


Abstract
Although 18F-FDG PET/CT is widely available and is increasingly being used to monitor response to immunotherapy and simultaneously identify immune-related adverse events, there are several challenges in interpreting the results of this investigation, especially early in the course of treatment. It also has limited utility in selecting the optimal type of immunotherapy. As knowledge about immune contexture increases, new targets that may be amenable to imaging are being defined. These exciting advances, coupled with increasingly sophisticated methods for generating radiopharmaceuticals, provide the potential for either replacing or complementing 18F-FDG PET/CT in the selection and monitoring of immunotherapy. Approaches include imaging specific characteristics of immune cell infiltrates or aspects of the tumor microenvironment that are known to be associated with suppression of the innate and adaptive immune response. Following a large body of preclinical work, promising agents that are entering into early clinical evaluation are discussed. We suggest a speculative algorithm as to how these might be used in routine practice, subject to validation in clinical trials.
There are many steps required to mount an immune response to cancer (1). These include release of antigens from cancer cells and their recognition by antigen-presenting cells, particularly dendritic cells, followed by priming and activation of T cells, which then traffic to and infiltrate tumor deposits before recognizing and killing cancer cells. T-cell–mediated killing of cancer cells is accomplished by binding T-cell receptors of peptides with class I major histocompatibility complexes on the cell membrane of cancer cells, with the subsequent release of perforin and granzymes by CD8+ cells, or alternatively, by inducing death receptor–ligand interactions. There are many ways in which this process can fail, leading to immune escape and cancer progression. The development of therapeutic approaches to reinvigorate immune killing of cancer cells is one of the most exciting developments of modern oncology. Unfortunately, despite its promise, a minority of cancers currently respond to available immunotherapies, and many patients either fail to respond at all or respond incompletely, with resistance occurring.
Predictors of response to various immunotherapy approaches are being actively investigated at a pathologic level (2). Those reliant on tissue sampling suffer from the heterogeneity that may occur between and within tumor deposits and over time, both spontaneously and under selective pressure of therapeutic intervention. Some, but not all, of these phenomena can be imaged using molecular probes, either directly by demonstrating increased expression or indirectly by lack of signal or overexpression of factors known to be associated with immunosuppression. Imaging these targets by PET technology, also known as immune-PET, may provide information that is an alternative to, or complements, that already available from the more widely available technique of 18F-FDG PET/CT, with the common advantage of whole-body sampling that can be repeated during the course of treatment. In particular, immune-PET may address some of the recognized challenges of 18F-FDG PET/CT (3).
IMAGING OF IMMUNE CELLS
Imaging of Effector CD8+ T Cells
The importance of CD8+ tumor-infiltrating lymphocytes (TILs) to the outcome of many cancers has been recognized in numerous studies (4). Several different immune environments have been described. These include immune-desert, immune-excluded, and immune-inflamed (2). Mismatch-repair pathways play an important role in tumor immunogenicity (5). Defects in DNA mismatch-repair proteins and subsequent microsatellite instability-high cancer cells lead to the accumulation of tumoral mutation burden and generation of neoantigens, which stimulate a dense infiltration of CD8+ TILs and result in a durable antitumor immune response, particularly to anti-programmed death receptor-1 and its ligand (PD-1/PD-L1) monoclonal antibodies (6). However, the immune-desert phenotype leads to immune escape due to low tumor immunogenicity, impaired dendritic cell maturation, and suboptimal T-cell activation and is characterized by low CD8+ TILs in the tumor. T-cell activation and trafficking can be inhibited by cytotoxic T-lymphocyte 4 (CTLA-4) expression, which can be therapeutically abrogated by use of anti-CTLA-4 monoclonal antibodies. Ipilimumab is the most widely investigated of this class of agents and has been used as monotherapy or in combination with other immune checkpoint inhibitors (ICIs) (7). Accordingly, imaging of CD8+ T cells would be anticipated to be most useful for identifying failure of the early phases of immune surveillance and may provide complementary information to 18F-FDG PET/CT in selecting patients who may benefit most from immunotherapy regimens, including anti-CTLA-4 agents.
