


Abstract
The currently available therapeutic radiopharmaceutical for high-risk neuroblastoma, 131I-metaiodobenzylguanidine, is ineffective at targeting micrometastases because of the low–linear-energy-transfer (LET) properties of high-energy β-particles. In contrast, Auger radiation has high-LET properties with nanometer ranges in tissue, efficiently causing DNA damage when emitted near DNA. The aim of this study was to evaluate the cytotoxicity of targeted Auger therapy in preclinical models of high-risk neuroblastoma. Methods: We used a radiolabled poly(adenosine diphosphate ribose) polymerase (PARP) inhibitor called 125I-KX1 to deliver Auger radiation to PARP-1, a chromatin-binding enzyme overexpressed in neuroblastoma. The in vitro cytotoxicity of 125I-KX1 was assessed in 19 neuroblastoma cell lines, followed by in-depth pharmacologic analysis in a sensitive and resistant pair of cell lines. Immunofluorescence microscopy was used to characterize 125I-KX1–induced DNA damage. Finally, in vitro and in vivo microdosimetry was modeled from experimentally derived pharmacologic variables. Results: 125I-KX1 was highly cytotoxic in vitro across a panel of neuroblastoma cell lines, directly causing double-strand DNA breaks. On the basis of subcellular dosimetry, 125I-KX1 was approximately twice as effective as 131I-KX1, whereas cytoplasmic 125I-metaiodobenzylguanidine demonstrated low biological effectiveness. Despite the ability to deliver a focused radiation dose to the cell nuclei, 125I-KX1 remained less effective than its α-emitting analog 211At-MM4 and required significantly higher activity for equivalent in vivo efficacy based on tumor microdosimetry. Conclusion: Chromatin-targeted Auger therapy is lethal to high-risk neuroblastoma cells and has the potential to be used in micrometastatic disease. This study provides the first evidence for cellular lethality from a PARP-1–targeted Auger emitter, calling for further investigation into targeted Auger therapy.
Neuroblastoma is the most common extracranial solid tumor in childhood, with only up to a 50% 5-y survival rate in high-risk patients (1). Relapsed cases are often managed with 131I-metaiodobenzylguanidine (131I-MIBG) radiopharmaceutical therapy, but its suboptimal therapeutic efficacy and very low complete response rate remain unsatisfactory (2). The main limitation of 131I-MIBG therapy is the low linear energy transfer (LET) of β-particles, which are unable to produce sufficient radiation fields for lethal DNA damage in micrometastatic disease (3).
Our group has recently developed a 211At-labeled radiopharmaceutical, 211At-MM4, which has shown strong antitumor efficacy in neuroblastoma (4). In contrast to MIBG, which targets the norepinephrine transporter, 211At-MM4 binds to poly(adenosine diphosphate ribose) polymerase 1 (PARP-1), a chromatin-associated protein overexpressed in neuroblastoma (4).
PARP-targeted radiopharmaceutical therapeutics not only enable efficient delivery of radiation to DNA but also allow using other types of high-LET therapeutic radiation that otherwise would not penetrate cells. Auger electrons are emitted in cascades and have a low energy of up to 100 keV with less than a 0.5-μm range (5). The most extensively investigated Auger emitter is 125I, with a 60-d half-life and 20–25 Auger emissions per decay (5), exhibiting high relative biological effectiveness (RBE) and a low oxygen-enhancement ratio characteristic of high-LET radiation when integrated into DNA (6).
Therefore, targeting PARP-1 with an Auger-emitting 125I-labeled small molecule is an attractive strategy to deliver high-LET radiation to neuroblastoma. One such radiopharmaceutical is 125I-KX1, an analog of 211At-MM4 in which 211At is replaced by 125I (4,7). Although 125I-KX1 has previously been described as a biomarker to quantify PARP-1 expression in ovarian cancer models (7), its therapeutic use has not been investigated in any cancer models.
In the present study, we used 125I-KX1 to examine the therapeutic potential of a PARP-1–targeted Auger emitter in preclinical models of high-risk neuroblastoma (Fig. 1). After characterizing its PARP-dependent in vitro cytotoxicity, we made a dosimetry-based comparison of 125I-KX1 to α-, β-, γ-, and cytoplasmic Auger radiation to evaluate its effectiveness relative to other conventional and experimental therapies. Finally, a comparison with its α-emitting analog 211At-MM4 was made using in vivo tumor dosimetry to explore the feasibility of using Auger-emitting PARP-targeted radiopharmaceutical therapeutics for in vivo therapy.
