


Visual Abstract

Abstract
Asymmetries of amyloid-β (Aβ) burden are well known in Alzheimer disease (AD) but did not receive attention in Aβ mouse models of Alzheimer disease. Therefore, we investigated Aβ asymmetries in Aβ mouse models examined by Aβ small-animal PET and tested if such asymmetries have an association with microglial activation. Methods: We analyzed 523 cross-sectional Aβ PET scans of 5 different Aβ mouse models (APP/PS1, PS2APP, APP-SL70, AppNL-G-F, and APPswe) together with 136 18-kDa translocator protein (TSPO) PET scans for microglial activation. The asymmetry index (AI) was calculated between tracer uptake in both hemispheres. AIs of Aβ PET were analyzed in correlation with TSPO PET AIs. Extrapolated required sample sizes were compared between analyses of single and combined hemispheres. Results: Relevant asymmetries of Aβ deposition were identified in at least 30% of all investigated mice. There was a significant correlation between AIs of Aβ PET and TSPO PET in 4 investigated Aβ mouse models (APP/PS1: R = 0.593, P = 0.001; PS2APP: R = 0.485, P = 0.019; APP-SL70: R = 0.410, P = 0.037; AppNL-G-F: R = 0.385, P = 0.002). Asymmetry was associated with higher variance of tracer uptake in single hemispheres, leading to higher required sample sizes. Conclusion: Asymmetry of fibrillar plaque neuropathology occurs frequently in Aβ mouse models and acts as a potential confounder in experimental designs. Concomitant asymmetry of microglial activation indicates a neuroinflammatory component to hemispheric predominance of fibrillary amyloidosis.
Alzheimer disease (AD) is the most frequent neurodegenerative disease, with burgeoning incidence rates due to the rising life expectancy in most of the world (1). The neuropathology of AD is histologically characterized by the triad of accumulation of amyloid-β (Aβ) peptide as extracellular plaques, aggregation of fibrillary tau protein within neurons, and activation of multiple neuroinflammatory pathways, as mediated by activated microglia expressing high levels of the marker 18-kDa translocator protein (TSPO) (2). Animal models that accurately reflect this complex pathology are indispensable for contemporary preclinical research into the molecular mechanisms of AD. In this context, a range of different overexpressing and knock-in Aβ mouse models has been established for molecular imaging with PET. In recent PET studies, increased binding of the Aβ tracer 18F-florbetaben and the TSPO tracer 18F-GE-180 was firmly established by longitudinal in vivo quantification of cerebral amyloidosis and microglial activation in various Aβ mouse models of AD (3–5). In humans, an asymmetric spatial distribution of neuropathologic AD hallmarks is frequently discovered by PET studies in vivo (6–8). A recent human PET study has already shown that asymmetric spatial distributions of Aβ plaques are positively correlated with ipsilateral neurodegeneration (8). However, no study has hitherto systematically investigated the asymmetry in Aβ mouse models of AD. Although a large-scale investigation of this phenomenon by histopathologic investigations would be costly and difficult in terms of standardization, in vivo PET imaging methods should afford the means to readily compare the Aβ plaque burden in both hemispheres of individual animals.
Given this background, our aim was to investigate the occurrence of asymmetric fibrillar Aβ deposition in the well-established Aβ mouse models APP/PS1, PS2APP, APP-SL70, AppNL-G-F, and APPswe. Using a large series of historical 18F-florbetaben Aβ PET recordings, we tested for asymmetric Aβ deposition while considering age as a predictive variable. We also estimated sample sizes for detecting asymmetric Aβ and tested the hypothesis that Aβ asymmetry is associated with ipsilateral microglial activation as assessed by 18F-GE-180 TSPO PET.
MARTERIAL AND METHODS
Experimental Design
All experiments were performed in compliance with the German National Guidelines for Animal Protection and with the approval of the regional animal committee (Regierung Oberbayern) and were overseen by a veterinarian. Animals were housed in a temperature- and humidity-controlled environment with a 12-h light–dark cycle, with free access to food (Sniff) and water. A detailed overview of the investigated mouse cohorts is given in Supplemental Table 1 (supplemental materials are available at http://jnm.snmjournals.org). All PET raw data originated from previous in-house studies (cited below) conducted on the same Inveon small-animal PET scanner under identical acquisition parameters. Of the mice investigated, 87% were female. APP/PS1 and APPswe comprised only female mice, whereas PS2APP, APP-SL70, and AppNL-G-F included both sexes. All raw data were reprocessed to guarantee optimal agreement of spatial and radioactivity normalization. Either descriptive datasets or control groups of therapy and genotype studies were included. From each investigated mouse, the degree of asymmetry in Aβ PET and TSPO PET was assessed by volume-of-interest–based quantification in both cerebral hemispheres.
Animal Models
APP/PS1 (APPPS1-21)
This transgenic mouse model was generated on a C57BL/6J genetic background that coexpresses KM670/671NL mutated amyloid precursor protein (APP) and L166P mutated presenilin (PS) 1 under the control of a neuron-specific Thy1 promoter. Cerebral amyloidosis in this model starts at 6–8 wk of age (9). Historical 18F-florbetaben data from 41 scans of APP/PS1 mice imaged at 4 different ages (3, 6, 9, and 12 mo) were reprocessed (10). Twenty-seven contemporaneous 18F-GE-180 scans were available.
PS2APP (APPswe/PS2)
The transgenic B6.PS2APP (line B6.152H) is homozygous both for human PS2, the N141I mutation, and for the human APP K670N/M671L mutation (11). Homozygous B6.PS2APP mice first show plaques in the cerebral cortex and hippocampus at 5–6 mo of age (12). Historical 18F-florbetaben data from 147 scans of PS2APP mice imaged at 4 different age ranges (6–8, 9–10, 11–14, and 15–17 mo) were reprocessed (13,14). Twenty-three contemporaneous 18F-GE-180 scans from these mice were likewise reprocessed by standard methods.
