


Visual Abstract
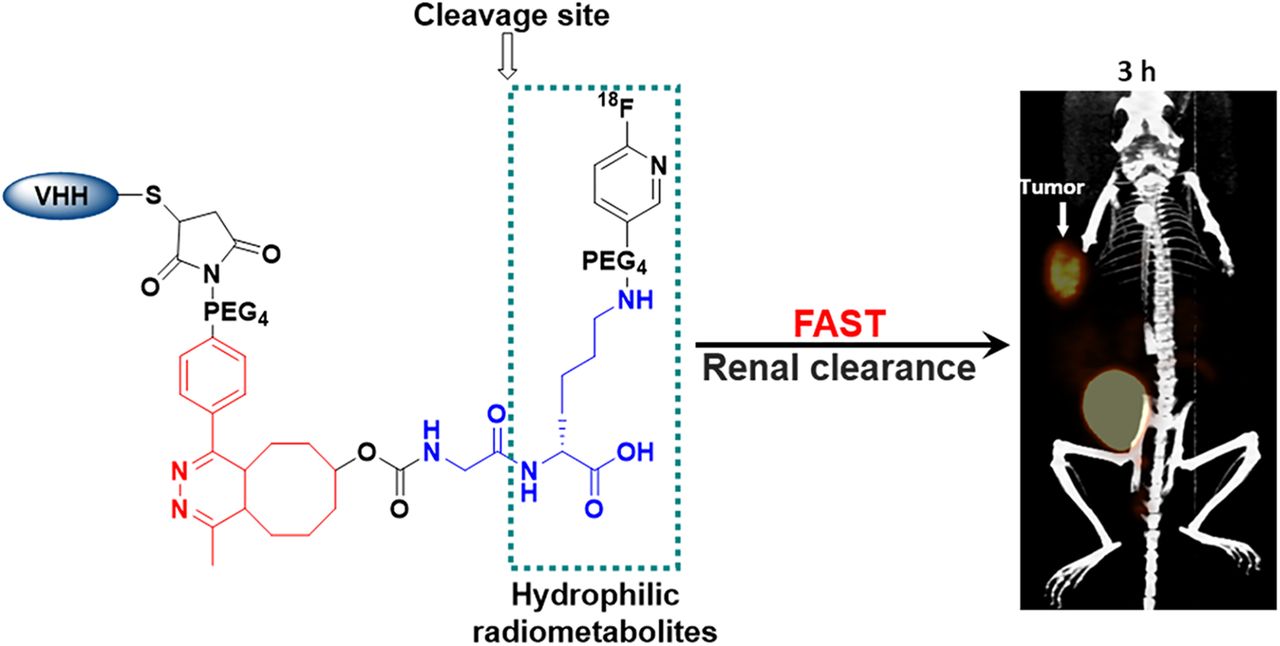
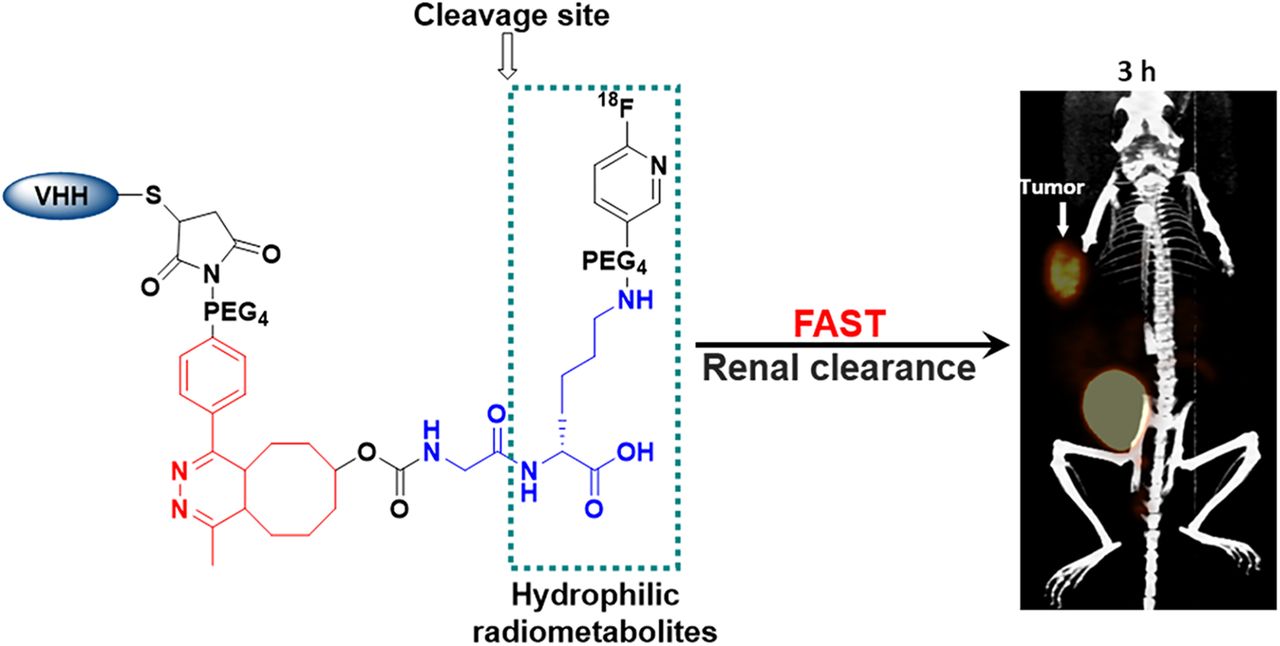
Abstract
Single-domain antibody fragments (sdAbs) are promising vectors for immuno-PET; however, better methods for labeling sdAbs with 18F are needed. Herein, we evaluate a site-specific strategy using an 18F residualizing motif and the anti–epidermal growth factor receptor 2 (HER2) sdAb 5F7 bearing an engineered C-terminal GGC tail (5F7GGC). Methods: 5F7GGC was site-specifically attached with a tetrazine-bearing agent via thiol-maleimide reaction. The resultant conjugate was labeled with 18F by inverse electron demand Diels–Alder cycloaddition with a trans-cyclooctene attached to 6-18F-fluoronicotinoyl moiety via a renal brush border enzyme-cleavable linker and a PEG4 chain (18F-5F7GGC). For comparisons, 5F7 sdAb was labeled using the prototypical residualizing agent, N-succinimidyl 3-(guanidinomethyl)-5-125I-iodobenzoate (iso-125I-SGMIB). The 2 labeled sdAbs were compared in paired-label studies performed in the HER2-expressing BT474M1 breast carcinoma cell line and athymic mice bearing BT474M1 subcutaneous xenografts. Small-animal PET/CT imaging after administration of 18F-5F7GGC in the above mouse model was also performed. Results: 18F-5F7GGC was synthesized in an overall radiochemical yield of 8.9% ± 3.2% with retention of HER2 binding affinity and immunoreactivity. The total cell-associated and intracellular activity for 18F-5F7GGC was similar to that for coincubated iso-125I-SGMIB-5F7. Likewise, the uptake of 18F-5F7GGC in BT474M1 xenografts in mice was similar to that for iso-125I-SGMIB-5F7; however, 18F-5F7GGC exhibited significantly more rapid clearance from the kidney. Small-animal PET/CT imaging confirmed high uptake and retention in the tumor with very little background activity at 3 h except in the bladder. Conclusion: This site-specific and residualizing 18F-labeling strategy could facilitate clinical translation of 5F7 anti-HER2 sdAb as well as other sdAbs for immuno-PET.
Smaller protein scaffolds exemplified by 6-kDa Affibody molecules (Affibody AB) and 12- to 15-kDa single-domain antibody fragments (sdAbs) have emerged as promising platforms for theranostics because they possess properties intermediate between those of monoclonal antibodies and peptides. A recent review (1) has summarized efforts with these delivery vehicles to image and treat cancers overexpressing epidermal growth factor receptor 2 (HER2), an internalizing receptor that is overexpressed in breast, ovarian, gastric, and other cancers (2). Because only about 20%–25% of breast cancers are HER2-positive and metastatic disease frequently exhibits heterogeneities in HER2 expression, assessment of HER2 status is imperative before embarking on HER2-targeted therapies (3).
