


Visual Abstract

Abstract
Bispecific T-cell engager (BiTE) molecules are designed to engage and activate cytotoxic T cells to kill tumor cells. Little is known about their biodistribution in immunocompetent settings. Methods: To explore their pharmacokinetics and the role of the immune cells, BiTE molecules were radiolabeled with the PET isotope 89Zr and studied in immunocompetent and immunodeficient mouse models. Results: PET images and ex vivo biodistribution in immunocompetent mice with [89Zr]Zr-DFO-N-suc-muS110, targeting mouse CD3 (dissociation constant [KD], 2.9 nM) and mouse epithelial cell adhesion molecule (EpCAM; KD, 21 nM), and with [89Zr]Zr-DFO-N-suc-hyS110, targeting only mouse CD3 (KD, 2.9 nM), showed uptake in the tumor, spleen, and other lymphoid organs, whereas the human-specific control BiTE [89Zr]Zr-DFO-N-suc-AMG 110 showed similar tumor uptake but lacked spleen uptake. [89Zr]Zr-DFO-N-suc-muS110 spleen uptake was lower in immunodeficient than in immunocompetent mice. After repeated administration of nonradiolabeled muS110 to immunocompetent mice, 89Zr-muS110 uptake in the spleen and other lymphoid tissues decreased and was comparable to uptake in immunodeficient mice, indicating saturation of CD3 binding sites. Autoradiography and immunohistochemistry demonstrated colocalization of [89Zr]Zr-DFO-N-suc-muS110 and [89Zr]Zr-DFO-N-suc-hyS110 with CD3-positive T cells in the tumor and spleen but not with EpCAM expression. Also, uptake in the duodenum correlated with a high incidence of T cells. Conclusion: [89Zr]Zr-DFO-N-suc-muS110 biodistribution is dependent mainly on the T-cell–targeting arm, with a limited contribution from its second arm, targeting EpCAM. These findings highlight the need for extensive biodistribution studies of novel bispecific constructs, as the results might have implications for their respective drug development and clinical translation.
- bispecific T-cell engager (BiTE) molecule
- PET imaging
- syngeneic mouse model
- oncology
- cancer immunotherapy
Impressive results from immunotherapy across various cancer types have greatly increased confidence in modulation of the immune system as a treatment for cancer. Currently, several immune-checkpoint–inhibiting monoclonal antibodies are registered for cancer treatment. Also, numerous modified monoclonal antibodies are being studied with the aim of improving antitumor efficacy (1). Among them are bispecific T-cell engager (BiTE; Amgen) molecules, which are 53-kDa antibody constructs consisting of 2 single-chain variable fragments on 1 peptide chain (2). One single-chain variable fragment binds to T cells via the CD3 receptor, and the other single-chain variable fragment binds to the tumor cell via a tumor-associated antigen. When the BiTE molecule binds to both cell types, a cytolytic synapse is formed resulting in perforin and granzyme-B release and eventually tumor cell killing (3–5).
Blinatumomab, a BiTE molecule targeting CD19 and CD3, is approved by the Food and Drug Administration and European Medicine Agency for the treatment of Philadelphia-chromosome–negative B-cell acute lymphoblastic leukemia. In a phase 1 trial, patients with relapsed or refractory acute myeloid leukemia were treated with AMG 330, a BiTE molecule targeting CD33 and CD3. Two of 35 patients had a complete response and 2 patients had a complete response with incomplete hematologic recovery (6).
In addition, several BiTE molecules for both solid tumors and hematologic malignancies are currently in development (7,8). In solid tumors, few clinical results have been published to date (7,9). Preclinical PET-imaging studies with BiTE molecules showed specific tumor uptake in immunodeficient mice bearing solid human tumors (10,11). No uptake in lymphoid organs was seen. However, evaluating the role of the CD3-targeting arm on the distribution or accessibility was not possible since human-specific BiTE molecules were used. Recently, a first small clinical PET-imaging study was performed with 89Zr-labeled AMG 211, a BiTE molecule targeting CD3 and carcinoembryonic antigen. Here, heterogeneous tumor uptake within and between patients as well as uptake in lymphoid tissue was shown (12).
Epithelial cell adhesion molecule (EpCAM) is expressed by many epithelial tumors (13), and muS110, a BiTE molecule targeting mouse EpCAM and mouse CD3, reduced tumor growth in mice (14). This murine BiTE molecule mus110 allowed us to study the biodistribution of muS110 in a syngeneic mouse model, best reflecting the human situation. To evaluate the influence of the T-cell–targeting arm and the EpCAM-targeting arm on the whole-body biodistribution of this BiTE molecule, we performed a PET imaging study with [89Zr]Zr-DFO-N-suc-BiTE molecules in immunocompetent and immunodeficient mouse models.
MATERIALS AND METHODS
BiTE Molecules and Cell Lines
BiTE molecules were provided by Amgen. MuS110 binds to mouse CD3ε and mouse EpCAM, hyS110 to mouse CD3ε and human EpCAM, and AMG 110 to human CD3ε and human EpCAM. Production of these BiTE molecules has been described previously (14,15). Their molecular weight is approximately 53 kDa. The dissociation constants (KD) of muS110 are 2.9 nM and 21 nM for mouse CD3 and mouse EpCAM, respectively (14). The mouse CD3-targeting arm of muS110 is used for hyS110 as well.
MuS110, hyS110, and AMG 110 were purified from buffer excipients using a Vivaspin-500 30-kDa filter (Sartorius), and the formulation buffer was changed to 0.9% NaCl (Braun). Tetrafluorophenol-N-succinyldesferrioxamine-B (ABX) was conjugated to muS110, hyS110, and AMG 110, resulting in a protein-to-chelator end ratio of 1:6, 1:6, and 1:3, respectively. Conjugation has been described previously (16). Conjugation efficiency and formation of aggregates were evaluated by size-exclusion high-performance liquid chromatography (Waters) using a Superdex 75 column (GE Healthcare) and phosphate-buffered saline (140 mM NaCl, 9 mM Na2HPO4, and 1.3 mM NaH2PO4; pH 7.4) as the mobile phase. No visible particles were detected.