APP-SL70
The PS1 knock-in line was generated by introducing 2-point mutations in the wild-type (WT) mouse PSEN1, corresponding to the mutations M233T and L235P. The APP751SL mouse overexpresses human APP751 carrying the London (V717I) and Swedish (K670N/M671L) mutations under the control of the Thy1 promoter. Aβ deposits appear as early as 2.5 mo of age in these mice (15). Historical 18F-florbetaben data from 208 scans of APP-SL70 mice imaged at 4 different ages (4–6, 7–9, 10–12, and 13–15 mo), deriving from a descriptive observational study (16), along with control scans from an as-yet-unpublished therapy study were reprocessed. Twenty-six contemporaneous 18F-GE-180 scans were available in this group.
AppNL-G-F(AppNL-G-F/NL-G-F)
The knock-in mouse model AppNL-G-F carries a mutant APP gene encoding the humanized Aβ sequence (G601R, F606Y, and R609H) with 3 pathogenic mutations, namely Swedish (KM595/596NL), Beyreuther/Iberian (I641F), and Arctic (E618G). Homozygotic AppNL-G-F mice progressively exhibit widespread Aβ accumulation from 2 mo of age (17,18). Historical 18F-florbetaben data from 55 scans of homozygotic AppNL-G-F mice imaged at 4 different ages (2.5, 5.0, 7.5, and 10 mo) were reprocessed (3). Fifty-five contemporaneous 18F-GE-180 scans were available in this dataset.
APPswe
Transgenic mice overexpressing human APP with the Swedish double mutation (K670N, M671L) driven by the mouse Thy1.2 promoter were generated as described earlier (11). Mice heterozygous for the transgene begin accumulating β-amyloid at approximately 9 mo of age and develop β-amyloid plaques at 12 mo of age, mainly in the cortical mantle. Historical 18F-florbetaben data from 72 scans of APPswe mice imaged at 3 different age ranges (9–12, 13–16, and 17–20 mo) were reanalyzed (19,20). Contemporaneous 18F-GE-180 scans were not available for these mice.
C57BI/6
Historical and unpublished 18F-florbetaben data from 27 scans of C57BL/6 mice (WT) were reprocessed and served as control material (age, 2.5–16 mo).
PET Imaging
PET Data Acquisition, Reconstruction, and Postprocessing
For all PET procedures, radiochemistry, data acquisition, and image preprocessing were conducted according to an established, standardized protocol (4,21). In brief, 18F-florbetaben Aβ PET recordings (average dose, 11.4 ± 2.0 MBq) with an emission window of 30–60 min after injection were obtained to measure fibrillar cerebral amyloidosis. 18F-GE-180 TSPO PET recordings (average dose, 11.1 ± 2.0 MBq) with an emission window of 60–90 min after injection were performed for assessment of cerebral TSPO expression. Anesthesia was maintained from just before tracer injection to the end of the imaging time window.
PET Image Analysis
We performed all analyses using PMOD (version 3.5; PMOD technologies). Emission images were normalized to SUV ratio (SUVR) images using previously validated white matter reference regions for transgenic amyloid mouse models (APP/PS1, PS2APP, APP-SL70, and APPswe) (4,21). For the knock-in mouse line AppNL-G-F, the mesencephalic periaqueductal gray was used as a reference region, as recently published (3). Two bilateral telencephalic volumes of interest (containing cortex and hippocampus) comprising 50 mm3 each were used for calculation of the forebrain–to–white matter SUVR or the forebrain–to–periaqueductal gray SUVR. For each scan, the hemispheric asymmetry index (AI) was calculated for 18F-florbetaben or 18F-GE-180 scans using the following formula:
Statistical Analysis
We calculated 95% and 99% confidence intervals (CIs) for 18F-florbetaben AIs in normal C57BL/6 mice. Aβ mouse model 18F-florbetaben scans were judged as asymmetric when they exceeded the 95%CI (moderate asymmetry) or the 99%CI (strong asymmetry) of C57BL/6 mice. Significant 18F-florbetaben |AIs| (absolute magnitude) were correlated with age for each Aβ mouse model to evaluate the age dependency of asymmetric plaque distribution. For each Aβ mouse model, age-independent lateralized plaque distributions were compared by a χ2 test to test for left or right predominance of Aβ deposition. The frequency of strong asymmetries was calculated in groups of comparable age for Aβ mouse models and correlated with the coefficient of variance for SUVR in the same groups of mice. Pearson coefficients of correlation were calculated for the latter analyses and for correlation analyses between 18F-florbetaben AIs and age, as well as between 18F-florbetaben AIs and 18F-GE-180 AIs. Hypothetic 2-sided t tests of independent measures were done to calculate sample sizes for comparisons of SUVR in single hemispheres with SUVR in combined hemispheres using G*Power (version 3.1.9.2). We used a given 5% therapy effect on SUVR at a power (1 − β) of 0.80 and type 1 error with an α value of 0.05. A P value of less than 0.05 was considered to be significant for rejection of the null hypothesis. SPSS 25 statistics (IBM Deutschland GmbH) was used for all statistical tests.