The enhanced metabolic activity in the tumor tissue, depicted by 18F-FDG uptake, is a composite of multiple factors, including competitive interaction for glycolysis between cancer and immune cells, limiting the ability of this modality to characterize tumor microenvironment (TME) composition or its temporal plasticity under immunotherapeutic strategies (8). For example, there are reports that, in lung and bladder cancer, higher 18F-FDG uptake is associated with higher CD8+ TILs and PD-L1 expression, hence predicting response to ICIs (9,10). In contrast, a recent paper evaluating the outcomes of patients with oral squamous cell carcinoma found that low infiltrating CD8+ TILs and low PD-L1 expression correlated with higher 18F-FDG uptake and poorer outcome (11), suggesting that immune-desert tumors might be better identified by CD8+ imaging since there was a significant overlap in 18F-FDG uptake in this study, limiting the discriminatory value of SUV alone. A first-in-humans trial of a CD8 minibody imaging agent, 89Zr-Df-IAB22M2C, has been reported by Pandit-Taskar et al. (12). This demonstrated tumors both with and without significant CD8+ T-cell infiltration, potentially differentiating immune-inflamed or immune-excluded tumors from tumors that would be classified as immune-desert. A similar imaging approach has been described in preclinical models using a PEGylated single-domain antibody fragment, 89Zr-PEGylated VHH-X118, with extremely low uptake in normal organs, including the kidneys (13). This study highlighted the potential ability of T-cell imaging in predicting the response to ICIs, as the tumor xenografts with homogeneous distribution of the CD8+ T-cell PET signal responded to anti-CTLA-4 agent whereas more heterogeneous infiltration of CD8+ T cells correlated with poor responses. Use of such agents would potentially enable selection of ipilimumab as either monotherapy or in combination with other ICIs.
In the later phases of the immune response, T cells accumulate but are unable to kill cancer cells. These tumors are considered to be either immune-excluded or immune-inflamed. It would be expected that these patients could be identified either by biopsy with analysis of the number of CD8+ TILs or by demonstration of CD8+ T cells within the TME by imaging. Accordingly, imaging of T cells within tumor deposits might be of value both prognostically and in determining the optimal therapeutic strategy. The ability to longitudinally evaluate treatment-induced dynamic changes in the immune cell composition and CD8+ TILs in TME has been demonstrated in preclinical models and remains one of the great advantages of CD8+ TIL imaging (14).
In this regard, it is important to recognize that, besides immunotherapy itself, other therapeutic interventions can modulate the immune environment. For example, radiotherapy is increasingly being used for its potential ability to positively impact the TME toward enhanced immune cell killing, even at remote sites through so-called abscopal effects (15). Key to this concept is the development of damage-associated molecular patterns, which include moieties such as calreticulin, heat-shock proteins, and cell membrane phospholipids. Damage-associated molecular patterns dictate whether cell death is immunogenic rather than tolerogenic. Combination therapies with ICIs and various DNA-damage response–modifying agents are in progress to investigate these phenomena (16). In theory, increased presentation of tumor-associated antigens acts to prime and activate T cells to traffic to lesions beyond the radiation volume. Demonstration of increased CD8+ T-cell numbers in remote sites could be helpful to confirm induction of abscopal effects. By irradiating multiple sites of disease simultaneously, the spectrum of tumor-associated antigens generated by radionuclide therapy might exceed that achieved by external-beam radiotherapy of a limited number of lesions but may also itself alter the spectrum of immune cell infiltrates. Information from CD8+ T-cell imaging may thus inform the optimal sequencing of radionuclide therapy and immunotherapy.
Imaging of T-Cell Function
The presence of CD8+ TILs in a tumor deposit does not, however, indicate whether these are functional or whether they are confined to the stromal compartment in the immune-excluded context. T-cell exhaustion is a key feature of immune escape. Accordingly, tracers that reflect their activation might provide further insight into the immune response. Reinvigoration of exhausted T cells is one of the mechanisms of both anti-CTLA-4 and anti-PD-1/PD-L1 monoclonal antibodies (17). Activation of T cells has been shown to be associated with altered nucleotide transport and phosphorylation. A fluorinated nucleotide, 2′-deoxy-2′-18F-fluoro-9-β-d-arabinofuranosylguanine (18F-Ara-G), is entering clinical evaluation to assess activation of T cells (18). An alternative approach is the use of a 68Ga-labeled peptide for PET imaging designed to target granzyme B, which is a serine-protease released by CD8+ T cells and natural killer cells involved in T-cell mediation of cancer cell death (19). Either of these agents may help to differentiate pseudoprogression from hyperprogression as a cause for increased 18F-FDG-uptake early during ICI therapy (Figs. 1 and 2).