Combining an anti-HER2 sdAb with a radiolabeling strategy tuned to the properties of small proteins could provide a promising immuno-PET approach for global and repeatable evaluation of HER2 status. Given the high and prolonged uptake of radiometal-labeled sdAbs in kidneys (1,4), 18F is a promising radionuclide for sdAb-based immuno-PET. Moreover, 18F has a half-life compatible with the rapid clearance of sdAbs, positron emission characteristics that are ideal for imaging, and a readily available supply chain. Although several 18F protein labeling methods have been developed (5), N-succinimidyl 4-18F-fluorobenzoate (18F-SFB) (6) remains the most widely used reagent for this purpose. Both our group (7) and others (8) have labeled sdAbs with 18F-SFB; however, the nonresidualizing capacity of 18F-SFB compromised tumor retention of 18F after receptor-mediated internalization.
Studies with 131I- and 211At-labeled HER2-targeted sdAbs have demonstrated that very effective trapping of radioactivity in tumor can be achieved using charged prosthetic groups (9–11). In recent years, we have developed conceptually similar prosthetic agents for labeling sdAbs with 18F (7,12,13). Two tactics that we investigated were the very fast tetrazine (Tz)-trans-cyclooctene (TCO) inverse electron demand Diels–Alder cycloaddition reaction (IEDDAR) and exploiting a renal brush border enzyme (RBBE)–cleavable linker to reduce kidney activity (12,14). In these studies, 18F was introduced on the Tz-bearing moiety. Although kidney activity could be reduced considerably, activity in hepatobiliary organs was elevated, presumably because of the high lipophilicity of Tz-bearing radiocatabolites (15,16).
Herein we describe a new linker for 18F labeling of the HER2-targeted sdAb 5F7 that also could be applied in principle to site-specific labeling of other proteins with engineered cysteine residues via thiol-maleimide conjugation (17). Precise control of the 18F-labeling site on the C terminus of an sdAb can avoid chemical modification of amino acids involved in receptor/antigen recognition and provide a uniform product more amenable to clinical translation (18). Other design features of this linker include the use of the 6-18F-fluoronicotinoyl group for residualization (13), and a GlyLys RBBE-cleavable linker strategically positioned within its structure (Fig. 1) to create rapid, high contrast between tumor and both kidneys and hepatobiliary organs.
