial called the PEACE III trial, aimed to assess an alternative novel antiandrogen, enzalutamide, with and without 223Ra, was performed (47). Given the outcomes of the ERA 223 trial, the PEACE III trial mandated the use of bone-modifying agents such as denosumab or zoledronic acid, which resulted in reduced fracture rates (47).
177Lu-PSMA-617
More recently, 177Lu PSMA-617 (Pluvicto, Novartis) achieved FDA approval after a priority review for metastatic CRPC (48). 177Lu-PSMA-617 is a β-particle emitter, targeting PSMA-positive cells and the associated tumor microenvironment (49). PSMA is a transmembrane protein of intense interest for PCa, initially discovered 25 y ago (49). Physiologically, the function of PSMA is related to the hydrolysis of C-terminal residues of small peptides in the extracellular space, and PSMA is implicated in folate metabolism. PSMA represents an ideal target in PCa, given that PSMA expression increases 1,000-fold in malignant cells with increasing dysplasia (50). In the setting of diagnostics, PSMA PET imaging with 68Ga or 18F has shown utility in accurate localization (51,52).
In the setting of radiopharmaceuticals, early feasibility and tolerability data for 177Lu-PSMA-617 were produced in a retrospective German multicenter review (22). This retrospective review examined 145 patients with metastatic CRPC who were treated off clinical trials in a compassionate-use program and had PSMA expression on PSMA PET, excluding patients who experienced progression under second-line antiandrogens or chemotherapy. 177Lu-PSMA-617 was administered every 8–12 wk, up to 4 cycles. A PSA response of more than 50% was observed in 45% of patients after the first 177Lu-PSMA-617 cycle and 57% after a second cycle. Overall, 177Lu-PSMA-617 demonstrated acceptable toxicity, with 12% of patients experiencing grade 3–4 hematotoxicity.
Initial phase II data were produced in the LuPSMA trial based in Australia (53). This single-arm trial recruited 30 men with progressive CRPC after prior treatment with taxane-based chemotherapy and second-generation antiandrogen therapy. Patients underwent pretreatment PSMA PET screening to confirm high PSMA expression. Up to 4 cycles of 177Lu-PSMA-617 were administered at 6-wk intervals, with doses varying from 6 to 8.5 GBq based on tumor burden. The primary endpoint of a PSA decline of more than 50% was achieved in 17 of 30 patients. A coprimary endpoint was imaging response (on conventional imaging or PSMA PET), for which 40% of patients had nonprogressive disease. This group subsequently published TheraP, an open-label randomized, phase II trial comparing 177Lu-PSMA-617 with cabazitaxel (54). TheraP enrolled 200 men with metastatic CRPC and PSMA PET–positive disease, who were previously treated with docetaxel and had progressive PSA based on the PCWG3 criteria. Patients were randomized 1:1 to receive either cabazitaxel (20 mg/m2 every 3 wk for a maximum of 10 cycles) or 177Lu-PSMA-617 (8.5 GBq, decreasing by 0.5 GBq per cycle, every 6 wk for a maximum of 6 cycles). The primary endpoint for TheraP was PSA response rate (PSA reduction of ≥50% from baseline), and secondary endpoints included progression-free survival, which was denoted by either PSA progression or rPFS as defined by the PCWG3. The 177Lu-PSMA-617 group had a greater PSA response (66% vs. 37%, P < 0.0001), delayed radiographic progression (HR, 0.64; 95% CI, 0.46–0.86; P = 0.0070), and PSA progression (HR, 0.60; 95% CI, 0.44–0.83; P = 0.0017). The toxicity of 177Lu-PSMA-617 was acceptable when compared with the cabazitaxel control group. More recently, phase II data assessing 177Lu-PSMA-617 within the United States confirmed the previous findings of tolerability and therapeutic dosing schedules (55,56).
VISION, an international phase III prospective, randomized, open-label trial, reported results in late 2021 (57). The VISION trial design was derived from the design of ALSYMPCA with respect to the control and treatment groups (similarities defined in Table 1). In total, 831 men with metastatic CRPC and previous chemotherapy exposure were randomized 2:1 to receive a protocol-defined standard-of-care treatment with or without 177Lu-PSMA-617. Eligible patients had to have demonstrated PSMA-positive metastatic disease and no PSMA-negative lesions based on 68Ga-PSMA-11 PET. The liberal standard-of-care inclusion criteria allowed patients to have access to concurrent treatments, excluding cytotoxic chemotherapy, systemic radioisotopes (e.g., 223Ra), immunotherapy and investigational drugs. These exclusions regarding concurrent therapy existed given the lack of safety data on combining 177Lu-PSMA-617 with these agents. The VISION trial had alternate primary endpoints, meaning the trial would be positive if either or both endpoints were reached. These endpoints were rPFS (as defined by PCWG3) and overall survival. The VISION trial met both of its primary endpoints, prolonging rPFS (8.7 vs. 3.4 mo; HR, 0.40; 95% CI, 0.29–0.57; P < 0.001) and overall survival (15.3 vs 11.3 mo; HR, 0.62; 95% CI, 0.52–0.74; P < 0.001). Unlike the ALSYMPCA trial, the VISION trial included radiographic outcome measures, including objective response and disease control according to RECIST; 9.2% of patients achieved a complete response in the 177Lu-PSMA-617 group, compared with 0% in the control group. The VISION trial also met other secondary endpoints, including delay to first symptomatic skeletal event (11.5 vs. 6.8 mo; HR, 0.50; 95% CI, 0.40–0.62; P < 0.001) in the 177Lu-PSMA-617 group.