RESULTS
Asymmetric Plaque Distribution Is Frequent in Aβ Mouse Models
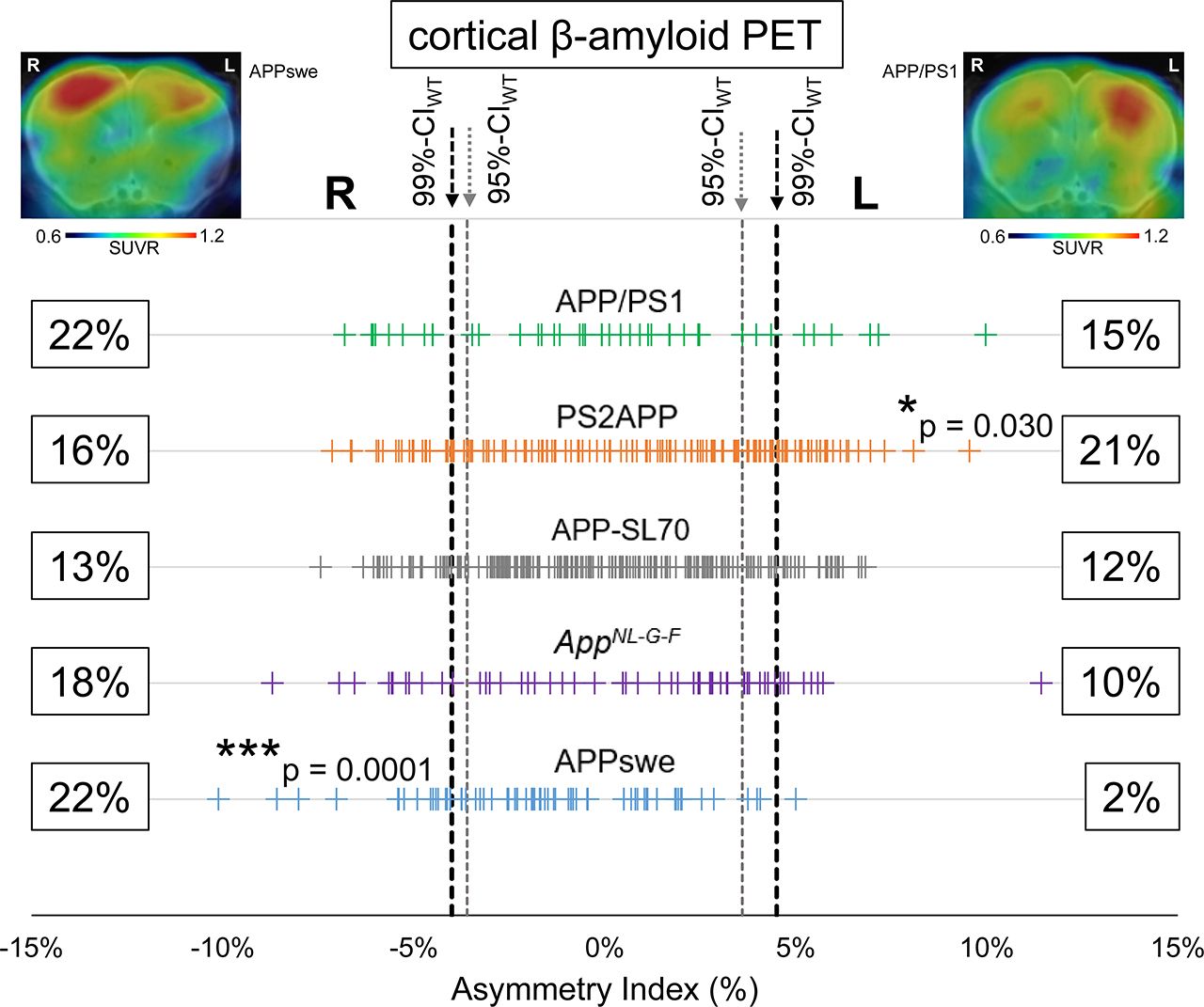
First, we defined an asymmetry threshold based on PET measurements in WT mice to establish real Aβ asymmetry, without bias in the spatial normalization or by physiologic variability in tracer uptake. The 95%Cl of 18F-florbetaben AIs in C57BL/6 mice was −3.6% (right lateralization) to 3.6% (left lateralization) and defined the threshold for moderate Aβ asymmetry. The 99%Cl of 18F-florbetaben AIs in C57BL/6 was −4.0% (right lateralization) to 4.5% (left lateralization) and defined the threshold for strong Aβ asymmetry. Using these thresholds, 40% (left, 21%; right, 19%; 95%CI) of all amyloid-accumulating mice showed moderate asymmetry of 18F-florbetaben forebrain uptake and 30% (left, 14%; right, 16%; 99%CI) showed strong asymmetry (Fig. 1). There was no significant hemispheric predominance across the whole cohort of different Aβ mouse models. A detailed overview is provided in Supplemental Table 1.
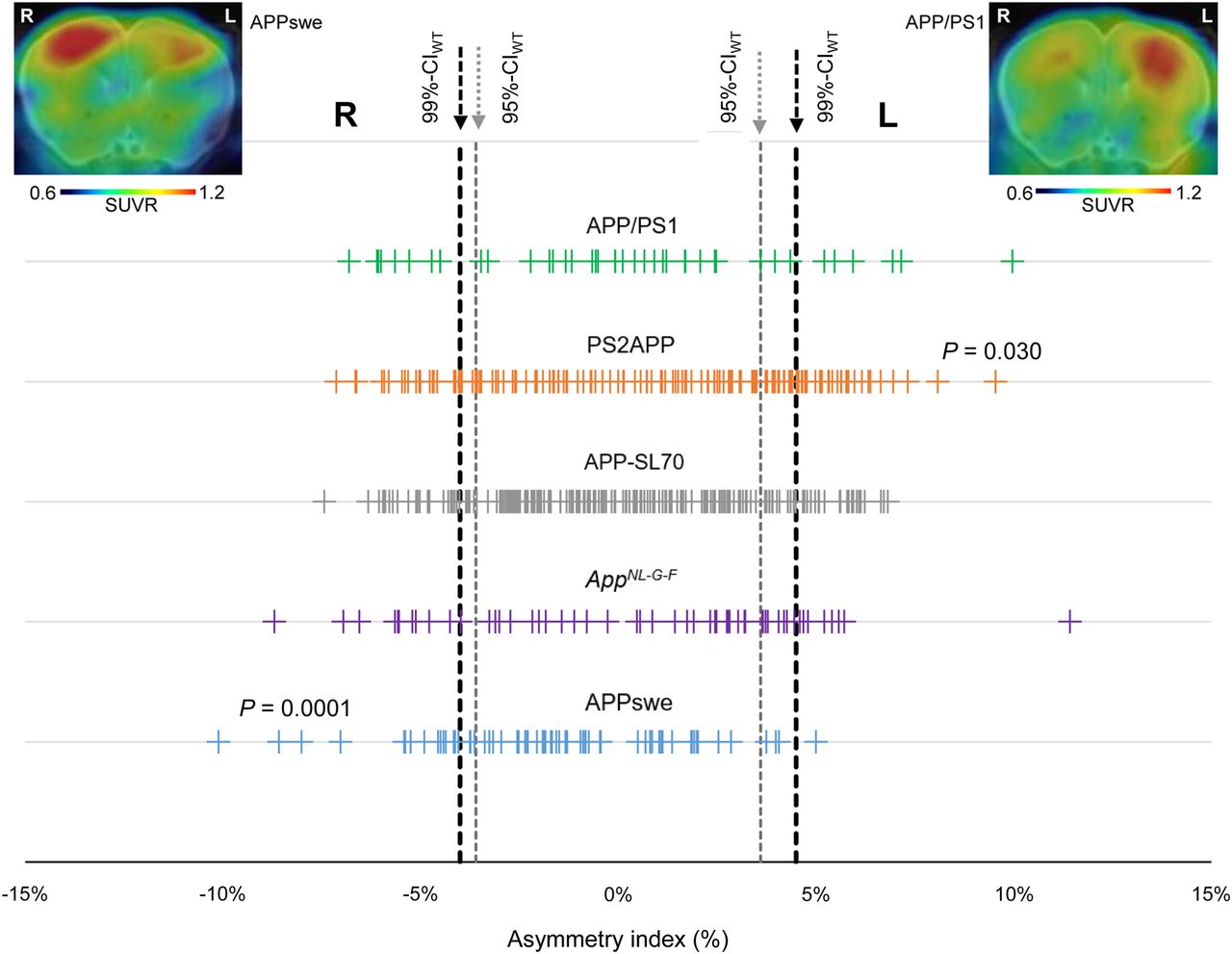