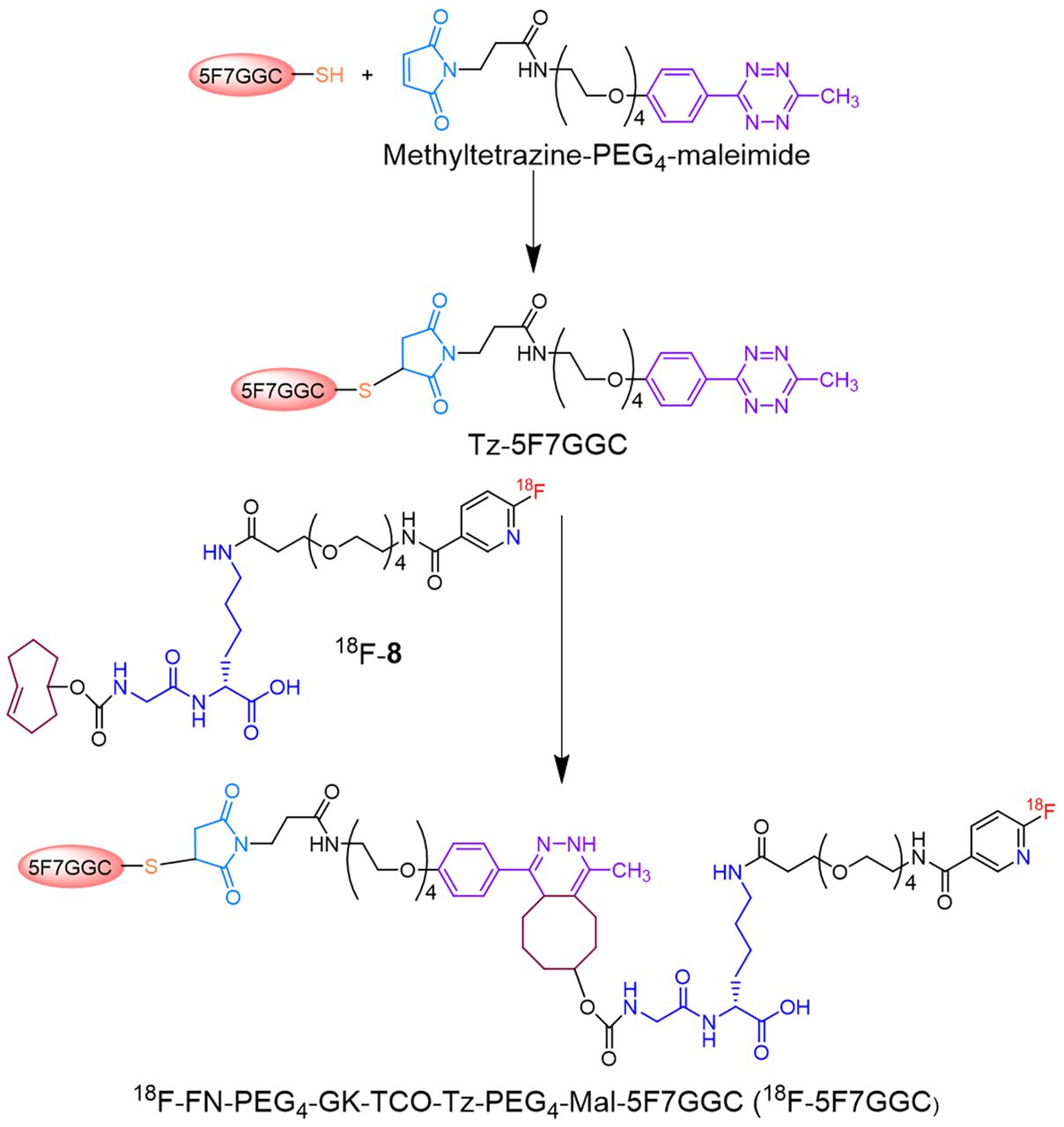
Scheme for synthesis of 18F-FN-PEG4-GK-TCO-Tz-PEG4-Mal-5F7GGC (18F-5F7GGC).
MATERIALS AND METHODS
sdAbs, Cells, Culture Conditions, and Animal Model
The anti-HER2 sdAb 5F7 and 5F7GGC, its analog with an engineered cysteine at the C terminus (19,20), were purchased from ATUM.
Cell culture reagents were purchased from Thermo Fisher Scientific and InvivoGen. HER2-expressing BT474 human breast carcinoma cells and its more metastatic version, BT474M1 (21), were cultured in RPMI 1640 medium containing 10% fetal bovine serum, 1% penicillin-streptomycin, 5 μg/mL plasmosin, 1% sodium pyruvate, 1% 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, and 0.4 μg/mL insulin.
All procedures involving animals were approved by the Duke University institutional animal care and use committee. Female nude mice were obtained from an internal breeding colony at Duke or from Charles River Laboratories, and subcutaneous BT474M1 xenografts were established as previously described (7,11). Biodistribution and imaging studies were initiated when tumors were 150–350 mm3 in volume.
Synthesis, Radiosynthesis, and 18F Labeling of 5F7GGC
Details of the synthesis of the precursor (7), standard (8), and 18F-8, as well as the synthesis of N-succinimidyl 3-(guanidinomethyl)-5-125I-iodobenzoate (iso-125I-SGMIB), are provided in the supplemental materials (available at http://jnm.snmjournals.org). First, the sdAb was reduced to convert any dimer present to monomer. For this, a slurry of immobilized tris-[2-carboxyethyl] phosphine hydrochloride gel (Thermo Fisher Scientific; 300 μL) was added to a solution of 5F7GGC (1.5 mg, 115 nmol) in 300 μL of 0.2 M NH4OAc containing 5 mM ethylenediaminetetraacetic acid, pH 6.3, and the mixture was stirred at 37°C for 1 h. The suspension was centrifuged, and the supernatant containing monomeric 5F7GGC was immediately added to a vial containing methyltetrazine-PEG4-maleimide (0.18 mg, 345 nmol) and the mixture stirred at 37°C for 1 h. The Tz-5F7GGC product was isolated from the mixture by size-exclusion (SE) high-performance liquid chromatography (HPLC). For this, an Agilent PL Multisolvent 20 (7.8-mm internal diameter × 150 mm) SE-HPLC column was eluted in isocratic mode with water at a flow rate of 0.9 mL/min. The pooled HPLC fractions containing the Tz-5F7GGC (tR = 3.3 min) were lyophilized to obtain 0.5 mg of a pink solid. Its molecular weight was determined by liquid chromatography–mass spectrometry and binding affinity to HER2-Fc by surface plasmon resonance (supplemental materials). A solution of Tz-5F7GGC in phosphate-buffered saline, pH 7.4 (50 μL, 2 mg/mL), was added to a vial containing dried 18F-8 (60–280 MBq), and the mixture was incubated at 37°C for 10 min. 18F-FN-PEG4-GK-TCO-Tz-PEG4-Mal-5F7GGC (18F-5F7GGC) was isolated by gel filtration over a PD-10 column eluted with phosphate-buffered saline (20). Radiochemical purity, HER2 binding affinity, and immunoreactivity were determined as described previously (12), with details provided in the supplemental materials.