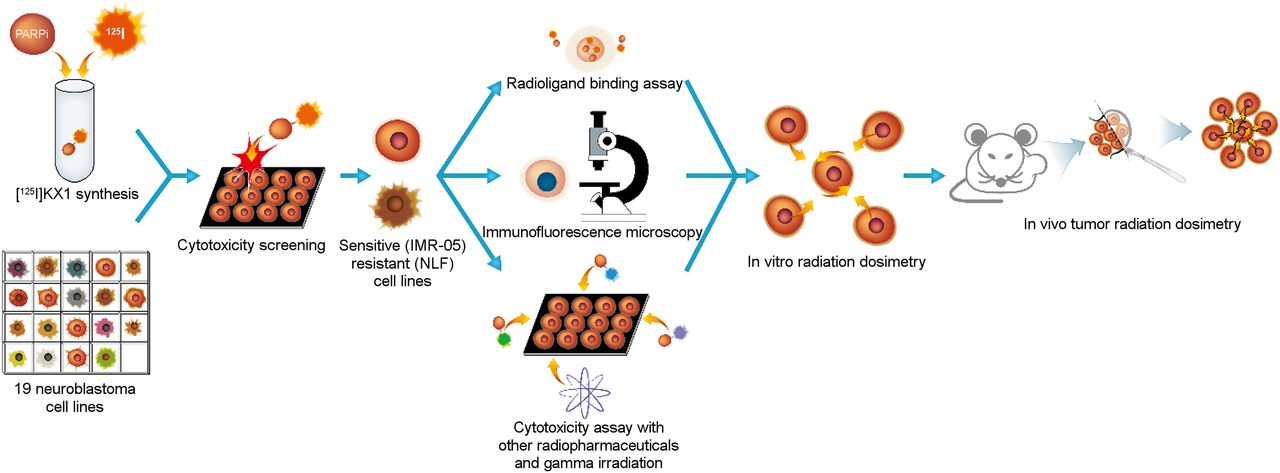
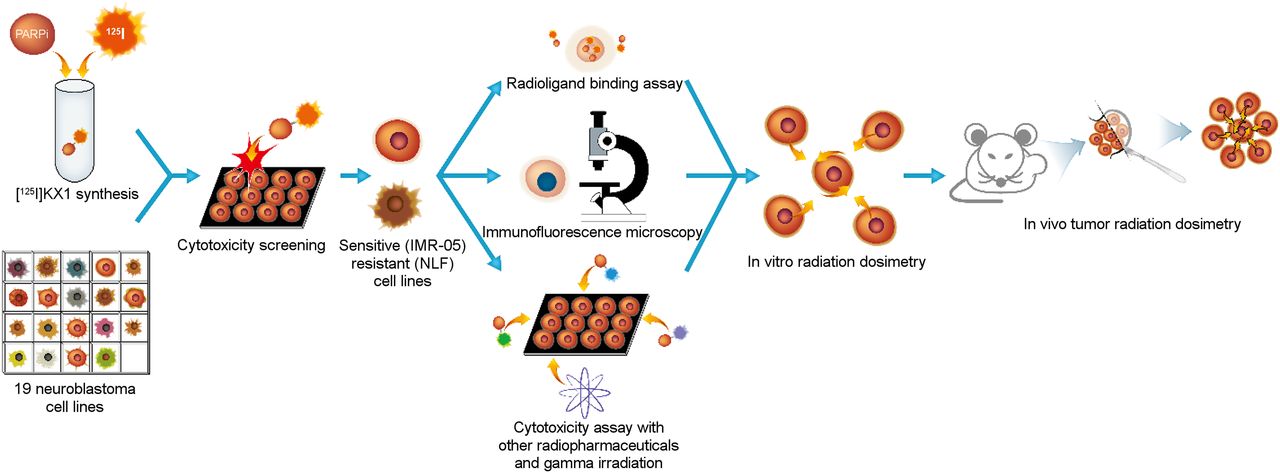
Workflow for preclinical evaluation of 125I-KX1 in high-risk neuroblastoma. Nineteen human neuroblastoma cell lines were screened for cytotoxic sensitivity to 125I-KX1. IMR-05 and NLF were chosen as representative cell lines for downstream experiments. Radioligand binding assay, immunofluorescence microscopy, and further cytotoxicity screening were followed by in vitro and in vivo radiation dosimetry.
MATERIALS AND METHODS
The overall experimental design is shown in Figure 1.