Asymmetry of plaque distribution in amyloid mouse models. Forest plot shows AI for total of 523 amyloid PET scans in APP/PS1, PS2APP, APP-SL70, APPswe, and AppNL-G-F mice. Lateralized plaque distributions were compared by χ2 test to test for left or right predominance in each mouse model. Representative PET SUVR images show exemplary mice with right (APPSwe) and left (APP/PS1) asymmetry.
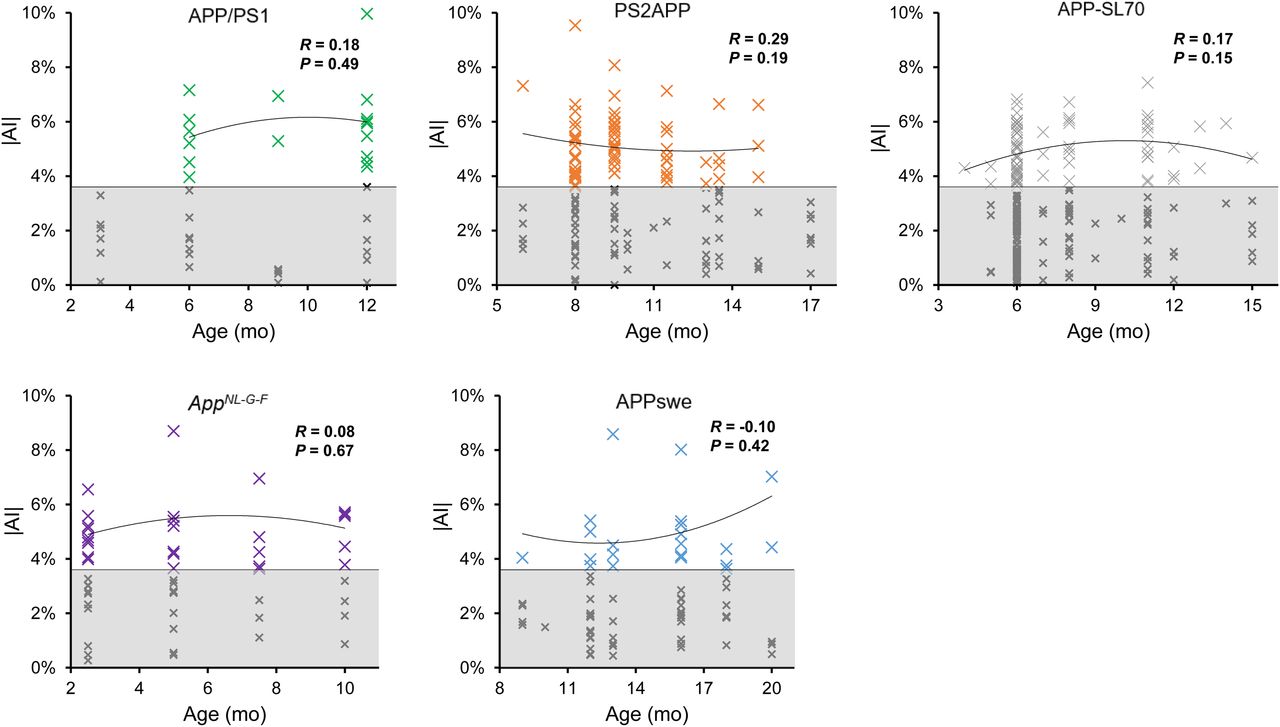
The highest frequency of moderate Aβ PET asymmetry was observed in PS2APP and AppNL-G-F mice (49% each). Strong Aβ PET asymmetry was most frequently observed in PS2APP and APP/PS1 mice (37% each). The lowest frequency of Aβ PET asymmetry was present in APPswe mice, in which 32% of scans indicated moderate and 24% strong asymmetry. A significant left-hemispheric predominance of Aβ deposition was detected in the PS2APP mice (χ2 = 4.7; P = 0.030), whereas a significant right-hemispheric predominance of Aβ deposition was seen in APPswe mice (χ2 = 15; P = 0.0001). There was no significant association between age and asymmetric Aβ distribution in any Aβ mouse model (Fig. 2). In summary, asymmetry of plaque burden was frequently observed in all studied Aβ mouse models, but with different magnitudes and side predilections.