After purification by dialysis with Slide-A-Lyzer cassettes (10-kDa filter; ThermoFisher Scientific), the conjugate was stored at a 1 mg/mL concentration at −80°C. Stability and immunoreactivity were evaluated by high-performance liquid chromatography and by cell-based binding assays, respectively. In binding assays, increasing concentrations of conjugated or unmodified BiTE molecules were incubated with human or murine T cells or human or murine EpCAM-positive B16/F10 tumor cells. Maintained immunoreactivity was further evaluated by adding increasing concentrations of conjugated or unmodified muS110 to fixed concentrations of murine T cells in the presence of murine EpCAM-positive B16/F10 tumor cells in a ratio of 1:10. Readouts were propidium iodide–positive tumor cells for cytotoxicity, and CD69-positive (553237; BD Biosciences) and CD25-positive (553237; BD Biosciences) T cells for T-cell activation. Murine and human T cells were obtained by negative selection with the Pan T Cell Isolation Kit II (mouse; Miltenyi Biotec) or the Pan T Cell Isolation Kit (human; Miltenyi Biotec), respectively. Data were acquired by FACS Canto II (BD Biosciences), and binding curves were read out by median fluorescence intensity on FACS Diva (BD Biosciences) software.
The conjugated BiTE molecules were labeled with 89Zr (16) with a specific activity of 400–500 MBq/mg. Radiochemical purity was evaluated by a trichloroacetic acid precipitation assay; all tracers had a radiochemical purity of more than 95%.
The murine mammary carcinoma cell line 4T1 (American Type Culture Collection) is EpCAM-positive and was cultured in RPMI-1640 medium (Invitrogen) containing 10% fetal calf serum (Bodinco BV). B16/F10 tumor cells transfected with human EpCAM were cultured in Dulbecco modified Eagle medium (Biochrom) containing 10% fetal calf serum (Invitrogen). B16/F10 tumor cells transfected with murine EpCAM were cultured in Dulbecco modified Eagle medium containing 10% fetal calf serum (Invitrogen) and a 600 U/mL concentration of G418 (Millipore).
Cells were used between passages 5–20 after thawing and were routinely tested for Mycoplasma. All cells were cultured under aseptic conditions at 37°C in an incubator providing a humidified atmosphere of 5% CO2 in air.
Animal Experiments
Animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Groningen. Female BALB/c mice (8–10 wk old, BALB/cOlaHsd; Envigo)—from now on referred to as immunocompetent—were acclimatized for 1 wk, as were female nude BALB/c mice (BALB/cOlaHsd-Foxn1nu; Envigo)—from now on referred to as immunodeficient. All mice were housed in groups of 3–6 per cage. Immunocompetent mice were housed in compliance with the 2014 guidelines of the Federation for Laboratory Animal Science Associations. Immunodeficient mice were housed in individually ventilated cages. When possible, mice were allocated randomly to tracer groups. The mice were injected in the lower mammary fat pad with 5 × 104 4T1 tumor cells in 50 μL of RPMI-1640, containing phenol red. To prevent toxicity from the tracer dose (10 μg), the mice received low-dose muS110 (0.2 μg) intravenously daily for 5 d before tracer injection. Previously observed muS110 side effects, leading to cytokine release, appeared dependent on EpCAM-positive B and T cells in the circulation, and depleting these cells with low-dose muS110 increased the muS110 tolerance of these mice (17). In addition, after the low-dose muS110 administration, a group of BALB/c mice (BALB/cOlaHsd) received a 10-μg muS110 bolus intravenously daily for another 5 d to assess possible target saturation. This group will be referred to as the repeated-muS110-administration group.
[89Zr]Zr-DFO-N-suc-BiTE molecules (10 μg) were injected intravenously in the tail vein when tumors reached ±200 mm3. The mice were anesthetized with an isofluorane and medical air mixture (5% induction, 2.5% maintenance) during injections and PET imaging. First, the in vivo biodistribution of 10 μg of [89Zr]Zr-DFO-N-suc-muS110 over time in 4T1-bearing immunocompetent mice was visualized by small-animal PET scanning performed at 0.5, 3, 6, 24, 48, and 72 h after injection. Small-animal PET scans were acquired as described before with the Focus220 rodent scanner (CTI Siemens) (10).
Second, the biodistribution of 10 μg of [89Zr]Zr-DFO-N-suc-muS110, [89Zr]Zr-DFO-N-suc-hys110, and [89Zr]Zr-DFO-N-suc-AMG 110 was compared in 4T1-bearing immunocompetent mice 24 h after intravenous tracer injection by PET imaging followed by ex vivo biodistribution. This procedure was repeated for 10 μg of [89Zr]Zr-DFO-N-suc-muS110 and [89Zr]Zr-DFO-N-suc-AMG 110 in 4T1-bearing immunodeficient mice and to the repeated-mus110-administration mice. Again, all mice underwent PET scans 24 h after intravenous tracer administration followed by ex vivo biodistribution.
Organs of interest were weighed and measured in a calibrated Wizard γ-counter (PerkinElmer). Counts of known standards were used to convert counts into injected dose. Tissue activity is expressed as percentage injected dose per gram. Relevant tissues were fixed in formalin (4% paraformaldehyde in phosphate-buffered saline) for 1–3 d or stored at −80°C for further analysis. PET scans were analyzed with PMOD (version 3.8; PMOD Technologies). Volume of interests were drawn as spheres based on weight of organs found in the biodistribution. Data are expressed as SUVmean. PET scans were visualized as a coronal projection containing the tumor or as a maximum-intensity projection scaled to 30% as maximal intensity.