Chemistry and Radiochemistry
Nonradioactive KX1, 125I-KX1, 211At-MM4, and 125I-MIBG were synthesized as previously described, with greater than 95% chemical and radiochemical purity (4,7,8). 131I-KX1 was synthesized in an identical manner to 125I-KX1. The specific activities of 125I-KX1/125I-MIBG, 131I-KX1, and 211At-MM4 were 81.4, 592, and 16.021 GBq/μmol, respectively.
Cell Culture
Nineteen human neuroblastoma cell lines were obtained from the Children’s Hospital of Philadelphia Cell Bank (Supplemental Table 1; supplemental materials are available at http://jnm.snmjournals.org) and were cultured as previously described (4).
Immunofluorescence Microscopy
To characterize the specificity of 125I-KX1 Auger-induced DNA damage, IMR-05 and NLF cells were treated with a 1.85 MBq/mL concentration of 125I-KX1 with or without 500 nM veliparib for 1 h and were subsequently analyzed by assessing γH2AX (05-636; Millipore) and PARP-1 (46D11; Cell Signaling Technology) fluorescence under manufacturer-recommended conditions. Secondary antibodies were used at a 1:200 dilution (A32723 and A32794; Invitrogen). The slides were mounted using Prolong Glass with NucBlue stain (P36985; Invitrogen), and images were acquired at ×40 magnification on a Leica STED 3X super-resolution confocal microscope. γH2AX fluorescence was quantified on mapped nuclei by intensity and normalized to nucleus area using the Speckle Counting pipeline of the CellProfiler software (9). Three random fields of view were quantified per treatment condition.
In Vitro Cytotoxicity
The cytotoxicity of 125I-KX1 was screened in 19 human neuroblastoma cell lines as previously described (4), with 72-h treatments of 0.925 Bq/mL to 925 kBq/mL concentrations of 125I-KX1 compared with 100 pM to 100 μM KX1 as nonradioactive controls. Half-maximal effective concentrations (EC50) of 125I-KX1 were correlated with EC50 values of KX1 and published EC50 values of 211At-MM4 (4), as well as with PARP-1 messenger RNA (mRNA) expression levels measured by reverse transcription polymerase chain reaction.
For all downstream experiments, IMR-05 and NLF were chosen as examples of a sensitive and a resistant cell line, respectively (Fig. 1). External γ-radiation was delivered with a 137Cs source at a dose rate of 1.5 cGy/s, up to a 2-Gy total dose for IMR-05 and 6 Gy for NLF. The cells were seeded immediately after the irradiation, and survival fraction was quantified after 72 h as in the 125I-KX1 cytotoxicity assay. Survival fraction was plotted against radiation dose using the linear-quadratic model (10).
As a model for targeted low-LET β-therapy, a cytotoxicity assay with 131I-KX1 was performed on IMR-05 and NLF cell lines with a dose range of 2.4 kBq/mL to 37 MBq/mL. Cytotoxicity data for 211At-MM4 and unconjugated free 211At (211At-NaAtx) were obtained from the literature as examples of targeted and nontargeted α-particle therapy, respectively (4). Finally, a cytotoxicity assay with 125I-MIBG was performed at a dose range of 0.925 Bq/mL to 925 kBq/mL to model cytoplasmic Auger emitter therapy. 125I-MIBG served as a strict negative radioactive control in lieu of free 125I, which is not taken up by neuroblastoma cells and therefore deposits only an extracellular Auger dose (11). Experiments were completed in triplicates 3 independent times.
Radiopharmacology
A radioligand saturation binding assay was performed with 125I-KX1 on IMR-05 and NLF cell lines as previously published (4). The maximum number of binding sites (Bmax) was expressed as targets per cell by normalizing to the cell number using a Countess II Automated Cell Counter (Thermo Fisher Scientific). The binding affinity (Kd) of 211At-MM4 was obtained indirectly by comparison to 125I-KX1 binding under nonsaturating concentrations up to 37 kBq/mL (Supplemental Eq. 1). 125I-MIBG uptake was measured after a 2-h incubation at the nonsaturating concentrations used in the cytotoxicity assay (≤925 kBq/mL) with or without blocking with 10 μM of desipramine (12).
Subcellular Radiation Dosimetry
To calculate the radiation dose to the cell nucleus from 125I-KX1, 131I-KX1, 125I-MIBG, and 211At-MM4, on-target cumulated activity was derived from the radiopharmacology data (Supplemental Eq. 2). Then, the radiation dose to the cell nucleus was obtained using Monte Carlo simulation with Medical Internal Radiation Dosimetry Cell (MIRDcell), version 2.1, as described previously (13). The radii of the cell and its nucleus were measured with phase-contrast and fluorescence microscopy with 4′,6-diamidino-2-phenylindole, respectively.