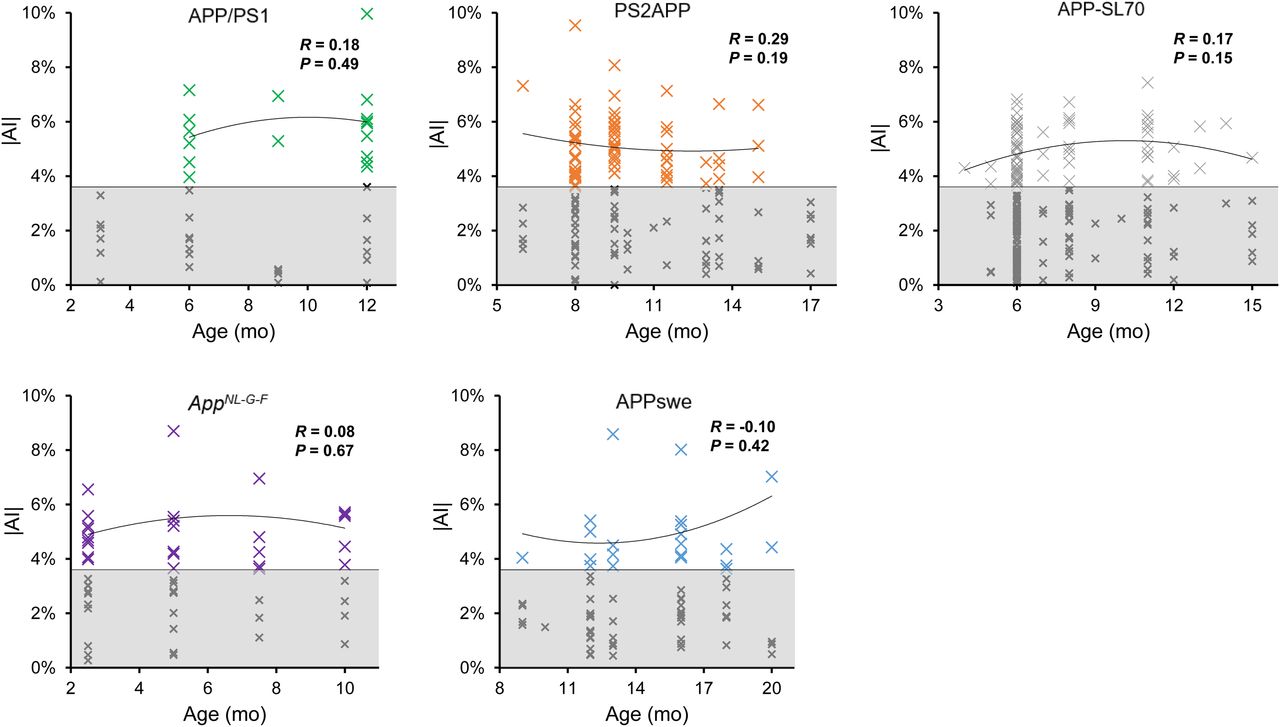
Age dependency of asymmetric amyloid deposition. Asymmetry (|AI|) is shown as function of age for APP/PS1, PS2APP, APP-SL70, APPswe, and AppNL-G-F mice. Datapoints with significant asymmetric 18F-florbetaben uptake (|AI| > 95%CIWT; white area) indicate no relevant dependency of asymmetric plaque distribution on age in any mouse models. Values with symmetric distribution (gray area) were excluded from correlation analysis.
Asymmetric Plaque Burden Impacts the Sufficient Sample Sizes in Preclinical Trials
Given the observed asymmetries in all Aβ mouse models studied, we hypothesized that measures in single hemispheres (as are typically examined by histologic methods) would suffer from higher variance, subsequently leading to increased required sample sizes in preclinical trials when compared with combined measures of both hemispheres, as are obtained by PET. Coefficient of variance was positively associated with the frequency of plaque burden asymmetry (99%CI) in groups of comparable age in different Aβ mouse models (R = 0.380, P = 0.027, Fig. 3A). Coefficient of variance by groups of comparable age in the different Aβ mouse models was 4.3% ± 1.2% for separate measures of left and right hemispheres and significantly lower for the combined quantification of both hemispheres (3.9% ± 1.2%; P = 0.0003, left vs. both; P = 0.0007, right vs. both; paired t test). For detection of a 5% therapy effect on SUVR at a power (1 − β) of 0.80 and type 1 error with an α value of 0.05, calculated sample sizes were 14.1 for separate measures for the left hemisphere, 13.9 for separate measures of the right hemisphere, and 11.9 for combined quantification of both hemispheres (P = 0.0020 and 0.0016 for left vs. both and right vs. both, respectively; paired t test). Required sample sizes as a function of power were consistently increased for calculation with left (Fig. 3B) and right (Fig. 3C) hemispheric values when compared with combined quantification of both hemispheres. The average reductions in required sample sizes for combined quantification of both hemispheres were 2.1 ± 0.6 (vs. left) and 1.8 ± 0.5 (vs. right). These results indicate that asymmetry of plaque burden in Aβ mouse models considerably increases required sample sizes when hemispheres are analyzed separately.
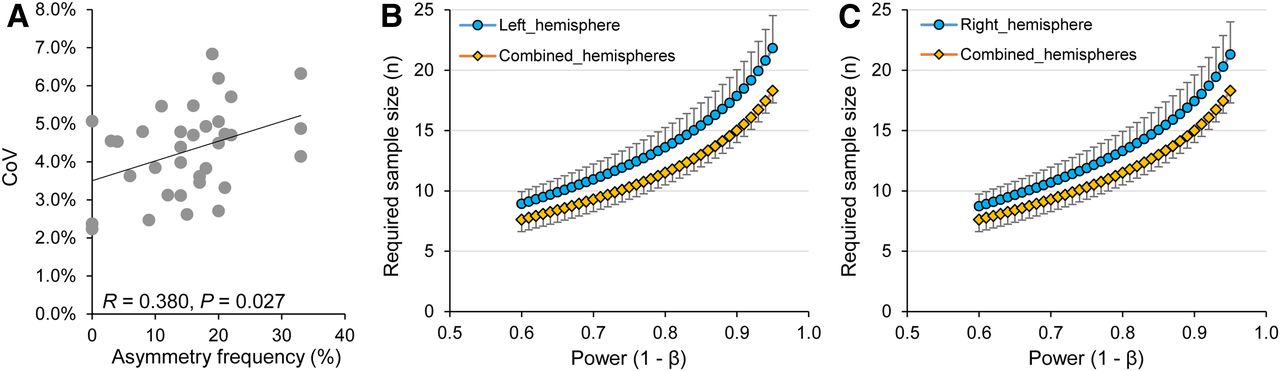
Statistical relevance of asymmetric plaque distribution in amyloid mouse models. (A) Association of higher coefficients of variation (CoV) in SUVR with higher frequency of asymmetry in age-related groups of amyloid mouse models (Supplemental Table 1). (B and C) Required sample sizes as function of power in comparison of analyses in single hemispheres and combined hemispheres (given effect of 5%, α = 0.05, hypothetical 2-sided t test of independent measures).