Ex Vivo Tissue Analysis
Formalin-fixed paraffin-embedded tissues were sliced to 4-μm sections and mounted on tissue slides. For autoradiography, phosphor imaging screens (PerkinElmer) were exposed to the tissue slides in x-ray cassettes for 24–72 h and digitized by the Cyclone Phosphor System (PerkinElmer). Autoradiography images were analyzed with ImageJ, version 1.52p. Tissues were stained with hematoxylin and eosin for tissue morphology. When possible, in subsequent 4-μm sections T-cell presence was visualized immunohistochemically for mouse CD3 with a rabbit antimouse CD3 antibody, clone SP7 (ab16669; Abcam). EpCAM presence was confirmed by a rabbit antimouse EpCAM antibody (ab71916; Abcam). For both stainings, antigen retrieval was 15 min at 95°C in citrate buffer at pH 6. For the CD3 staining, the primary antibody dilution was 1:50, and for the EpCAM staining, a 1:250 dilution was used. Next, a peroxidase conjugated goat antirabbit antibody (p0448; Dako) was used, at a 1:100 dilution. For EpCAM staining, a tertiary antibody was used, a peroxidase-conjugated rabbit antigoat antibody with 1:100 dilution (Dako; p0449). 3-3′-diaminobenzidine was added to visualize peroxidase activity. For CD3 and EpCAM immunohistochemistry, any membrane staining was considered positive. Slides were viewed with NDP.view2 (Hamamatsu).
Lysates of tumor, liver, and spleen were prepared using radioimmunoprecipitation assay buffer (ThermoFisher Scientific) including 1% protease blocker (ThermoFisher Scientific) and 1% phosphatase blocker (ThermoFisher Scientific). Together with plasma and intact [89Zr]Zr-DFO-N-suc-muS110, they were loaded into mini-PROTEAN TGX precast gels (Bio-Rad). Gel-exposed phosphor imaging screens were read out by the Cyclone Phosphor System to identify the molecular weight of the [89Zr]Zr-DFO-N-suc-BiTE. The molecular weight of bands was identified using ProSieve color protein marker (Lonza).
Statistical Methods
Data are presented as median with interquartile range. A Bonferroni-corrected Mann–Whitney U test was performed to test differences between groups (Prism 7; GraphPad). P values of 0.05 or less were considered statistically significant. Blood half-life was calculated using a 2-phase decay (Prism 7).
RESULTS
Conjugation and Labeling of BiTE Molecules with 89Zr
N-suc-DFO-BiTE molecules were labeled with 89Zr with a radiochemical purity of more than 95% (Supplemental Fig. 1A; supplemental materials are available at http://jnm.snmjournals.org). Immunoreactivity was maintained for muS110, targeting mouse CD3ε and murine EpCAM, and for hyS110, targeting mouse CD3ε and human EpCAM, after conjugation to N-suc-DFO (Supplemental Fig. 1B), despite the formation of 10%–14% aggregates for muS110 and hyS110 as revealed by size-exclusion high-performance liquid chromatography (Supplemental Fig. 1A). Moreover, conjugated muS110 maintained similar in vitro cytotoxicity (Supplemental Fig. 1C) and T-cell activation (Supplemental Fig. 1D) to those of parental muS110.
[89Zr]Zr-DFO-N-suc-muS110 PET-Imaging over Time in 4T1-Bearing Immunocompetent Mice
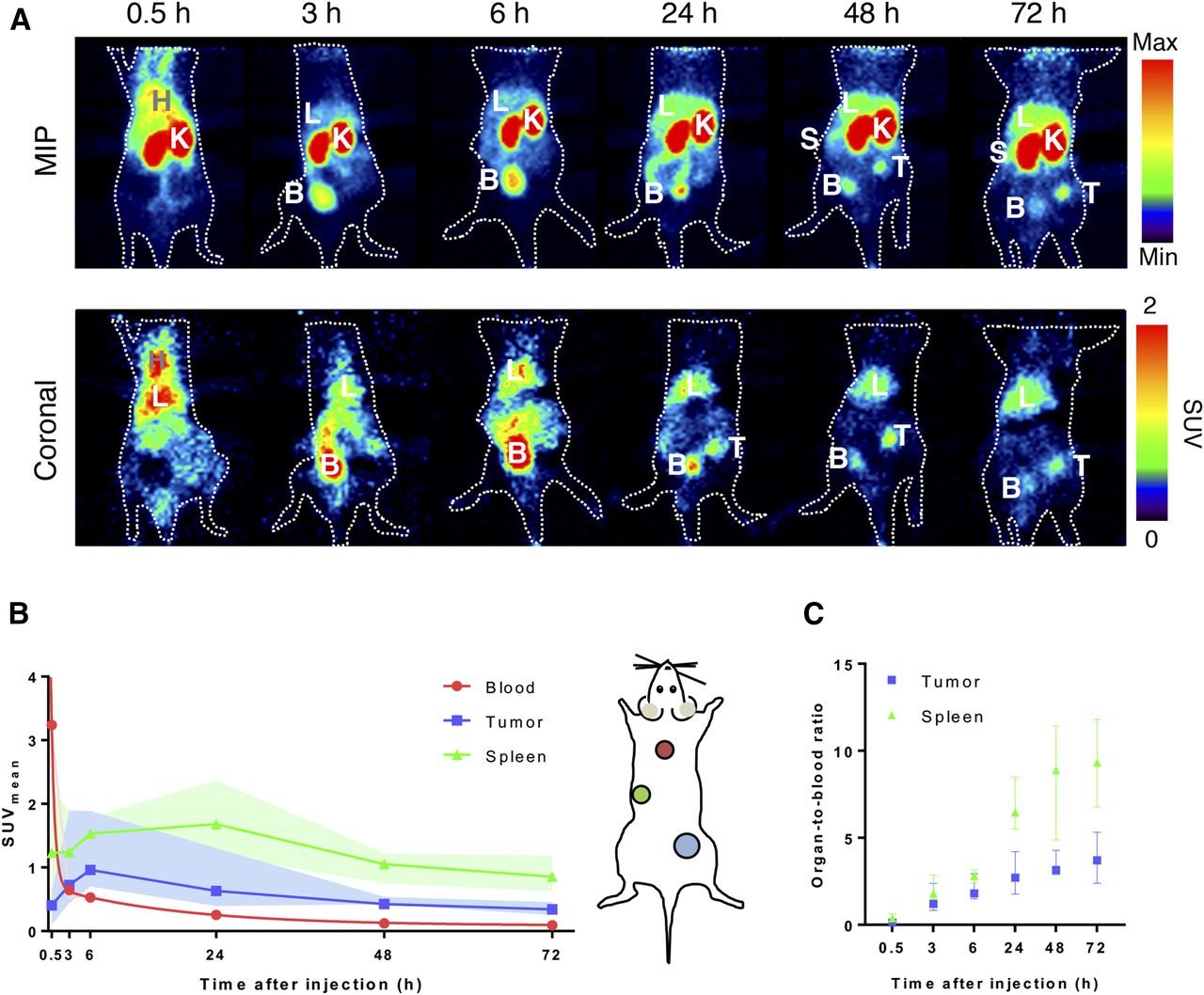
To evaluate the role of binding to EpCAM on distribution of the BiTE molecules, immunodeficient mice were used. Accumulation of signal over time was seen in the tumor, spleen, and liver in the maximum-intensity projections of PET scans of [89Zr]Zr-DFO-N-suc-muS110. High tracer signal was observed in the kidneys and bladder, indicating renal clearance (Fig. 1A). [89Zr]Zr-DFO-N-suc-muS110 tumor and liver uptake over time was confirmed by coronal PET images (Fig. 1A). Blood half-life, based on the SUVmean of the heart, was 0.4 h (distribution) and 12.8 h (elimination) (Fig. 1B). Uptake in the spleen increased until 24 h, with an SUVmean of 1.7 (range, 1.5–1.9), and uptake in the tumor increased until 6 h after injection, with an SUVmean of 1.0 (range, 0.8–1.3; Fig. 1C). After 24 h, tumor- and spleen-to-blood ratios for SUVmean were 2.7 (range, 2.3–3.3) and 6.5 (range, 5.8–7.5).