To calculate the cross-dose from neighboring cells, the growth rates of IMR-05 and NLF cell lines were measured by counting the cells daily with a Countess II Automated Cell Counter. Then, the cell numbers, cumulated activity, radionuclide information, and dimensions of the wells in the cytotoxicity assay were applied to the 2-dimensional dosimetry model on MIRDcell to obtain cross-dose values. For nonspecific α-therapy with 211At-NaAtx, the radiation dose to the culture medium at the bottom of the wells was used to represent cellular radiation dose.
With the dosimetric calculations, the cytotoxic dose–response curves for the radiopharmaceuticals were transformed to radiation dose–response curves based on the linear-quadratic model. Using 50% survival as the reference endpoint, the RBE among the different types of radiation was calculated.
In Vivo Tumor Dosimetry with 3-Dimensional Modeling
To compare the therapeutic efficacy of 125I-KX1 and 211At-MM4 for in vivo therapy, radiation dosimetry was performed with densely packed 3-dimensional face-centered cubic modeling of a solid xenograft tumor consisting of spheric IMR-05 tumor cells. Radiation dosimetry was then performed both macroscopically and microscopically. First, per decay event in every tumor cell, the radiation dose to the macroscopic tumor volume due to the entire particulate energy absorption was calculated. In addition, the subcellular radiation dose to the tumor cell nuclei was calculated by accounting for the self- and cross-dose contributions based on the geometric distances among tumor cells in the 3-dimensional model. The S value for cross-dose calculation was obtained for each cell–cell pair on MIRDcell, version 2.1 (13).
Finally, the nuclear radiation dose per decay event was adjusted by differences in RBE and Kd to PARP-1, as well as effective half-lives for 125I-KX1 and 211At-MM4 from previously published in vivo biodistribution data (4,7). The adjusted dosimetry results enabled comparison of equivalent therapeutic administered doses between 125I-KX1 and 211At-MM4.
Statistical Analysis
The log(EC50) values between 125I-KX1 and KX1 for each cell line were compared using an unpaired t test. Linear regression was performed to determine the variation in log(EC50) of 125I-KX1 with respect to log(EC50) of KX1 and 211At-MM4, as well as with respect to PARP-1 mRNA level. The γH2AX fluorescence intensity of the cells treated with 125I-KX1 was compared with those of other treatment conditions using an unpaired t test. The Bmax, Kd, growth rate, log(EC50), and dose for 50% cell survival were compared between IMR-05 and NLF cell lines using an unpaired t test. All statistical comparisons were 2-tailed, and a P value of less than 0.05 was considered statistically significant. Prism (version 7; GraphPad Software) was used to perform all statistical tests and data fitting. Numeric values are reported as mean ± SE of measurement (SEM).
RESULTS
Immunofluorescence Microscopy
In IMR-05 and NLF cells, treatment with 125I-KX1 caused double-strand DNA breaks as visualized with γH2AX on immunofluorescence microscopy (Fig. 2). γH2AX was substantially reduced when 125I-KX1 binding to PARP-1 was blocked by 500 nM of veliparib.

Dependency of 125I-KX1–induced DNA damage on PARP-1. (A) Immunofluorescence microscopy images with γH2AX show 125I-KX1–induced double-strand DNA breaks, which were substantially blocked with veliparib. (B) Quantification of fluorescence intensity revealed statistically significant γH2AX signal increase with 125I-KX1 relative to other conditions. ****P < 0.0001.
In Vitro Cytotoxicity of 125I-KX1
Compared with the nonradioactive analog KX1, 125I-KX1 demonstrated 104–106 times greater cytotoxicity than KX1 on a molar scale (Figs. 3A [P < 0.0001] and 3B). Sensitivity to 125I-KX1 and KX1 was moderately correlated (R2 = 0.58, P < 0.001, for nonzero slope), with an exponential increase in sensitivity to 125I-KX1 (slope, 2.15 ± 0.47 on log–log plot) (Fig. 3C). Sensitivity to 125I-KX1 and 211At-MM4 showed a strong positive correlation (R2 = 0.93, P < 0.0001, for nonzero slope) with a 1-to-1 relationship (slope, 1.07 ± 0.08) (Fig. 3D). PARP-1 mRNA level was only minimally correlated with sensitivity to 125I-KX1 (R2 = 0.23, P < 0.05, for nonzero slope) (Fig. 3E).