Asymmetric Plaque Burden Is Associated with Ipsilateral Glial Activation
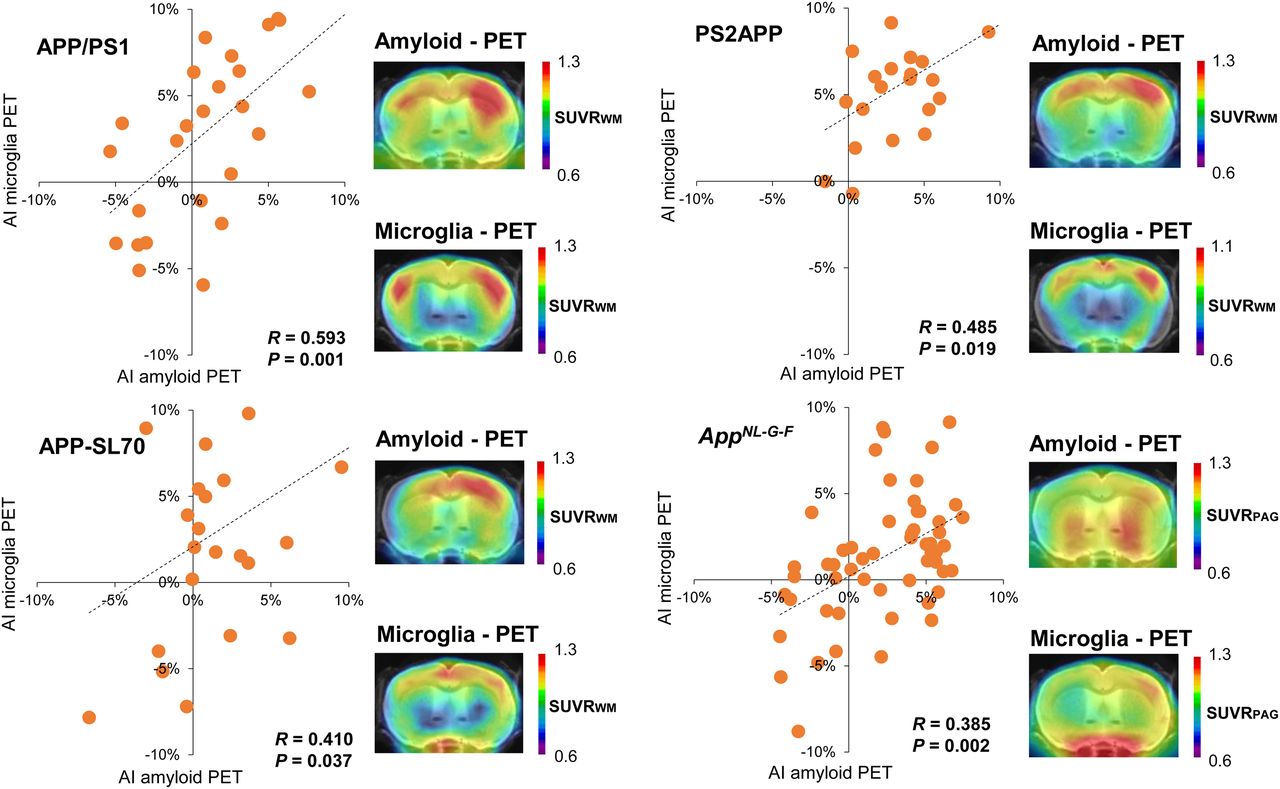
Several studies have revealed associations between amyloid deposition and microglial activation in Aβ mouse models (3,4,16). However, it has not hitherto been investigated if microglial activation follows any asymmetry of plaque burden or if the microgliosis is globally distributed. Hence, we made use of contemporaneous TSPO PET data for correlation analysis with lateralization to Aβ PET. Significant positive associations between asymmetric Aβ deposition and ipsilateral lateralization of TSPO expression were observed in all 4 Aβ mouse models (Fig. 4). The magnitude of correlation between asymmetric Aβ PET and ipsilateralized TSPO PET uptake was similar among APP/PS1 (R = 0.593; P = 0.001; n = 27; Pearson correlation), PS2APP (R = 0.485; P = 0.019; n = 23; Pearson correlation), APP-SL70 (R = 0.410; P = 0.037; n = 26; Pearson correlation), and AppNL-G-F (R = 0.385; P = 0.002; n = 60; Pearson correlation) mice. Taken together these results clearly indicate a spatial association between asymmetric distribution of fibrillar Aβ plaques and ipsilateral microglial activation.