Cellular Uptake and Internalization Assays
Paired-label uptake and internalization assays were performed on BT474M1 cells to compare the behavior of 18F-5F7GGC and iso-125I-SGMIB-5F7 sdAbs as described in the supplemental materials.
Biodistribution Studies
The paired-label biodistribution of 18F-5F7GGC and iso-125I-SGMIB-5F7 was evaluated in athymic mice bearing subcutaneous BT474M1 xenografts. Groups of 5 mice received 166 kBq (0.64 μg) of iso-125I-SGMIB-5F7 and 666 kBq (3.6 μg) of 18F-5F7GGC in 100 μL of phosphate-buffered saline via the tail vein. It was necessary to use a larger amount of activity and hence a larger mass for 18F-5F7GGC to compensate for the more rapid decay of 18F. Ex vivo biodistribution was determined at 1 and 3 h, and percentage injected dose (%ID)/g were calculated as previously described (7).
Small-Animal PET/CT Imaging
Three BT474M1 tumor-bearing mice were imaged on a Siemens Inveon PET/CT system at 1, 2, and 3 h after injection of 18F-5F7GGC (1.6–2.2 MBq, 19–23 μg). The mice were anesthetized using 2%–3% isoflurane in oxygen, and a 5-min static PET acquisition followed by a 5-min CT scan was performed. List-mode PET data were histogram-processed, and the images were reconstructed using a standard algorithm consisting of 2 ordered-subsets-expectation-maximization 3-dimensional iterations and 18 maximum-a-posteriori iterations with a cutoff (Nyquist) of 0.5. Images were corrected for attenuation (CT-based) and radioactive decay. Image analysis was performed using Inveon Research Workplace software.
Statistical Analysis
Results are reported as mean ± SD. The statistical significance of differences between coincubated or coadministered tracers was determined by a 2-tailed, paired Student t test (GraphPad QuickCalcs). A P value of less than 0.05 was considered to be significant.
RESULTS
Synthesis, sdAb Conjugation, and Radiochemistry
As shown in Supplemental Figure 1, the precursor 7 was synthesized from the known compound 1 (22) in 6 steps. Compound 8 was synthesized by treatment of 7 with tetra-n-butylammonium fluoride in 75.4% yield. The 1H–nuclear magnetic resonance and mass spectrometry data for compounds 2–8 were consistent with their structures. Figure 1 shows the synthesis scheme for Tz-5F7GGC and its labeling via IEDDAR with 18F-8. SE-HPLC–mass spectrometry analysis of the reaction mixture for 5F7GGC conjugation with methyltetrazine-PEG4-maleimide indicated complete conversion of 5F7GGC to Tz-5F7GGC (Supplemental Fig. 2). The observed molecular weight was consistent with its structure. Surface plasmon resonance indicated a Kd of 0.3 nM (ka = 1.58 × 106 M−1s−1; kd = 4.60 × 10−4 s−1) for Tz-5F7GGC versus 0.2 nM for 5F7, demonstrating that attaching the Tz-PEG4-Mal moiety had minimal effect on HER2 binding affinity (Fig. 2).

Sensorgrams showing dose–response curves and kinetic profiles for binding of 5F7 (A) and Tz-5F7GGC (B) to HER2-Fc extracellular domain.
The radiochemical yield (RCY) for the synthesis of 18F-FN-PEG4-GK-TCO (18F-8) from 7 via SNAr reaction was 47.4% ± 9.0% (n = 11). With 100 μg of protein at 2 mg/mL, the RCY for IEDDAR between Tz-5F7GGC and 18F-8 was 27.3% ± 8.2% (n = 4). Although performed only twice, use of 200 μg of protein at 4 mg/mL increased conjugation yields to 46.1% ± 4.5%. Based on initial aqueous 18F-fluoride activity, the total synthesis time for 18F-5F7GGC was 90 min in an overall RCY of 8.9% ± 3.2% (n = 6) (7.3% ± 3.3% [n = 4] and 11.3% ± 0.4% [n = 2] for IEDDAR with 100 μg and 200 μg of sdAb, respectively). Although higher IEDDAR yields were obtained in the 200-μg syntheses, RCYs for 18F-8 were low because of an HPLC malfunction. The molar activity for 18F-5F7GGC was 5.2 ± 2.7 MBq/nmol (n = 6). Sodium dodecyl sulfate polyacrylamide gel electrophoresis (Supplemental Fig. 3A) and SE-HPLC (Supplemental Fig. 3B) of 18F-5F7GGC showed a single radioactive band/peak (∼100%) corresponding to the molecular weight of an sdAb. The Kd for binding of 18F-5F7GGC to BT474 cells was 3.37 ± 0.36 nM (Supplemental Fig. 3C) and its immunoreactive fraction 70.8% (Supplemental Fig. 3D), demonstrating that HER2 reactivity was not compromised with this 18F-labeling strategy.