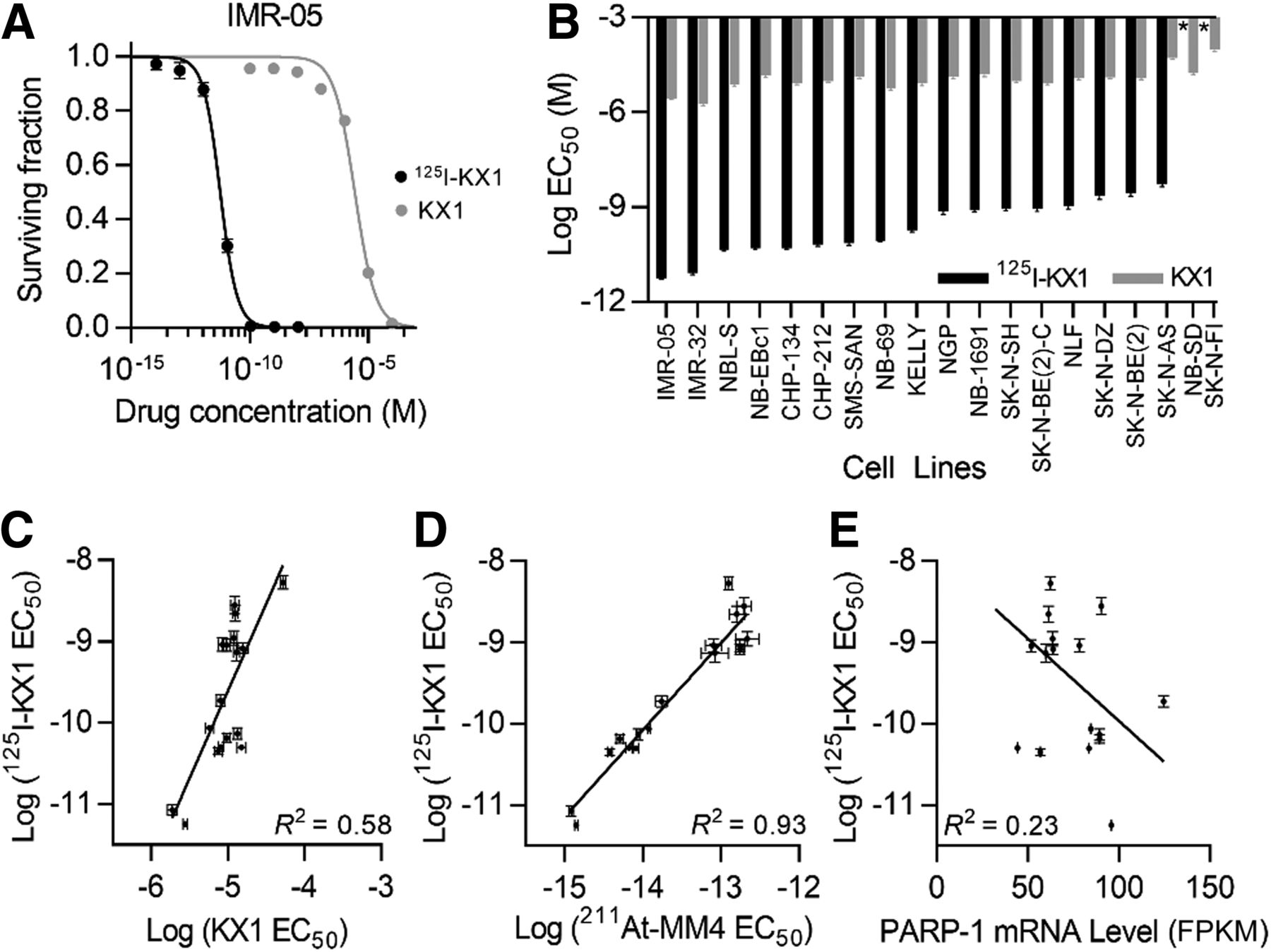
In vitro studies evaluating 125I-KX1–induced cytotoxicity in neuroblastoma cell lines. (A) Representative cytotoxic dose–response curves for 125I-KX1 and nonradioactive KX1 in neuroblastoma cell line (IMR-05). (B) Waterfall plot of EC50 values for 125I-KX1 and KX1 in panel of 19 neuroblastoma cell lines. *Less than 50% cell kill in dose range tested (P < 0.0001 for 125I-KX1 vs. KX1 in all 17 cell lines with >50% cytotoxicity). (C–E) Sensitivity to 125I-KX1 was correlated with sensitivity to KX1 (C) and 211At-MM4 (D), but minimally with PARP-1 mRNA level (E). FPKM = fragments per kilobase million.