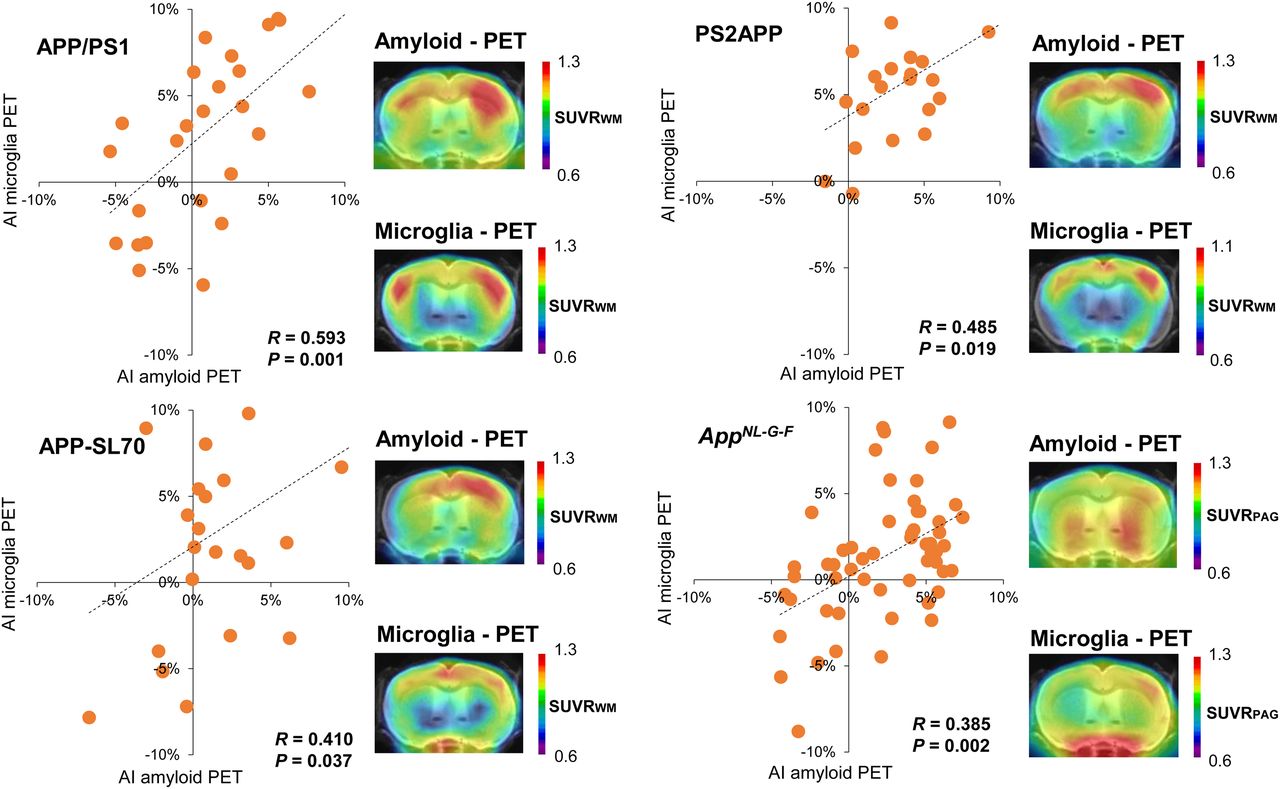
Association between lateralized amyloid deposition and microglia activation. Correlations between AIs of amyloid and microglia PET in APP/PS1, PS2APP, APP-SL70, and AppNL-G-F mice show congruent asymmetry of both biomarkers. PAG = periaqueductal gray; WM = white matter.
DISCUSSION
In contrast to human investigations on asymmetric Aβ distribution in AD (6,8,22), only scant evidence is available for the presence of Aβ asymmetry in mouse models (19,23). We present the first large-scale preclinical in vivo investigation of fibrillar plaque burden asymmetry by standardized evaluation of PET data. With respect to animal welfare guidelines, in particular reduction of animal numbers in accordance with the 3R principle (replacement, reduction, and refinement), we used scans from various earlier studies, thus avoiding any requirement for additional animal experiments to test our hypotheses.
First, we endeavored to establish a reasonable threshold of lateralized Aβ PET signal to exclude asymmetry findings driven by reasons other than Aβ pathology. To this end, we used Aβ PET data of C57BL/6 WT mice, as they are not known to manifest any Aβ accumulation. Minor asymmetry of florbetaben tracer uptake in WT mice could be attributed to factors such as differences in cerebral blood flow, differing hemispheric volumes, or methodologic issues such as lateralized spillover of bone uptake, imperfect attenuation correction, or bias in spatial normalization. Hence, we used the 95%CI and 99%CI of 18F-florbetaben AIs in WT to discern moderate and strong asymmetry in the groups of Aβ-accumulating mice. By these criteria, 40% of all Aβ-accumulating mice revealed moderate asymmetry, and 30% showed strongly asymmetric Aβ deposition, but without evidence for a general lateralization across all AD models. Nevertheless, 2 of 5 investigated amyloid models revealed significant lateralization of Aβ plaque distribution to Aβ PET. There was a significant left-hemispheric predominance of Aβ deposition in PS2APP mice, but a significant right-hemispheric predominance in APPswe mice. Although molecular explanations and causal mechanisms giving rise to this phenomenon are presently unknown, we contend that this is a real phenomenon requiring special consideration when comparing data from different Aβ mouse models of AD. For example, a comparison of exclusively right-hemisphere readouts, as might be obtained by histologic analysis, between APPswe and PS2APP could cause false-negative findings, and likewise for the left hemisphere. The highest frequency of asymmetry was observed in Aβ models with a presenilin mutation (PS2APP and APP/PS1), indicating that involvement of this gene might increase the probability of asymmetric plaque burden. Variable expression of APP messenger RNA across different PS2APP mice is already postulated to be a key determinant of variance in individual Aβ deposition (12); therefore, we speculate that this phenomenon could likewise hold true for differences between hemispheres.
By making sample-size estimations, we established that the observed asymmetries of fibrillar plaque burden are potentially relevant to the design of preclinical trials. Importantly, the calculated sample sizes sufficient to detect relevant therapeutic effects, which are comparable to those of earlier drug trials in these Aβ mouse models (13,20), were significantly higher when only single hemispheres were analyzed, as opposed to combined measurement of both hemispheres. As Aβ PET and histology markers for fibrillar Aβ were strongly intercorrelated in previous studies (10,19,24), we assume that asymmetry effects on required sample sizes should also hold true for stand-alone histologic or biochemical analyses. This conjecture remains to be demonstrated, since usual practice is to process 1 hemisphere for histology and 1 for biochemistry. Aβ PET findings at the terminal time-point could help to identify mice with asymmetric plaque burden, which would allow consecutive adjustment of measures by different modalities in separate hemispheres.