Comparison of 223Ra and 177Lu-PSMA-617 Phase III Trial Designs
After achievement of the primary outcome measures of the VISION trial, an NDA priority review was submitted to the FDA, and approval was confirmed in April 2022 (48). Given the inclusion of PSMA PET in the trial protocol, the FDA approval stipulated that 177Lu-PSMA-617 eligibility was contingent on PSMA-positive expression on PSMA-11 PET–based imaging (48).
FUTURE OF RADIOPHARMACEUTICALS IN PCa
The FDA approval of 223Ra-dichloride and the success of the recent phase III VISION trial assessing 177Lu-PSMA-617 provide a pathway and a model for future development of radiopharmaceuticals in PCa. Careful adherence to published FDA regulations pertaining to radiopharmaceuticals (7), the principles for trial conduct outlined in the PCWG3 recommendations, and considerations in recommended FDA trial design are critical.
From the recent experiences, important lessons have been learned that may optimize future radiopharmaceuticals. Given the variation in regulatory processes globally, improved international collaboration may improve efficiency in the conduct of phase II and III trials. Many patients may be treated outside clinical trial protocols, particularly in countries with less restrictive regulatory oversight; however, publishing such retrospective data provides limited value in the context of regulatory approval. Thus, it is paramount that early experience with novel agents be published in the context of trial protocols to add to the body of knowledge while providing robust clinical data that are beneficial for regulatory approval. With respect to study design, there is a need to reach consensus on an optimal control arm with which to compare radiopharmaceuticals. The respective control arm treatment should be commensurate with the risks faced by the treated population. Indeed, in some scenarios, the use of passive control groups (e.g., placebo) may be suitable, though in other trial designs a suitable active control will be needed. For example, there is some role for chemotherapy control groups (e.g., docetaxel or cabazitaxel), although not all patients require chemotherapy.
At present, most studied agents have assessed the role in late PCa, typically metastatic CRPC. The role of radiopharmaceuticals at earlier times in the PCa course is of interest, though it has a unique set of challenges (Fig. 2). For example, the PSMAfore trial is a phase III, open-label, randomized trial assessing the role of 177Lu-PSMA-617 in taxane-naïve patients versus a change in the androgen receptor pathway inhibitor in metastatic CRPC (NCT04689828) (58). Use of such agents in even earlier disease states, such as localized disease, requires the use of more intermediate endpoints, given the prolonged follow-up required to demonstrate differences in overall survival. The fact that the optimal endpoint is currently not clear may be disruptive from a regulatory perspective. In particular, molecular imaging endpoints—be they based on response or progression—have been defined, have undergone clinical qualification as clinically relevant, and are not recognized by regulatory agencies as meritorious for drug approval. Additionally, combination therapies with radiopharmaceuticals and alternate systemic agents are being investigated, such as immunotherapy agents. Table 2 lists current radiopharmaceutical trials in PCa.
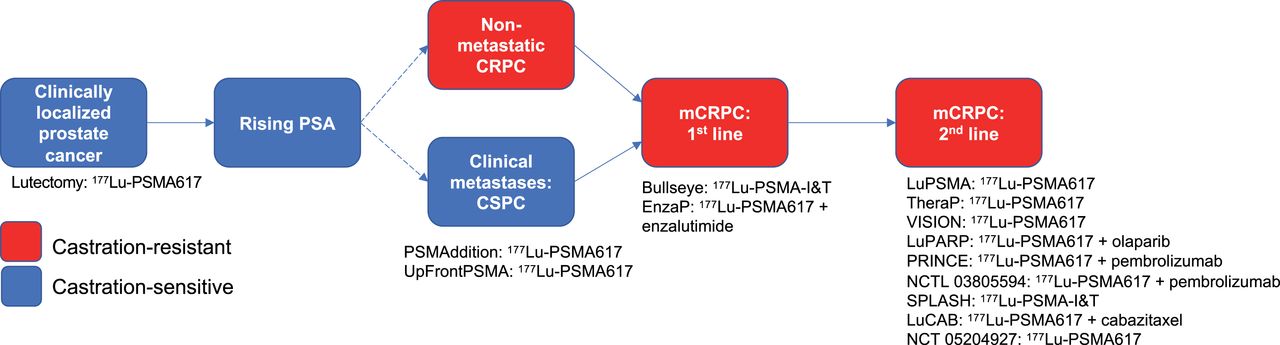
Trials assessing 177Lu-PSMA-617 therapy in various stages of PCa.
Current Radiopharmaceutical Trials in PCa
It is likely that novel radiopharmaceuticals will soon be developed and enter clinical-phase trials. Such agents may be directed to new targets that are overexpressed in PCa tumor cells or the tumor microenvironment (59). In addition to novel pathways and targets, variation in therapeutic radionuclides is a likely avenue of development. Although the most common radioisotopes are frequently β-particle emitters, α-emitters such as 225Ac, 213B, 212Pb, and 211At are attractive because they provide more energy across a shallower depth of penetration (25). Future trial designs may require demonstration of superiority or noninferiority compared with previously approved radiopharmaceutical agents.
CONCLUSION
Although radiopharmaceuticals are currently in their infancy, they represent a promising treatment pathway that warrants careful interrogation. Presently, significant barriers to entry exist in the realms of radiopharmaceutical development and progression through to clinical trials and eventual FDA approval. Despite these barriers, agents have been approved largely because of the clear regulatory guides published by the FDA. In metastatic CRPC, adherence to the framework provided by PCWG facilitated the successful designs of the phase III ALYSMPCA and VISION trials. As such, these recent approvals provide optimism and highlight a clear pipeline toward NDA application for novel radiopharmaceuticals. The roles of radiopharmaceuticals using alternative targeting ligands and radioactive nuclides, and at different disease points in PCa, are an area of future research.
- © 2022 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication June 17, 2022.
- Revision received November 3, 2022.





{kind=link}
{kind=link}