Although IMR-05 cells were more sensitive to all forms of radionuclide therapy than NLF (P < 0.0001), different radiopharmaceuticals maintained the same order of potency relative to one another in both cell lines (Fig. 4A). The most potent radiopharmaceutical was 211At-MM4, followed by 125I-KX1, 211At-NaAtx, 131I-KX1, and 125I-MIBG (Supplemental Table 2). As with radiopharmaceuticals, external γ-irradiation was more lethal, with a lower D50 in IMR-05 (0.71 ± 0.02 Gy) than in NLF (3.7 ± 0.1 Gy) (P < 0.0001) (Fig. 4B).

Response to various types of radiotherapy with radiation dosimetry. (A) Cytotoxic dose–response curves for 211At-MM4, 125I-KX1, 211At-NaAtx, 131I-KX1, and 125I-MIBG based on radioactivity concentration. (B) Cell survival curves for nontargeted radiotherapy with external γ-irradiation, 211At-NaAtx, and 125I-MIBG based on radiation dose to cell nuclei. (C) Cell survival curves for PARP-targeted radiopharmaceutical therapeutics (125I-KX1, 131I-KX1, and 211At-MM4) based on radiation dose to cell nuclei.
Radiopharmacology
Radioligand binding assays revealed that IMR-05 had higher 125I-KX1 uptake (P < 0.0001) but lower 125I-MIBG uptake (P < 0.0001) than NLF, whereas the Kd of 125I-KX1 (P = 0.40) and 211At-MM4 (P = 0.38) was similar between the 2 cell lines (Supplemental Figs. 1A–1C), suggesting different expression levels of structurally identical target proteins. IMR-05 demonstrated a higher growth rate than NLF (P < 0.0001) (Supplemental Fig. 1D), leading to a higher cross-dose deposition due to proximity between adjacent cells. On microscopy, the nuclear and cellular radii of IMR-05 cells were measured to the nearest whole number at 6 and 8 μm, respectively, compared with NLF with 9- and 12-μm radii (Supplemental Fig. 1E).
Subcellular Radiation Dosimetry
Despite the wide variation in EC50, dosimetric analysis revealed that the RBEs of the radiopharmaceuticals were within the same order of magnitude. Without PARP-1 targeting, α-particles from 211At-NaAtx demonstrated 3.6–3.7 times greater RBE than γ-irradiation (Fig. 4B). On the other hand, cytoplasmic Auger radiation from 125I-MIBG was slightly weaker than γ-radiation in IMR-05 (RBE, 0.65) and equally efficacious in NLF (RBE, 1.0).
Among the PARP-1–targeted radiopharmaceuticals, the α-emitting 211At-MM4 remained the most effective, with RBEs of 6.3–6.7, compared with the high-energy β-emitting 131I-KX1 (Fig. 4C). In contrast to 125I-MIBG, the Auger-emitting 125I-KX1 was more effective than 131I-KX1, with RBEs of 1.8–1.9. As expected from their high LET or low dose rate, all the targeted radiopharmaceuticals demonstrated linear (α-dominant) survival curves in the linear-quadratic model (Supplemental Table 3) (10). Therefore, the RBEs were maintained across a different choice of endpoints. A summary of the in vitro dosimetry results and RBEs is provided in Table 1.
In Vitro Dosimetry
In Vivo Tumor Dosimetry with 3-Dimensional Modeling
The face-centered cubic tumor model and target volumes are represented in Figures 5A and 5B, respectively. When the radiation dose to the entire tumor volume was considered, 211At-MM4 deposited 350 times more dose to the tumor per decay event than did 125I-KX1 because of the much higher energy carried by the α-particles than by Auger electrons. Compared with the entire tumor dose, the radiation dose only to the tumor cell nuclei was higher with 125I-KX1 because of its highly localized dose deposition centered on the nuclei, whereas it was unchanged with 211At-MM4 (Fig. 5C). Therefore, compared with 125I-KX1, 211At-MM4 deposited only a 150 times higher tumor-nucleus dose per decay event.

In vivo tumor radiation dosimetry with 3-dimensional modeling. (A) Diagram showing tumor cell and its nearest neighbors. Cell (red) receives self-dose as well as cross-dose from neighboring cells on same (green) and different (yellow) planes. (B) Target volume diagrams for conventional organ-level dosimetry (whole tumor) and microdosimetry (cell nuclei). (C) Dose-per-decay-event comparison of 211At-MM4 and 125I-KX1.