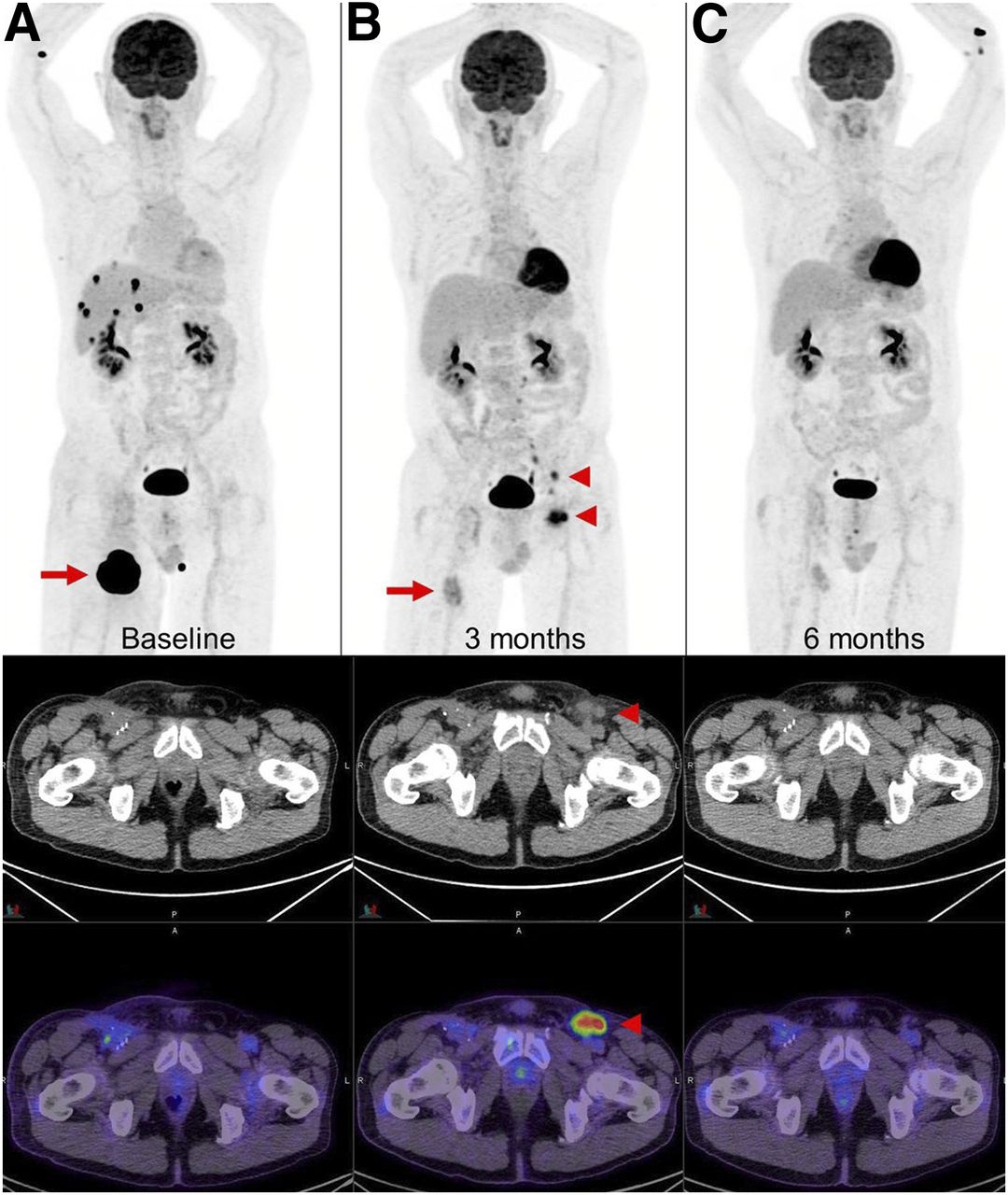
New lymph nodes in draining basin of regressing metastasis. (A and B) Metastatic melanoma (A, arrow) after 4 cycles of combination ipilimumab and nivolumab demonstrated marked regression of right thigh lesion and complete metabolic response of multiple liver and adrenal metastases (B, arrow); however, new 18F-FDG–avid lymph nodes were noted in left inguinal and iliac regions (B, arrowheads). (C) Biopsy of these lymph nodes showed reactive T cells that resolved on subsequent scan. Transient appearance of reactive lymph nodes contralateral to responding right thigh lesion was attributed to likely aberrant nodal drainage to contralateral nodes due to prior ipsilateral inguinal dissection. Immune-PET targeting CD8+ T cells, such as 18F-Ara-G/granzyme B, may allow noninvasive characterization of some cases with suspected pseudoprogression.