Cell Uptake and Internalization Assay
After incubation with BT474M1 cells at 37°C, total cell-associated activity (Fig. 3A) for 18F-5F7GGC was 24.3% ± 0.9%, 31.3% ± 0.9%, and 35.3% ± 2.5% of input activity at 1, 2, and 4 h, respectively, values that were slightly lower than those for coincubated iso-125I-SGMIB-5F7 (25.2% ± 0.6%, 32.5% ± 0.8%, and 37.1% ± 2.0%); but differences between the radioconjugates were not statistically significant (P > 0.05). Nonspecific uptake, determined at the 2-h time point by coincubation with excess trastuzumab, was less than 1% of input activity for both radiotracers. The percentage of input activity that was intracellularly trapped was 12.3% ± 0.5%, 17.3% ± 1.1%, and 20.8% ± 1.5% for 18F and 13.4% ± 4.4%, 18.2% ± 1.2%, and 22.3% ± 1.2% for 125I at 1, 2, and 4 h, respectively (Fig. 3B; P > 0.05 for all).
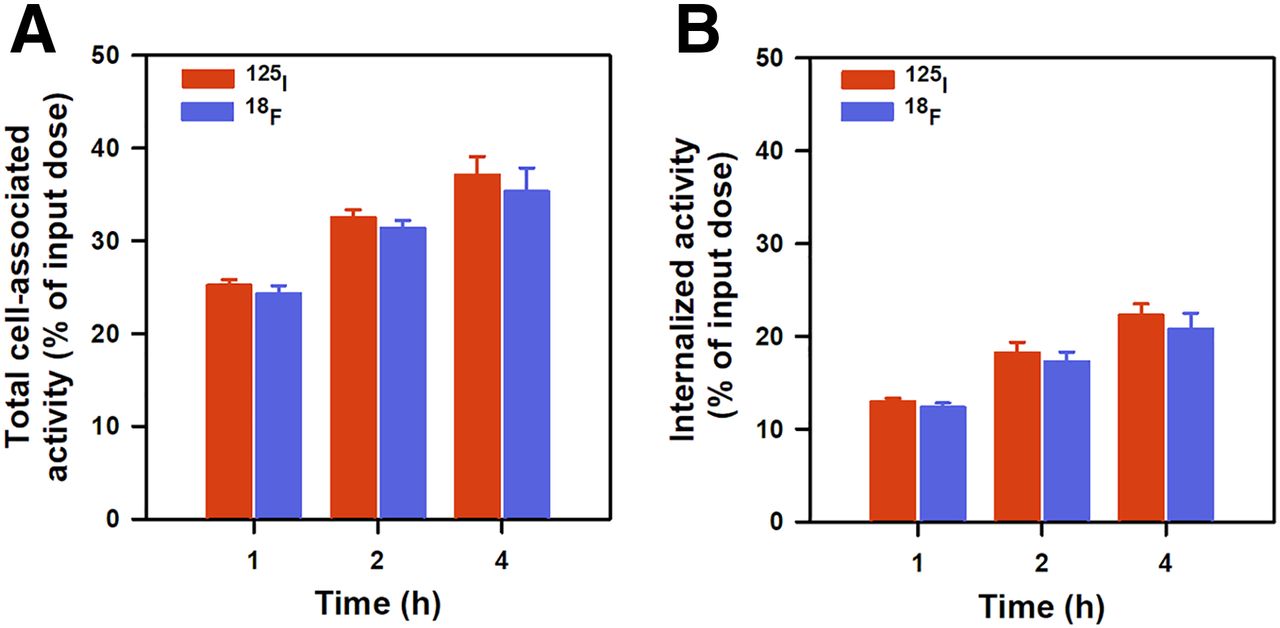
Paired-label uptake and internalization of iso-125I-SGMIB-5F7 and 18F-5F7GGC by HER2-positive BT474M1 breast carcinoma cells. Data (mean ± SD) are percentage of initially added activity that was bound to cells (membrane + internalized) (A) and internalized (B).
Biodistribution
The data from the paired-label biodistribution of 18F-5F7GGC and iso-125I-SGMIB-5F7 in athymic mice bearing BT474M1 xenografts are presented in Figure 4. Tumor uptake of 18F-5F7GGC was similar to that seen for iso-125I-SGMIB-5F7 both at 1 h (125I, 18.34 ± 1.62 %ID/g; 18F, 14.87 ± 1.41 %ID/g; P < 0.01) and 3 h (125I, 17.20 ± 5.09 %ID/g; 18F, 12.92 ± 3.73 %ID/g; P > 0.05). On the other hand, with a few exceptions, normal-tissue activity for 18F-5F7GGC was similar to or less than that observed for iso-125I-SGMIB-5F7. Remarkably, 18F-activity levels in kidneys were 4- and 6-fold lower than those for 125I at 1 and 3 h, respectively. Bone uptake of 18F was low, suggesting limited susceptibility of 18F-5F7GGC to defluorination in vivo. Tumor–to–normal-tissue ratios for the 2 radiolabeled sdAbs are shown in Figure 5. Tumor-to-kidney ratios for 18F (1.2 ± 0.4 and 12.2 ± 3.5) were 3.3- and 4.5-fold higher (P < 0.004) than those for 125I (0.37 ± 0.05 and 2.7 ± 0.7) at 1 and 3 h, respectively. Likewise, tumor-to-blood ratios were 2- to 3-fold higher for 18F. The tumor-to-liver ratio for 18F-5F7GGC was more than 5:1 at 1 h and lower than that for iso-125I-SGMIB-5F7 (P < 0.0003).

Paired-label biodistribution of 18F-5F7GGC and iso-125I-SGMIB-5F7 sdAb conjugates in athymic mice bearing subcutaneous HER2-expressing BT474M1 breast carcinoma xenografts.