Previously published in vivo biodistribution results for 211At-MM4 and 125I-KX1 have been remarkably similar because of their identical chemical structures except for the radionuclide (4,7). Although the effective half-life of 125I-KX1 (1.5 h) is approximately 36% longer than that of 211At-MM4 (1.1 h) because of the longer physical half-life of 125I, it has approximately 35% less target Kd (5.8 nM) than 211At-MM4 (4.3 nM) in the IMR-05 cell line. Given the small differences in biologic distribution between the 2 compounds, the primary difference between 211At-MM4 and 125I-KX1 for in vivo efficacy estimation lies in the RBE of 3.5. Taking into account the 3.5-fold higher RBE and the 150-fold greater nuclear radiation dose per decay—both being in favor of 211At-MM4—administration of a 530-fold greater activity of 125I-KX1 is required for in vivo efficacy equivalent to 211At-MM4.
DISCUSSION
The present study evaluated the feasibility of chromatin-targeted high-LET Auger electron therapy in high-risk neuroblastoma with the iodinated PARP-1 inhibitor 125I-KX1. We first validated the basic mechanism of 125I-KX1–induced cytotoxicity: 125I-KX1 causes double-strand DNA breaks dependent on binding to active PARP-1 (Fig. 2) and not associated with pharmacologic inhibition (Figs. 3A and 3B).
Since sensitivity to PARP inhibitors such as KX1 is strongly correlated with the number of pharmacologically active binding sites (Bmax) (7), the correlation between sensitivity to 125I-KX1 and KX1 (Fig. 3C) reflects the relationship between 125I-KX1–induced cytotoxicity and its degree of target binding. The weak correlation found between sensitivity to 125I-KX1 and PARP-1 mRNA should be interpreted with caution, as previous reports demonstrated a lack of correlation between PARP-1 mRNA expression and protein level (14). Although an association is evident between 125I-KX1 sensitivity and PARP-1 mRNA expression, future studies will be needed to identify the most appropriate method for quantifying PARP-1.
The RBEs of 125I obtained from subcellular dosimetry in IMR-05 and NLF cell lines were in accordance with the previous understanding of Auger therapy. The RBE of cytoplasmic 125I-MIBG was at best equal to γ-irradiation and was slightly lower, at 0.65, in IMR-05 cells because of the loss of quadratic (β) cell kill at low dose rates (Supplemental Table 3) (10). The RBE of 125I-KX1, at approximately 2, demonstrates the high radiotoxicity of Auger therapy but is slightly lower than the previously documented RBE of 4–5 for DNA-incorporated 5-125I-iodo-2′-deoxyuridine (125IdUrd) versus 131IdUrd (15). The lower RBE likely results from the distance of 50 Å from the PARP-1 active site to DNA (Supplemental Fig. 2) (16–18), which further increases when PARP-1 is indirectly bound to DNA via chromatin-associated proteins. Even an angstrom-level increase in distance from DNA leads to a precipitous decrease in the biological effectiveness of 125I, where 12 Å is the critical distance (19). Rather, the observed RBE of 125I-KX1 (≤4) against γ-irradiation matches those of nucleus-targeted Auger emitters noncovalently bound to DNA (20). Therefore, 125I-KX1 demonstrated a high RBE corresponding to its subcellular targeting, which serves as a key component of its toxicity.
Compared with previously described therapeutic Auger emitters, the major significance of 125I-KX1 is the combination of high RBE and target specificity. Attempts to specifically deliver Auger emitters with antibodies and small molecules such as 125I-MIBG suffered from low cytotoxicity due to nonnuclear localization (21,22). DNA-targeting Auger emitters, including 125IdUrd, have demonstrated high effectiveness, but the ubiquitous nature of DNA leads to poor target specificity and high normal-tissue toxicity (23). Although endocrine receptors have been targeted with Auger emitters (24,25), the receptors are not always bound to DNA and a specific endocrine receptor target has not been found in neuroblastoma. A previous study used 111In-labeled oligonucleotides complementary to N-myc in order to specifically deliver Auger radiation to neuroblastoma (26). Although reduced expression of N-myc led to slowed growth rate, the tumor cells remained viable because of the very limited nuclear uptake of oligonucleotides in unaltered form (26,27).