PET biodistribution of 10 μg of [89Zr]Zr-DFO-N-suc-muS110 in 4T1 tumor–bearing immunocompetent mice (n = 6). (A) Representative serial coronal and maximum-intensity-projection PET images up to 72 h after injection. (B) Image quantification of blood, tumor, and spleen. (C) Image quantifications expressed as organ-to-blood ratio for tumor and spleen. Data are presented as median with interquartile range. B = bladder; H = heart; K = kidney; L = liver; MIP = maximum-intensity projection; S = spleen; T = tumor.
[89Zr]Zr-DFO-N-suc-BiTE Molecules in 4T1-Bearing Immunocompetent Mice
High kidney uptake was seen with the CD3-targeting BiTE molecules 89Zr-muS110 and 89Zr-hyS110 and the control BiTE molecule [89Zr]Zr-DFO-N-suc-AMG 110 in the maximum-intensity projections 24 h after tracer administration (Fig. 2A). Uptake in the spleen and liver for [89Zr]Zr-DFO-N-suc-muS110 and [89Zr]Zr-DFO-N-suc-hyS110 but not for control [89Zr]Zr-DFO-N-suc-AMG 110 was visualized in the coronal projections (Fig. 2A).

Biodistribution of 10 μg of [89Zr]Zr-DFO-N-suc-muS110, [89Zr]Zr-DFO-N-suc-hyS110, or [89Zr]Zr-DFO-N-suc-AMG 110 (all n = 7) in 4T1 tumor–bearing immunocompetent mice 24 h after injection. (A) Representative coronal and maximum-intensity-projection PET images 24 h after injection of [89Zr]Zr-DFO-N-suc-BiTE molecules. (B) Ex vivo biodistribution. Data are presented as median with interquartile range. *P ≤ 0.05. **P ≤ 0.01. %ID = percentage injected dose; B = bladder; K = kidney; L = liver; LN = lymph node; MIP = maximum-intensity projection; S = spleen; T = tumor.
Ex vivo biodistribution confirmed that spleen uptake of [89Zr]Zr-DFO-N-suc-muS110 was higher than spleen uptake of [89Zr]Zr-DFO-N-suc-AMG 110, but no difference was observed between [89Zr]Zr-DFO-N-suc-muS110 and [89Zr]Zr-DFO-N-suc-hyS110 (Fig. 2B; Supplemental Table 1). Also, in other lymphoid tissues, such as the mesenteric lymph nodes, uptake of [89Zr]Zr-DFO-N-suc-muS110 and [89Zr]Zr-DFO-N-suc-hyS110 was increased compared with aspecific uptake of [89Zr]Zr-DFO-N-suc-AMG 110. Tumor uptake did not differ between [89Zr]Zr-DFO-N-suc-muS110 and [89Zr]Zr-DFO-N-suc-AMG 110 or [89Zr]Zr-DFO-N-suc-hyS110. [89Zr]Zr-DFO-N-suc-muS110 accumulated more in the duodenum than did [89Zr]Zr-DFO-N-suc-hyS110 or [89Zr]Zr-DFO-N-suc-AMG 110.
Sodium dodecyl sulfate polyacrylamide gel electrophoresis autoradiography showed intact [89Zr]Zr-DFO-N-suc-muS110 in the tumor lysates and plasma, whereas disintegrated tracer was found in the spleen and liver lysates (Supplemental Fig. 2).
[89Zr]Zr-DFO-N-suc-BiTE Molecules in 4T1-Bearing Immunodeficient Mice
PET scans showed no spleen uptake of [89Zr]Zr-DFO-N-suc-muS110 or [89Zr]Zr-DFO-N-suc-AMG 110 in immunodeficient mice (Fig. 3A). For [89Zr]Zr-DFO-N-suc-muS110, maximum-intensity projections showed, apart from the high uptake in the kidneys, little uptake in other organs.