Tumor-to-tissue ratios after injection of 18F-5F7GGC and iso-125I-SGMIB-5F7 sdAb conjugates in athymic mice bearing subcutaneous BT474M1 xenografts.
Small-Animal PET/CT Imaging
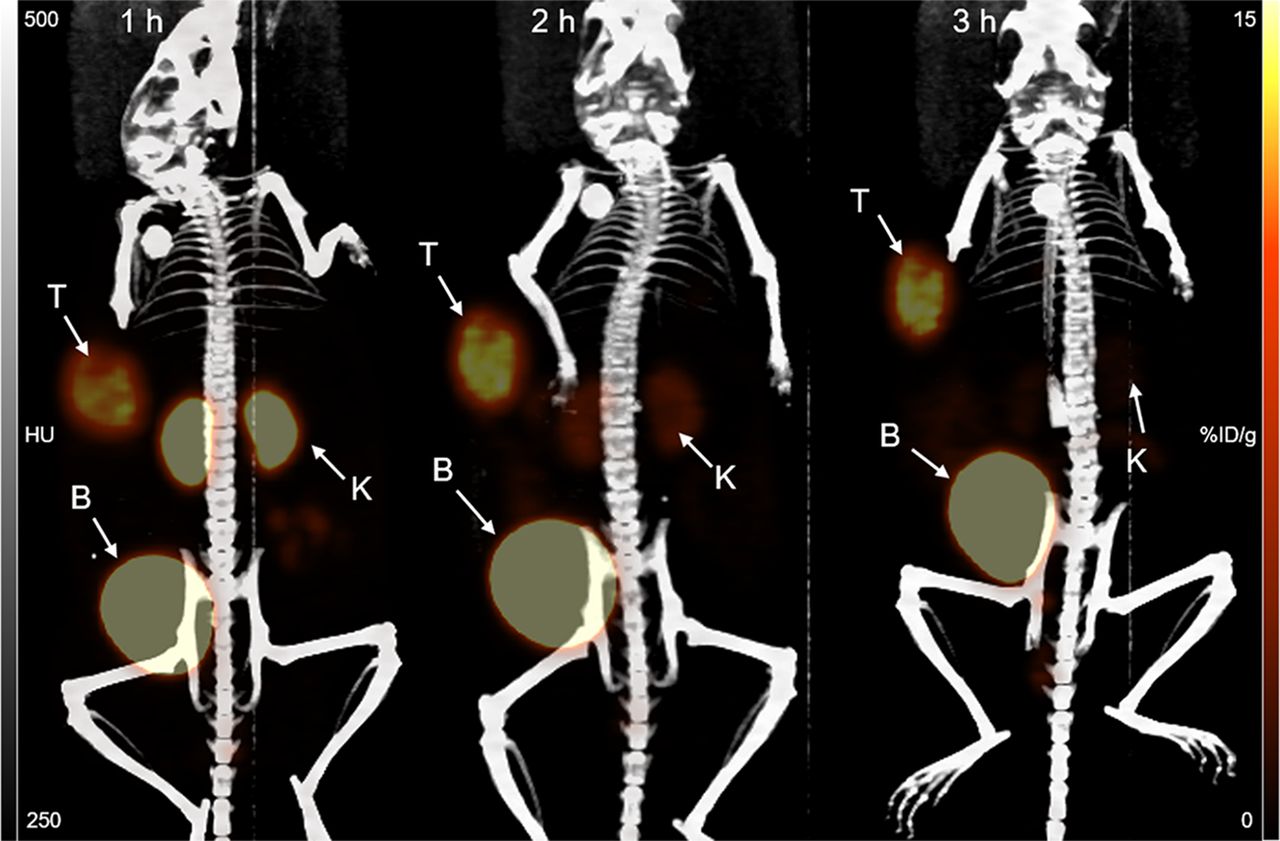
Small-animal PET/CT maximum-intensity-projection images of a representative mouse bearing a subcutaneous BT474M1 xenograft obtained 1, 2, and 3 h after administration of 18F-5F7GGC are shown in Figure 6. Consistent with the results from the necropsy study, pronounced tumor accumulation was observed at all time points, with minimal background activity except in kidneys and bladder. Kidney activity was quite low at 1 h, nearly undetectable by 2 h, and eliminated by 3 h, with bladder having prominent activity at all time points. The average tumor uptake values (n = 3) calculated from the PET imaging data, expressed as SUVmax followed by %ID/gmax in parentheses, were 4.6 ± 0.5 (18.0 ± 1.8), 4.7 ± 0.9 (17.9 ± 3.6), and 5.0 ± 0.8 (19.0 ± 3.1) at 1, 2, and 3 h, respectively, with corresponding values for kidneys of 2.9 ± 0.3 (11.2 ± 1.0), 1.0 ± 0.1 (4.3 ± 0.6), and 0.6 ± 0.1 (2.4 ± 0.2). Tumor-to-kidney ratios calculated from these PET imaging data were 1.6 ± 0.2, 4.2 ± 0.6, and 7.8 ± 0.8 at 1, 2, and 3 h, respectively.

Maximum-intensity-projection 18F-5F7GGC immuno-PET images of representative mouse bearing subcutaneous HER2-positive BT474M1 xenograft obtained 1, 2, and 3 h after injection. Positions of tumor (T), kidney (K), and bladder (B) are indicated.