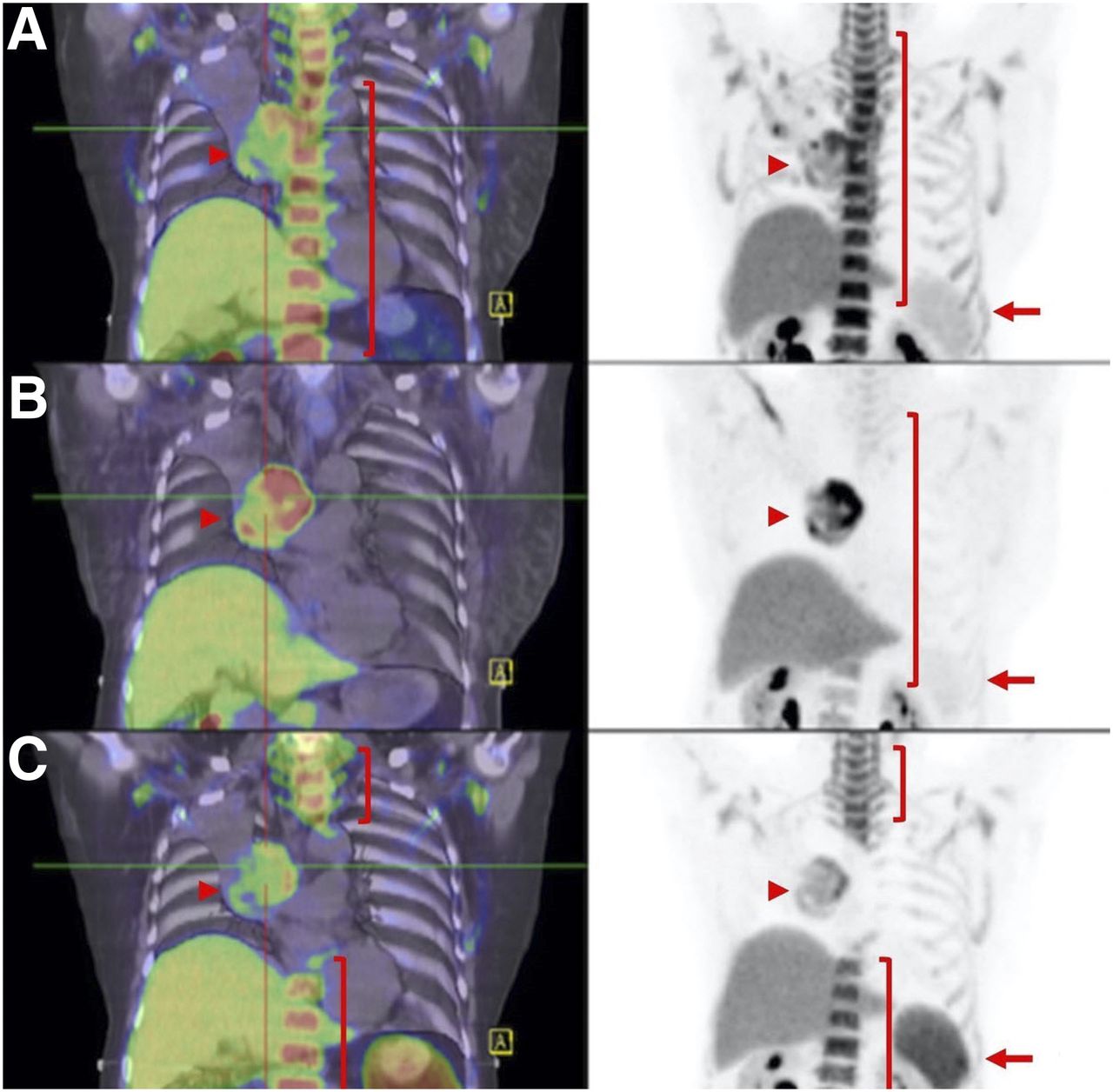
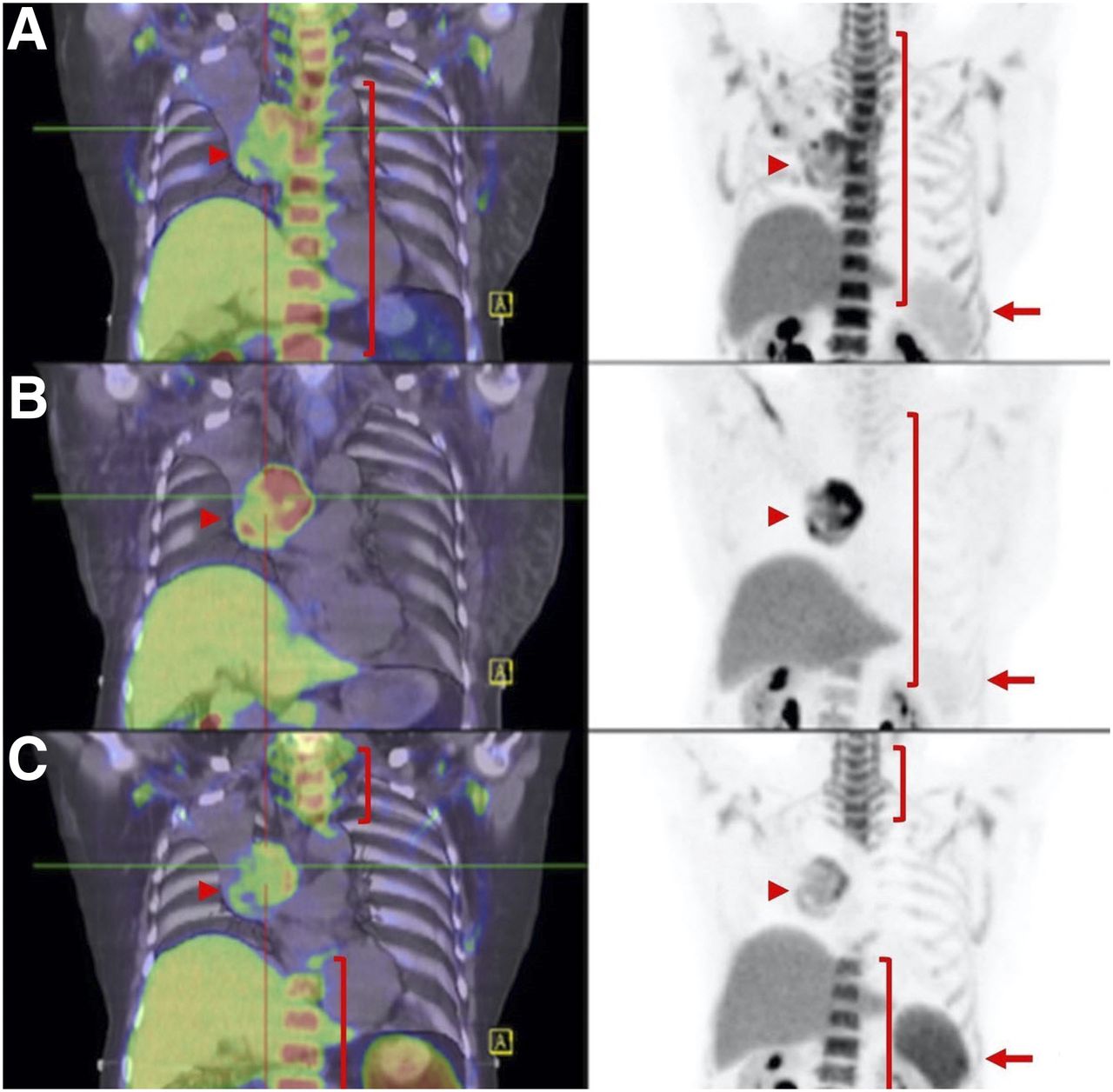
Hyperprogression vs. pseudoprogression. (A and B) Patient with metastatic melanoma at baseline (A, arrows) and 2 mo after start of combination ipilimumab and nivolumab demonstrates marked progression of liver metastasis (B, arrows). (C) As patient was clinically well, immunotherapy was continued, and further follow-up at 4 mo showed marked improvement of liver lesion (arrows). Immune-PET targeting CD8+ T cells, such as 18F-Ara-G/granzyme B, may allow pseudoprogression to be distinguished from hyperprogression in challenging cases.