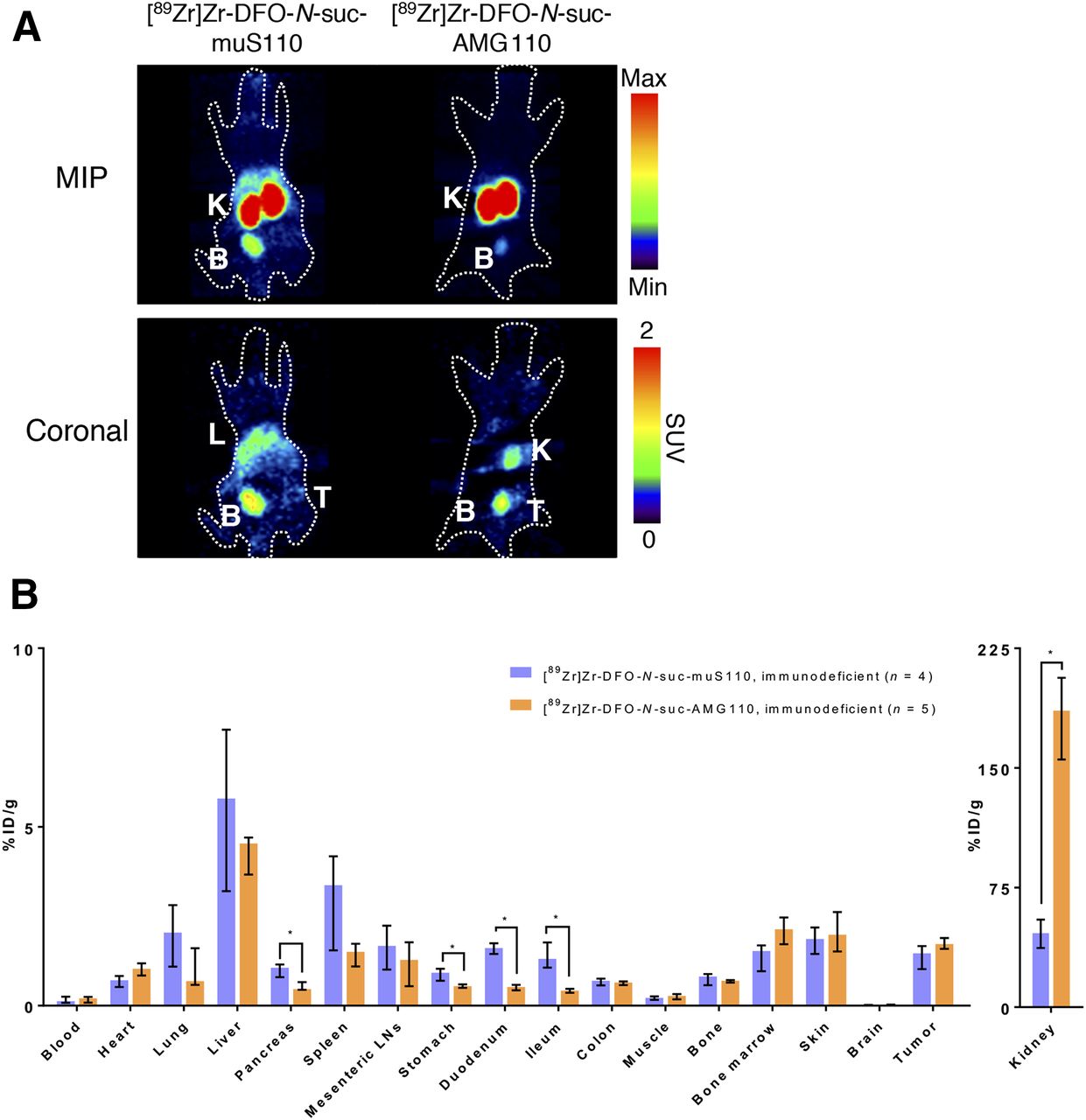
Biodistribution of 10 μg of [89Zr]Zr-DFO-N-suc-muS110 (n = 4) or [89Zr]Zr-DFO-N-suc-AMG 110 (n = 5) in 4T1 tumor–bearing immunodeficient mice 24 h after injection. (A) Representative coronal and maximum-intensity-projection PET images 24 h after injection. (B) Ex vivo biodistribution. Data are presented as median with interquartile range. *P ≤ 0.05. %ID = percentage injected dose; B = bladder; K = kidney; L = liver; LN = lymph node; MIP = maximum-intensity projection; T = tumor.
Similar tumor uptake of [89Zr]Zr-DFO-N-suc-muS110 and [89Zr]Zr-DFO-N-suc-AMG 110 was confirmed by ex vivo biodistribution in these immunodeficient mice (Fig. 3B; Supplemental Table 2). No differences in uptake in spleen and mesenteric lymph nodes were found. Uptake of [89Zr]Zr-DFO-N-suc-muS110 was higher than uptake of [89Zr]Zr-DFO-N-suc-AMG 110 in high-EpCAM-expressing tissues such as the pancreas, duodenum, and ileum.
[89Zr]Zr-DFO-N-suc-BiTE Molecules in 4T1-Bearing Immunocompetent Mice After Repeated MuS110 Administration
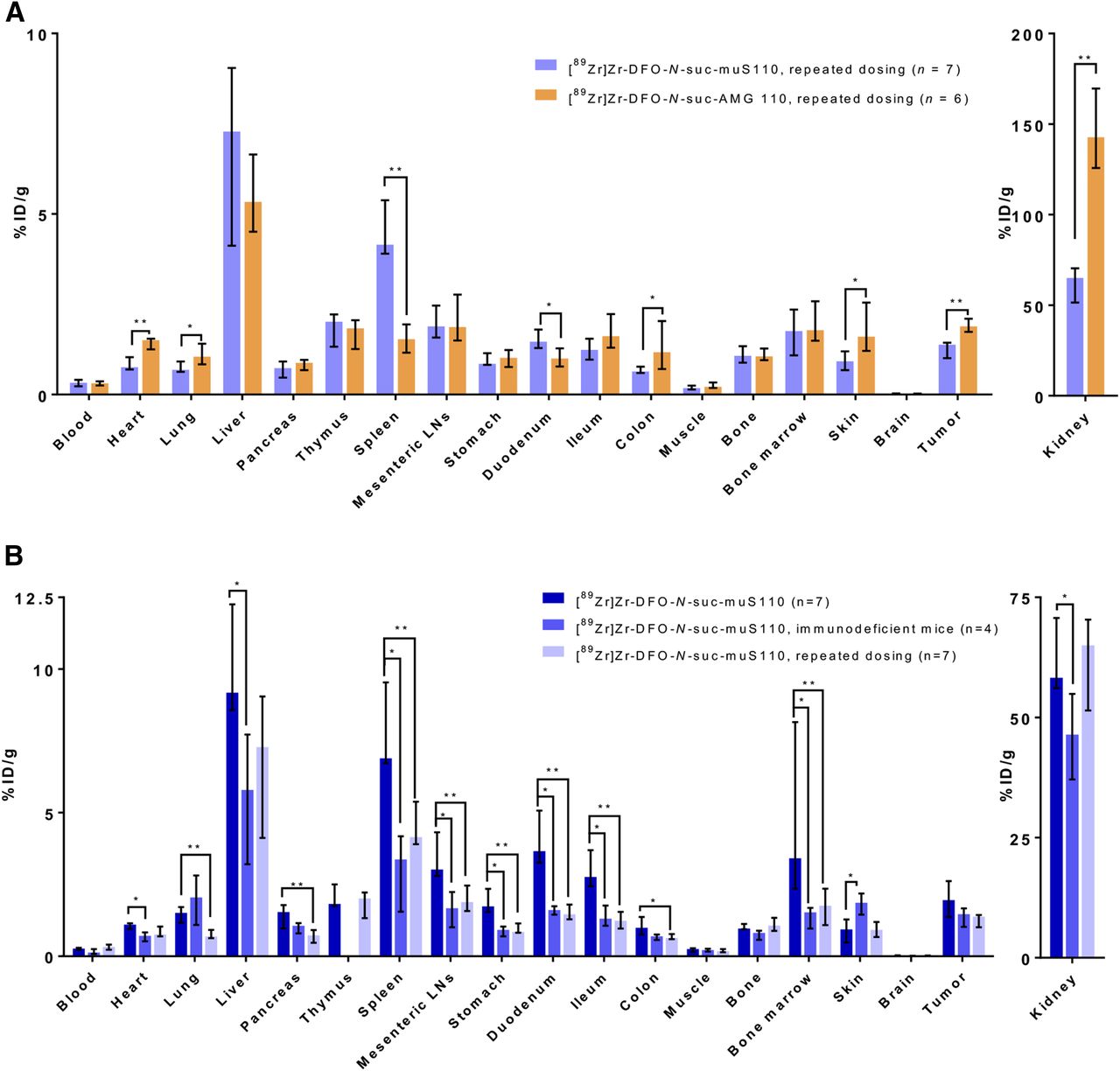
Ex vivo biodistribution of immunocompetent mice that received 10 μg of muS110 intravenously daily for 5 d showed lower tumor uptake of [89Zr]Zr-DFO-N-suc-muS110 than of [89Zr]Zr-DFO-N-suc-AMG 110 and higher spleen uptake of [89Zr]Zr-DFO-N-suc-muS110 (Fig. 4A; Supplemental Table 3).

(A) Ex vivo biodistribution 24 h after injection of 10 μg of [89Zr]Zr-DFO-N-suc-muS110 (n = 7) or [89Zr]Zr-DFO-N-suc-AMG110 (n = 6) in 4T1 tumor–bearing immunocompetent mice after receiving 10 μg of muS110 for 5 d. (B) Comparison of ex vivo biodistribution of 10 μg of [89Zr]Zr-DFO-N-suc-muS110 in immunocompetent mice (n = 7), immunodeficient mice (n = 4), and repeated-mus110-administration mice (n = 7) 24 h after injection. Data are presented as median with interquartile range. *P ≤ 0.05. **P ≤ 0.01. %ID = percentage injected dose; LN = lymph node.