DISCUSSION
Small protein platforms—exemplified herein by an sdAb—have favorable properties for immuno-PET including rapid tumor penetration and fast normal-tissue clearance and are ideally suited for use with 18F. However, because of their small size, they are much more likely than whole monoclonal antibodies to be adversely affected by chemical modification, resulting in unwanted normal-tissue retention or altered binding to their molecular target. Moreover, unlike monoclonal antibodies, retention of activity in the kidneys also can be problematic. To address these issues, we designed a labeling strategy (Fig. 1) comprising several synergistic components: a site-specific linker to control labeling location and stoichiometry (19), an RBBE-cleavable sequence to reduce kidney activity (12,14), a 6-fluoronicotinoyl group (13,14) to provide short-term residualization, and positioning the RBBE-cleavable sequence to avoid Tz- or TCO-bearing 18F-labeled catabolites in vivo.
Site-specific conjugation of the Tz-bearing maleimide with 5F7GGC was achieved without impairing HER2 binding of the sdAb (Fig. 2). The 48% RCY for TCO-bearing 18F-8 synthesis was reasonable given the presence of the free carboxyl group active hydrogens in compound 7, which is not generally conducive for labeling using nucleophilic 18F-fluoride (23). Of note, 18F labeling of TCO-bearing molecules was typically performed via the SN2 reaction, whereas in our case it was performed via the SNAr reaction, a first. RCYs for IEDDAR between Tz-5F7GGC and 18F-8 appeared to increase with increasing protein concentration but were lower than reported for 18F labeling using Tz-TCO IEDDAR in general (24) and for IEDDAR between TCO-modified sdAbs and 18F-labeled Tz derivatives in particular (12,14). A likely contributing factor is the number of prosthetic moieties per sdAb available for IEDDAR—when random conjugation was used, more than one TCO was attached per sdAb (12,14) versus just one Tz moiety in the current work. However, even with site-specific labeling, higher yields (53%–77%) have been reported for IEDDAR between 18F-TCO and a diabody site-specifically modified with a Tz moiety (25). Differences in solvent, protein concentration, and polarity of the reagents between the 2 studies are noted, and these variables will be evaluated in future optimization of the current labeling strategy.
With the current procedure, the overall decay-corrected RCYs for the synthesis of 18F-5F7GGC were 7.3% ± 3.3% and 11.3% ± 0.4% at 100 and 200 μg of Tz-5F7GGC, respectively, providing 154 and 237 MBq of 18F-5F7GGC from 3.7 GBq of aqueous 18F-fluoride in 90 min. On the basis of literature precedents (24), higher yields should be readily attainable by increasing protein levels beyond 200 μg, which should be compatible with patient use given that a 1-mg sdAb dose was used in a phase 1 trial with another anti-HER2 sdAb labeled with 68Ga (26). That study used an average administered activity of 107 MBq of 68Ga, suggesting that with our current radiochemistry procedures, clinical studies would be feasible.
Multiple methods for labeling proteins with 18F have been reported (5). Random labeling of constituent protein lysine residues can be accomplished using active esters such as N-succinimidyl 4-18F-fluorobenzoate, 2,3,5,6-tetrafluorophenyl 6-18F-fluoronicotinate (13,27) and their 4-nitrophenyl analogs (28), among others. The site-specific 18F labeling of proteins also has been described, including via oxime formation with an engineered aminoxy function on the protein with 4-18F-fluorobenzaldehyde or preconjugating the protein with maleimide-bearing agents containing moieties such as 4-fluorobenzaldehyde O-alkyl oxime, 4-SiFA, and AlF-chelate complex and subsequent 18F labeling of the conjugate (29,30). Of direct relevance to the current study, the last 3 approaches were evaluated for 18F labeling of an anti-HER2 Affibody. Although uptake in HER2-positive xenografts was reported, there were significant problems with each 18F-Affibody conjugate—either high uptake in the intestines or kidneys or excessive in vivo defluorination. From a radiochemistry perspective, it is important to note that the maleimide functionality is susceptible to SNAr and SN2 18F-labeling conditions and thus the maleimide-bearing part has to be conjugated to an 18F-bearing moiety, the synthesis of which often involves one or more steps. As mentioned above, an alternative approach of labeling proteins with 18F using maleimide agents involves conjugating them first with the maleimide-bearing agent and then performing the labeling under acidic pH and high temperatures (29); however, unfortunately, most proteins are not stable under these conditions. This led us to pursue the strategy described in the current work to exploit the very fast kinetics of IEDDAR under physiologic conditions. In addition, our strategy differs from those noted above in that it results in minimal background activity in the kidney and hepatobiliary organs as well as other tissues. Drawbacks of our method are the modest RCYs and relatively longer time for synthesis; however, systematic optimization of reaction conditions and other parameters should lead to a more efficient and streamlined procedure, which will be explored in future studies.
One of the main motivations for site-specific labeling is that it can provide a more homogeneous product and thereby facilitate clinical translation. In the conjugation of the prosthetic agent to the sdAb, a single product was obtained (Supplemental Fig. 2) whereas products with different levels (and likely sites) of substitution were observed with random lysine modification approaches (12,14). The other motivation is to avoid conjugation of the prosthetic agent on a lysine that could alter binding affinity. With 5F7 sdAb, this is a distinct possibility because 1 of its 5 lysines is located in CDR2 (20). However, a recent study showed no difference in binding affinity for radioiodinated 5F7 conjugates prepared by site-specific and random labeling (19), suggesting either lack of modification or involvement in HER2 recognition for the CDR2 lysine. On the other hand, improvements in binding affinity have been demonstrated for site-specific modification of other sdAbs (31), making the affinity advantage possible through this labeling strategy dependent on the characteristics of the particular sdAb.