Next, we investigated whether asymmetric Aβ distributions occur in an age-dependent manner. Our cross-sectional analysis of historical PET data did not indicate any significant association of AI with age among the 5 Aβ mouse models. This finding is consistent with our earlier longitudinal 18F-florbetaben PET findings in APPswe, were we incidentally noticed that some animals showed consistently right-sided plaque asymmetry between 13 and 20 mo of age. More precisely, the magnitude of asymmetry in SUVR increased with age, but with no temporal dependence of the AI per se (19). In conclusion, Aβ asymmetries, when present, are established at the onset of plaque deposition.
We suppose that there are hitherto few reports on asymmetric plaque burden in Aβ mouse models because of the logistic difficulty of conducting onerous histologic analysis of both hemispheres for sufficient numbers of animals. We performed a metaanalysis of the most recent 56 papers from journals with an impact factor of more than 4 published in the interval 2016–2019 with the key words “amyloid, mouse, model, AD.” Of these papers, 38% (21/56) provided detailed information about use of different hemispheres for histology and biochemistry; 81% among those (17/21) assigned a specific hemisphere to a given modality, whereas only 19% (4/21) performed randomization of hemispheres to different modalities. Most of the remaining 35 papers likewise split hemispheres to different modalities, but without detailed information about the selection process. Immunohistochemistry with Aβ antibodies such as 6E10 was most frequently used to assess fibrillar plaques in vitro, whereas other studies used histologic staining with methoxy-X04 or thioflavin S (14,25). These studies generally reported immunohistochemical and histologic findings for Aβ quantification from a few representative brain slices of a single hemisphere, whereas the other hemisphere was typically reserved for biochemical assays such as enzyme-linked immunosorbent assay or Western blotting, which are not compatible with tissue fixation. Therefore, evaluation of intraanimal asymmetry in vitro was not feasible because of allocation of the hemispheres for different kinds of analyses. In summary, potential asymmetries of fibrillar plaque burden have been only sparsely considered in published papers during recent years.
Contrary to the case in vitro, Aβ PET allows convenient quantification of amyloid pathology in both whole hemispheres, with the caveat that the PET method has inherent limitations in spatial resolution (26,27). Therefore, PET quantification of small brain areas can be challenging, although asymmetry assessment of Aβ plaque burden in large forebrain regions is a rather robust measure. Thus, conducting noninvasive PET examination before assignment of hemispheres to different terminal biochemical or histologic experiments could help to identify and adjust for relevant asymmetries of plaque burden. This possibility should encourage the combined use of PET together with immunohistochemistry and biochemistry readouts.
Another focus of our study was to investigate the relationship between lateralized Aβ deposition and microglial activation. Previous studies by our laboratory have already shown close correlations between fibrillar amyloidosis and TSPO expression in APP/PS1, PS2APP, APP-SL70, and AppNL-G-F mice (3,4,10,16). Although we acknowledge that our findings were anticipated from these earlier findings, we now show for the first time that microglial activation occurs concomitantly in the hemisphere ipsilateral to the predominant fibrillar amyloidosis. This association further strengthens the hypothesis that initial fibrillar Aβ accumulation triggers neuroinflammation mediated by activated microglia (28). Another recently published study has also demonstrated a link between amyloidosis and neuroinflammation based on comparative profiling of cortical gene expression in AD patients and an Aβ mouse model (29). Comparisons of gene expression between hemispheres of mice with asymmetric amyloidosis could give new insights into the molecular pathways and causal mechanisms underlying asymmetry in AD. PET screening could guide the selection for detailed study of mice with strong asymmetries.
CONCLUSION
Nearly a third of Aβ mice show distinct left or right asymmetry in the deposition of cerebral amyloid. This phenomenon is neglected in most current studies on Aβ mice and calls for consideration in the planning and design of preclinical trials, especially when single hemispheres are investigated by methods ex vivo. The lack of age dependency on asymmetric Aβ distribution implies that genetic factors underlie the development of lateralized amyloidosis in AD model mice. There is a clear association between asymmetries of glial activation and fibrillar amyloidosis in all Aβ mouse models investigated in this study, further strengthening the hypothesis that neuroinflammatory response to fibrillar Aβ contributes to the development of pathology in these mice.
DISCLOSURE
Christian Haass collaborates with Denali Therapeutics, participated on 1 advisory board meeting of Biogen, and received a speaker honorarium from Novartis and Roche. Christian Haass is chief advisor of ISAR Bioscience. Peter Bartenstein, Axel Rominger, and Matthias Brendel received speaking honoraria from Life Molecular Imaging and GE Healthcare. Matthias Brendel is an advisor of Life Molecular Imaging. Christian Haass is supported by the Koselleck Project HA1737/16-1 of the DFG, the Helmholtz-Gemeinschaft (Zukunftsthema “Immunology and Inflammation”(ZT-0027)), and the Cure Alzheimer’s fund. This work was supported by the Deutsche Forschungsgemeinschaft (Matthias Brendel and Axel Rominger BR4580/1-1 and RO5194/1-1). The APPPS1 colony was established from a breeding pair kindly provided by Mathias Jucker (Hertie-Institute for Clinical Brain Research, University of Tübingen, and DZNE-Tübingen). APPswe, PS2APP, and APP-SL70 mice were provided by Hoffmann-La Roche. APPNL-G-F mice were provided by RIKEN BRC through the National Bio-Resource Project of the MEXT, Japan. GE Healthcare made GE-180 cassettes available through an early-access model. No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Do amyloid mouse models have asymmetric plaque distribution and asymmetric neuroinflammation?
PERTINENT FINDINGS: Asymmetry in these amyloid mouse models is frequent and statistically relevant for planning of observational and interventional trials in these mice. Moreover, asymmetries of fibrillar plaque burden and glial activation are positively correlated.
IMPLICATIONS FOR PATIENT CARE: Lateralized distribution of fibrillar plaques is insufficiently considered in experimental studies with amyloid mouse models and a potential confounder in preclinical phases of drug development.
Footnotes
Published online May 15, 2020.
- © 2020 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication January 30, 2020.
- Accepted for publication April 3, 2020.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}