The comparison of 89Zr-muS110 biodistribution in the 3 different settings—the immunocompetent, immunodeficient, and repeated-administration groups—revealed lower uptake in the immunodeficient and the repeated-administration setting in lymphoid tissues such as the spleen and mesenteric lymph nodes (Fig. 4B; Supplemental Table 4). The biodistribution of control [89Zr]Zr-DFO-N-suc-AMG 110 in the 3 settings was comparable (Supplemental Fig. 3; Supplemental Table 5).
Additionally, uptake of [89Zr]Zr-DFO-N-suc-muS110 was higher in the digestive tract and the tumor in the immunocompetent model than in the other models (Fig. 4B), indicating specific uptake in the immunocompetent model and target saturation after repeated administration of muS110.
Microscopic Analysis of Tissues Ex Vivo to Determine the Cellular Source of BiTE Molecule Uptake
Autoradiography showed heterogeneous tumor uptake in immunocompetent mice (Fig. 5). High local uptake in the tumor colocalized with high expression of CD3 for [89Zr]Zr-DFO-N-suc-muS110 and [89Zr]Zr-DFO-N-suc-hyS110, whereas this finding was not seen for [89Zr]Zr-DFO-N-suc-AMG 110. Furthermore, no difference in EpCAM expression was observed between higher-uptake and lower-uptake areas in the tumor. In the spleen, areas of high uptake of [89Zr]Zr-DFO-N-suc-muS110 and [89Zr]Zr-DFO-N-suc-hyS110 colocalized with white pulp, where high numbers of T cells were found immunohistochemically (Supplemental Fig. 4). Although immunohistochemistry staining for EpCAM in spleen tissue can show nonspecific staining (14), we saw increased staining in the red pulp, where autoradiography detected less uptake of [89Zr]Zr-DFO-N-suc-muS110 and [89Zr]Zr-DFO-N-suc-hyS110.