In previous work, iso-*I-SGMIB emerged as the residualizing agent with the best combination of high tumor retention of radioactivity and low uptake in kidneys (9). Accordingly, iso-125I-SGMIB-5F7 was selected as the benchmark for use in these paired-label studies evaluating the HER2-targeting potential of 18F-5F7GGC. In vitro assays demonstrated HER2-specific uptake of 18F-5F7GGC on HER2-positive BT474M1 cells. Both total cell-associated and internalized activity for 18F-5F7GGC were not significantly different (P > 0.05; Fig. 3) from those for coincubated iso-125I-SGMIB-5F7 and increased with time, demonstrating that the 18F-prosthetic agent is residualizing. Moreover, with the caveat that assay conditions were not the same, the cellular uptake and internalization of 18F-5F7GGC were similar to or higher than that seen previously with 5F7 labeled with 18F using other prosthetic agents (7,13,14).
Necropsy and small-animal PET imaging studies demonstrated rapid, high-level accumulation of 18F-5F7GGC in BT474M1 xenografts, suggesting that same-day immuno-PET imaging would be feasible. This is an important practical advantage compared with intact monoclonal antibodies such as 89Zr-trastuzumab that require up to 6 d to achieve optimal imaging conditions (32). Tumor uptake of 18F-5F7GGC was about 20%–25% lower than that of coinjected iso-125I-SGMIB-5F7, which might reflect the greater residualizing capacity of the guanidino versus the pyridyl moiety (33) or the presence of the GK linker (14). Nonetheless, with the caveat that different xenograft models were used, 18F-5F7GGC tumor uptake was higher than uptakes reported for most 18F-labeled anti-HER2 sdAb conjugates (8,12–14,34,35). A notable exception is the results obtained with 18F-RL-I-5F7 in the same BT474M1 xenograft (7); however, even though the less favorable SGMIB isomer (9) was used for 5F7 labeling, tumor uptake of radioiodine also was almost 2-fold that observed in the current study for iso-125I-SGMIB-5F7. Differences in mouse strain (NOD/SCID vs. athymic) between the 2 studies could play a role (36), as could individual xenograft properties such as tumor size (11), emphasizing the importance of performing experiments in paired-label format when possible.
Importantly, achieving high uptake in tumor with 18F-5F7GGC was not associated with increased uptake in the kidneys or other organs, which might hamper repeated use in patients to monitor HER2 status or interfere with lesion detection. This is an important distinction with agents such as 18F-RL-I-5F7, noted above, that show excellent tumor uptake but at the expense of sustained kidney activity levels higher than 100 %ID/g (7). Indeed, renal activity levels for 18F-5F7GGC were about 4–5 times lower than those for coadministered iso-125I-SGMIB-5F7, possibly the radiolabeled sdAb exhibiting the most rapid clearance of those investigated to date (1,9,37). It is likely that this behavior reflects both the presence of an RBBE-cleavable linker and the rapidly clearing 6-18F-fluoronicotinoyl moiety–containing catabolites (13). Finally, in our recent study evaluating 5F7 iso-125I-SGMIB conjugates formed by different linkers, use of the maleimido linker as done herein resulted in significantly decreased kidney uptake compared with the N-succinimidyl linker (19).
Compounds with both TCO and Tz moieties, especially Tz, are known to accumulate in hepatobiliary organs (38,39). For this reason, one of our design considerations was to strategically position the RBBE-cleavable linker in such a way that likely radiolabeled catabolites would be devoid of Tz and TCO moieties. We hypothesized that this would reduce uptake of activity levels in hepatobiliary organs, especially in intestines and gallbladder. Gratifyingly, uptake of activity from 18F-5F7GGC in the gut was more than 5 times lower than observed with a similar construct expected to generate radiolabeled catabolites bearing a Tz moiety (14). Consistent with this, whereas intense gallbladder uptake was observed in the small-animal PET images with the previous 18F-labeled sdAb conjugate (14), intestinal and gallbladder uptake was minimal or not observed for 18F-5F7GGC (Fig. 6). Importantly, the ratios for activity in tumor to liver and to bone, 2 frequent sites for HER2-positive tumor metastases, were 9:1 and 40:1, respectively, at 1 h after injection.
CONCLUSION
A potentially widely applicable site-specific and residualizing 18F-labeling strategy for use with small proteins was validated using anti-HER2 sdAb 5F7. Strategic positioning of an RBBE-cleavable linker provided reduced activity levels in the kidneys and hepatobiliary organs. Moreover, 18F-5F7GGC demonstrated high potential as a probe for immuno-PET of HER2-expressing cancers.
DISCLOSURE
This work was supported by National Institutes of Health grants CA188177 and CA42324. Michael R. Zalutsky holds ownership interest and is a board member for Cereius, Inc. Ganesan Vaidyanathan is a consultant and shareholder for Cereius, Inc. No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Can a site-specific linker be devised for 18F labeling of small proteins that combines tumor-residualizing properties with rapid clearance of activity from kidneys and other normal tissues?
PERTINENT FINDINGS: An anti-HER2 sdAb with a C-terminal cysteine was site-specifically attached with a Tz derivative via maleimide-thiol conjugation and labeled with 18F by IEDDAR with a TCO agent attached to an 18F-fluoronicotinoyl moiety via a linker containing an RBBE-cleavable linker. The new 18F-sdAb conjugate exhibited high uptake in HER2-positive tumors with little activity in kidneys and other normal tissues on serial necropsy and small-animal PET imaging.
IMPLICATIONS FOR PATIENT CARE: This site-specific and residualizing 18F-labeling strategy could facilitate clinical translation of 5F7 anti-HER2 sdAb, as well as other sdAbs for immuno-PET.
ACKNOWLEDGMENTS
We thank Dr. Xiao-Guang Zhao, Thomas Hawk, and Brian Watts for excellent technical assistance with the animal studies, small-animal PET/CT imaging, and surface plasmon resonance, respectively.
Footnotes
Published online February 26, 2021.
- © 2021 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication December 6, 2020.
- Accepted for publication February 1, 2021.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}