Interestingly, the increase in RBE with PARP-1 targeting was observed not only with 125I but also with 211At, as can be explained by 2 properties specific to 211At. First, α-emission from 211At is accompanied by recoil of the parent nucleus with a range of 90 nm in the direction opposite to that which delivers high-LET radiation and therefore contributes to cytotoxicity only when the decay occurs in the nucleus (28). Second, 211At also has lesser-known emission of approximately 6.3 Auger electrons per decay, which deposit more radiation dose than α-particles within 10 nm (29). Despite the higher Auger yield of 125I, the combined effect of recoil and Auger emission from 211At decay makes PARP-1–targeted 211At therapy a more effective approach.
The benefit of 211At-MM4 therapy over 125I-KX1 was also demonstrated by in vivo tumor dosimetry. Despite 125I-KX1’s focused delivery of radiation to the cell nuclei, 211At-MM4’s much higher nuclear dose per decay and favorable RBE led to 2-orders-of-magnitude-higher therapeutic potency per decay. In the setting of irreversible DNA integration in which the number of 125IdUrd decays was 10-fold higher than that of 211AtdUrd, approximately 30 times more tumor dose was deposited by 211AtdUrd, congruent with the results of our organ-level dosimetry. The previously established lowest effective therapeutic dose of 211At-MM4 in IMR-05 tumor–bearing mice was 555 kBq (4), which would translate to 296 MBq of 125I-KX1. We predicted that this dose would not be safe in mice, in view of the toxicity of 2 different Auger-emitting therapeutics investigated in previous studies: a 125I-labeled monoclonal antibody had a median toxic dose of 111 MBq in mice, and the more cytotoxic DNA-incorporating 125IdUrd had a median toxic dose of 74 MBq in rats (15,30). In addition, 131I-MIBG has shown antitumor activity in neuroblastoma models at 9.4 MBq, which questions the utility of Auger therapy in macroscopic disease (VA Batra, January 6, 2019, unpublished data). Another limitation of 125I-KX1 is its remarkably long physical half-life of 60 d, compared with the biologic half-life of 1.5 h, as well as its significant target-specific uptake in the spleen and pancreas (7).
Although Auger therapy may be impractical for therapeutic evaluation in solid-tumor models, the present study suggests its potential value in micrometastatic disease. We demonstrated that a PARP-1–targeted Auger emitter has enhanced RBE over β-emitting analogs at the cellular level. The therapeutic ratio of Auger emitters would be maximized in a micrometastatic setting by providing single-cell lethality without compromising surrounding healthy tissue. Other aspects to improve the translatability of PARP-targeted radiopharmaceuticals for Auger therapy include increasing on-target drug retention at PARP-1 through medicinal chemistry approaches or using shorter-lived Auger emitters such as 77Br to overcome the challenges of using long-lived isotopes.
CONCLUSION
In this work, we showed that Auger emitters are cytotoxic to high-risk neuroblastoma, with high-LET properties when targeted to PARP-1. However, the utility of systemically administered Auger therapy with unfavorable in vivo kinetics remains questionable for evaluation in solid-tumor models.
DISCLOSURE
Funding support for the work was received though U.S. Department of Energy grant DE-SC0017646 (Robert Mach) and from the National Institutes of Health through Research Project Grant R01-CA219006 (Daniel Pryma). Robert Mach, Daniel Pryma, and Mehran Makvandi are designated as inventors on a patent held by the University of Pennsylvania that describes the intellectual property associated with 211At-MM4 (WO2018218025A1). Daniel Pryma discloses unrelated conflicts, including honoraria and research support from Siemens, Progenics, and 511 Pharma, as well as honoraria from Actinium and Nordic Nanovector. No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Do Auger emitters exhibit high-LET properties when targeted to PARP-1, and can they be used to treat radiosensitive cancers?
PERTINENT FINDINGS: PARP-1–targeted Auger emitters exhibit high-LET properties, with enhanced RBE in neuroblastoma over γ- and β-radiation but with lower RBE than α-radiation. Dosimetric comparisons between PARP-1–targeted α- and Auger radiation revealed that α-radiation deposits a much higher dose to the tumor than Auger radiation, primarily attributed to the low energy of Auger electrons.
IMPLICATIONS FOR PATIENT CARE: PARP-1 has emerged as a clinical drug target for treating cancers with DNA repair deficiencies, and our studies show that targeting PARP-1 with α- and Auger radiation is potently cytotoxic in neuroblastoma, warranting future clinical translation.
Footnotes
Published online Nov. 1, 2019.
- © 2020 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication July 16, 2019.
- Accepted for publication October 9, 2019.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}