Imaging of Other Immune Cell Populations
Preclinical studies suggest that CD8+ T lymphocytes are not the only cell types positively associated with response to ICIs, with group 2 innate lymphoid cells also appearing important, at least in some tumor types, as predictors of response to anti-PD1 agents (20). Therefore, imaging only 1 cell population may not necessarily be predictive of response.
Although there is a focus on imaging the effector limb of the immune response to cancer, it must be recognized that immune suppressive factors are important in allowing a cancer to develop, persist, and metastasize. Myeloid-derived suppressor cells, M2 macrophages, and regulatory T cells are important in this regard. Whether more specific markers on the surface of these cells may help to identify patients who may not benefit from current ICIs remains uncertain. FOXP3, which pathologically characterizes regulatory T cells, is expressed in the cell nucleus and is therefore unsuitable for imaging, and many cell-surface antigens are shared by several lymphocyte lineages, limiting their specificity. It is unclear if cells with negative immunomodulatory effects are present in sufficient density to allow imaging within the constraints of current PET imaging technology or whether there are appropriate cell-surface antigens that are specific enough to allow their detection. This will be a subject of further research.
Imaging of PD-1/PD-L1 Expression
Upregulation of PD-1/PD-L1 signaling is one of the key factors in suppressing cell killing in inflamed tumors and therefore a logical target for imaging cases most likely to respond to single anti-PD-1/PD-L1 agents (21). Although PD-L1 expression in tumors can be identified by immunohistochemistry, this has numerous pitfalls (22). Molecular imaging with PET/CT offers the possibility of imaging this marker, as well as other immune markers, noninvasively on a whole-body scale. Similar to immunohistochemistry, the aim of imaging the PD1/PD-L1 biomarker is to predict likely responders to ICIs and to provide the advantage of measuring inter- and intratumor heterogeneity of PD1/PD-L1 expression longitudinally under ICI treatment across all disease sites rather than a limited pathologic sampling. As discussed previously, whereas the predictive ability of 18F-FDG PET/CT in response to anti-PD-1/PD-L1 agents remains to be determined (9–11), there are consistent reports of its prognostic value by measuring metabolic tumor volume in patients undergoing treatment with anti-CTLA4 and anti-PD-1/PD-L1 monoclonal antibodies (23,24). The latter provides additional support for the rationale of assessing whole-body metabolic tumor volume on 18F-FDG PET/CT as a prognostic biomarker and potentially using PD-1/PD-L1 imaging, particularly in patients with a high whole-body metabolic tumor volume because patients with a low whole-body metabolic tumor volume are likely to respond a priori.