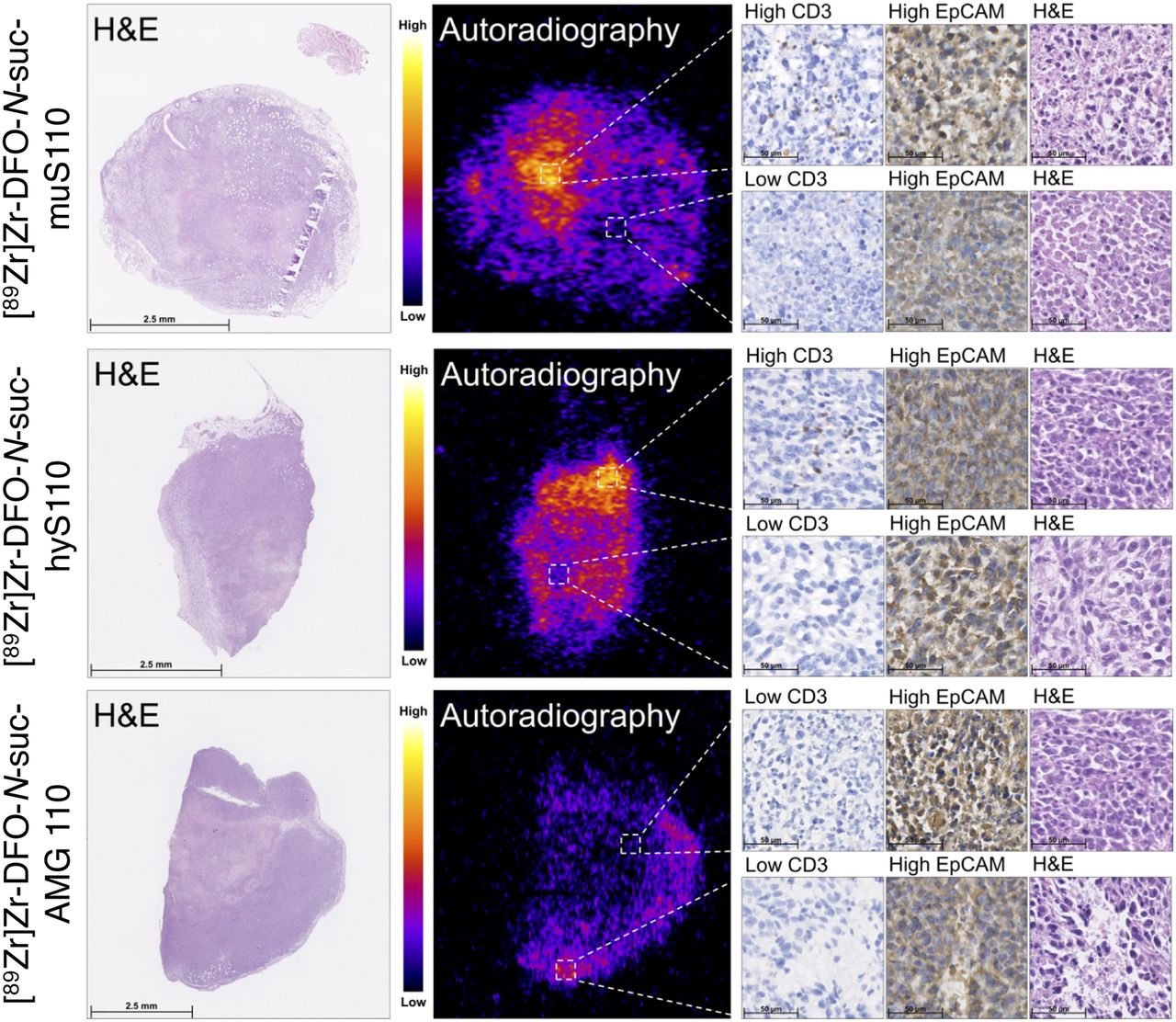
Ex vivo microscopic analysis of 4T1 tumor tissue from immunocompetent mice 24 h after injection of 10 μg of [89Zr]Zr-DFO-N-suc-BiTE molecule. From left to right are shown hematoxylin and eosin staining and corresponding autoradiography, followed by CD3 and EpCAM immunohistochemistry and hematoxylin and eosin staining.
Higher uptake of [89Zr]Zr-DFO-N-suc-muS110 and [89Zr]Zr-DFO-N-suc-hyS110 than of [89Zr]Zr-DFO-N-suc-AMG 110 in the duodenum of the immunocompetent mice was observed in the biodistribution study (Fig. 2B) and confirmed by autoradiography (Supplemental Fig. 5A). The uptake in the duodenum was homogeneous. Immunohistochemical staining confirmed a homogeneous presence of both CD3-positive T cells and EpCAM-positive cells in the duodenum (Supplemental Fig. 5B).
DISCUSSION
This study on immunocompetent mice showed that the [89Zr]Zr-DFO-N-suc-muS110 distribution was predominantly mediated by its increased affinity for CD3, compared with EpCAM. The CD3-mediated biodistribution results in specific uptake in tissues with high numbers of CD3-positive T cells, such as the spleen, mesenteric lymph nodes, and duodenum. In our immunocompetent mouse model, the CD3-targeting BiTE molecule [89Zr]Zr-DFO-N-suc-hyS110 behaved similarly to [89Zr]Zr-DFO-N-suc-muS110, despite the fact that [89Zr]Zr-DFO-N-suc-muS110 had an additional EpCAM-targeting arm. This finding demonstrates the limited influence of the EpCAM-targeting arm for this BiTE molecule. In contrast to the immunocompetent model, in our immunodeficient mouse model no difference in uptake between [89Zr]Zr-DFO-N-suc-muS110 and [89Zr]Zr-DFO-N-suc-AMG 110 was seen in lymphoid tissues.
This study therefore adds novel insight into the distribution of BiTE molecules by using an immunocompetent mouse model and murine BiTE molecules. Previously, preclinically, 89Zr-labeled BiTE molecules failed to show uptake in lymphoid organs, because human BiTE molecules were tested in immunodeficient mouse models (10,11). Recently, apart from uptake in tumor lesions, clear uptake was shown for the carcinoembryonic antigen/CD3 BiTE molecule 89Zr-labeled AMG 211 in the spleen in patients with advanced gastrointestinal adenocarcinomas (12). For AMG 211, the affinity for carcinoembryonic antigen was higher (KD, 5.5 nM) than the affinity for CD3 (KD, 310 nM). In transgenic mice expressing human CD3, a bispecific antibody targeting CD3 and HER2 with the same affinity for both antigens (KD, 0.5 nM) showed high accumulation in lymphoid tissues, in contrast to the same bispecific antibody with lower affinity for CD3 (KD, 50 nM). In parallel to the accumulation in the lymphoid tissues of the high-CD3-affinity bispecific antibody, there was reduced uptake in the tumor (18). Our findings contribute to the growing understanding that the interplay between binding arms of a bispecific antibody is complex and that, especially, the affinity for the immune component will greatly influence its biodistribution.
In cynomolgus monkeys, systemic cytokine release, indicative of immune-related adverse effects, depended on the affinity of the CD3 arm of a bispecific antibody targeting CD3 and CLL-1 (19). In transgenic mice, a high-affinity CD3-binding arm directed bispecific antibodies to lymphoid tissues (18), thus providing an additional rationale for molecular imaging in immunocompetent mouse models to assess lymphoid uptake when translating bispecific antibodies.
A limited role for the tumor-targeting arm directed against EpCAM of [89Zr]Zr-DFO-N-suc-muS110 was found in this study. This finding can be explained by the lower affinity for EpCAM than for CD3 (KD, 21 vs. 2.9 nM). However, in the absence of T cells in the immunodeficient model, little specific uptake was observed in high-EpCAM-expressing tissues. [89Zr]Zr-DFO-N-suc-muS110 did accumulate more in the high-EpCAM-expressing tissues pancreas and duodenum than did [89Zr]Zr-DFO-N-suc-AMG 110, but uptake was still lower than that of [89Zr]Zr-DFO-N-suc-muS110 in these tissues in the immunocompetent group. Higher EpCAM-mediated uptake in the immunodeficient model may been expected, because of the elimination of competition for CD3. In a human EpCAM-expressing transgenic mouse model in which the biodistribution of 125I-labeled anti-EpCAM antibodies was quantified, equal low tumor uptake was observed in an EpCAM-positive and EpCAM-negative tumor. High uptake in healthy EpCAM-expressing tissue was observed, such as the pancreas and ileum (20). Expression of EpCAM by numerous healthy tissues can serve as a sink and limit the availability of the anti-EpCAM antibody for the tumor. This effect can also explain the lack of evidence for EpCAM-specific tumor uptake of [89Zr]Zr-DFO-N-suc-muS110 compared with the 89Zr-AMG 110 tumor uptake in an immunodeficient mouse model bearing a human EpCAM-positive tumor (10).
Fast renal clearance and splenic uptake of 89Zr-muS110 might also have prevented tumor uptake. Higher doses could not be explored because of the low maximum tolerable dose of muS110 in mice (17). However, after repeated administration of muS110, a reduction in uptake of [89Zr]Zr-DFO-N-suc-muS110 in various tissues was noticed, indicative of target saturation. T cells in the circulation can redistribute after a single bolus injection of muS110 and return to base levels; meanwhile, splenic CD8-positive T-cell levels are unaffected (17). Target saturation seems in line with the clinical 89Zr-AMG 211 PET imaging. Here, 89Zr-AMG 211 imaging was performed on 2 patients at the end of the second 28-d continuous intravenous treatment with AMG 211. An approximately 2- to 3-fold higher presence in the blood pool and an approximately 2- to 3-fold lower kidney uptake but absence of tumor tracer uptake were seen in patients during BiTE treatment. The absence of tumor visualization is likely indicative of tumor target saturation (12).
In the tumor from the immunocompetent mouse group, uptake of [89Zr]Zr-DFO-N-suc-muS110 and [89Zr]Zr-DFO-N-suc-hyS110 was seen in areas with T cells. This finding proves that the tumors are accessible and that uptake is driven mainly by CD3. Nevertheless, [89Zr]Zr-DFO-N-suc-muS110 did not accumulate more in the tumor than did control 89Zr-AMG 110 in either the immunocompetent or the immunodeficient group. However, daily administration of muS110 led to a reduction in tumor uptake of [89Zr]Zr-DFO-N-suc-muS110, indicating that the tumor uptake in the immunocompetent mouse model is specific. Competition for [89Zr]Zr-DFO-N-suc-muS110 between the CD3-rich tissues, leading to its distribution to lymphoid tissues, could explain the similar accumulation of [89Zr]Zr-DFO-N-suc-muS110 and [89Zr]Zr-DFO-N-suc-AMG 110 in the tumor of the immunocompetent mice. Accumulation of [89Zr]Zr-DFO-N-suc-AMG 110 in the tumor will be nonspecific and therefore not hindered by any competition. Testing a range of affinities for both the CD3- and the EpCAM-targeting arms of muS110 would show whether we can shift the distribution away from the lymphoid tissues. However, such BiTE molecules were not developed.
CONCLUSION
The distribution of [89Zr]Zr-DFO-N-suc-muS110 is predominantly mediated by its affinity for CD3. This affinity results in uptake in tissues with high numbers of T cells, including immune-infiltrated tumors. A role for the arm targeting the tumor-associated antigen—here, in this specific example, EpCAM—was absent in the biodistribution of [89Zr]Zr-DFO-N-suc-muS110. These findings highlight the need for extensive biodistribution studies of novel bispecific constructs to support their specific drug development and determine their potential for clinical translation. Attention should be paid to the affinity of the CD3-targeting arm to prevent uptake in the lymphoid tissues and associated undesired effects.
DISCLOSURE
The work performed in this study was cofinanced by the PPP subsidy of the Top Consortia for Knowledge and Innovation of the Ministry of Economic Affairs. Grit Lorenczewski, Sabine Stienen, and Matthias Friedrich hold ownership interest (including patents) in Amgen. Elisabeth de Vries reports Institutional Financial Support from Sanofi, Pfizer, Daiichi Sankyo, NSABP, and Merck for her advisory role and institutional financial support from Amgen, Genentech, Roche, Chugai Pharma, Synthon, CytomX Therapeutics, Nordic Nanovector, Regeneron, G1 Therapeutics, AstraZeneca, Radius Health, and Bayer for clinical trials or contracted research. No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: What is the influence of the T-cell–targeting arm (CD3) and the tumor-targeting arm (EpCAM) of the BiTE molecule CD3/EpCAM on whole-body biodistribution in mice?
PERTINENT FINDINGS: This comparison biodistribution study with different 89Zr-labeled mouse BiTE molecules in immunocompetent and immunodeficient mouse models revealed that the biodistribution of the BiTE molecule muS110 (CD3/EpCAM) is dependent mainly on the T-cell–targeting arm, with a limited contribution from its tumor-targeting arm, which targets EpCAM.
IMPLICATIONS FOR PATIENT CARE: Extensive biodistribution studies of novel bispecific constructs are needed, with extra attention paid to the affinity of the CD3-targeting arm to prevent uptake in lymphoid tissues and associated undesired effects.
Acknowledgments
We thank Linda Pot-de Jong and Laura von Iven for immunohistochemical staining.
Footnotes
Published online Apr. 13, 2020.
- © 2020 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication January 10, 2020.
- Accepted for publication March 16, 2020.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.