Several different anti-PD-1/PD-L1 monoclonal antibodies in clinical use as therapeutic agents have been labeled with various radiotracers. Those entering clinical trials have recently been summarized (25). In a first-in-humans study on patients with non–small cell lung cancer by Niemeijer et al.—using an anti–PD-L1 adnectin labeled with 18F (18F-BMS-986192) and anti-PD1 monoclonal antibody labeled with 89Zr (89Zr-nivolumab)—tumor uptake heterogeneity was observed both within and between patients (26). A similar observation of intra- and intertumoral heterogeneity was made by Bensch et al. using the anti-PD-L1 monoclonal antibody 89Zr-atezolizumab, but this time also within various tumor types. Encouragingly, however, clinical response to atezolizumab correlated better with anti PD-L1 PET signal intensity than with immunohistochemistry or RNA-sequencing biomarkers (27). Smaller antibody fragments, including a single-domain agent labeled with 68Ga (68Ga-NOTA-Nb109), may have advantages for diagnostic application due to rapid blood clearance and high tumor binding (28).
However, there are certain inherent limitations that have made PD1/PD-L1 an imperfect predictive biomarker. PD-L1 is also expressed on various immunosuppressive cells, including myeloid-derived suppressor cells and regulatory T cells (29), limiting the potential positive predictive value of PD-L1 imaging strategies, whether using small target-binding proteins such as 18F-adnectins or radiolabeled monoclonal antibodies, which have been recently compared in human subjects (26). Furthermore, because the PD-1/PD-L1 axis is but one of many mechanisms for immune escape, it is not surprising that some patients fail to respond to anti-PD1/PD-L1 ICIs despite harboring an inflamed TME. Accordingly, imaging or immunohistochemical confirmation of the PD-1/PD-L1 axis may not necessarily predict response. How imaging might be guided by pathologic characterization of immune contexture (30) and be complementary to other biomarkers remains to be defined.
Imaging of Other Immunologic Targets
The field of immune-PET is rapidly advancing, accelerated by advances in radiochemistry and biomolecule design (31) and fueled by greater understanding of cancer immunity. In addition to the CTLA-4 and PD-1/PD-L1, other factors are involved in immune editing that allow escape of cancers from immune surveillance. These include immune-suppressive targets such as lymphocyte activation gene 3, T-cell immunoglobulin mucin receptor 3, and indoleamine-2,3-dioxygenase 1, which may suppress recognition of neoantigens that might otherwise induce an immune response (32). Indeed, T-cell immunoglobulin mucin receptor 3 upregulation has been demonstrated in anti-PD-1–resistant tumors (33). Another example is CD276 (also known as B7-H3), a member of the B7 costimulatory molecules, the same family that PD-1/PD-L1 belongs to, and similarly has a role in the 2-signal T-cell activation. In some cancers, B7-H3 overexpression is associated with more aggressive tumor behavior potentially by circumventing CD8+ T-cell–mediated immune surveillance (34). Anti-B7-H3 agents combined with anti-PD-1/PD-L1 ICIs are a promising approach for B7-H3–expressing cancers. Preclinical attempts to imaging CD276 with humanized anti-B7-H3 monoclonal antibody (89Zr-labeled 5573a) have found this agent to have high specificity, supporting further clinical investigation (35). The abovementioned targets for imaging may allow prediction of failure to respond to current immunotherapy paradigms, thereby sparing futile therapy. Alternatively, they may allow prescription of combination therapies targeting these factors.
Imaging the TME for Factors Known to Be Associated with Immunosuppression
Recognizing that the TME impacts immune function, indirect information assessable from molecular imaging probes might also become relevant to predicting outcomes from ICIs, as well as informing management decisions. For example, hypoxia is recognized to have immune-suppressive effects (36). Multiple hypoxia tracers have been evaluated and could be used to design new strategies to modulate the adverse effects of hypoxia (37). Upregulation of carbonic anhydrase IX is one of many features of hypoxia and itself has been found to impact cancer immunity by increasing the number of immunosuppressive regulatory T cells (38). The ability to image tissue expression of carbonic anhydrase IX has recently become possible (39) and, besides allowing carbonic anhydrase IX to become a potential theranostic target, may identify patients who are less likely to respond to ICIs. Similarly, tumor desmoplasia, which is related to cancer-associated fibroblasts, is also an adverse prognostic factor in many cancers (40) and has recently been shown to negatively impact immune surveillance through inducing myeloid-derived suppressor cells (41). Fibroblast-activating protein, which is elaborated by cancer-associated fibroblasts, has recently become a tractable imaging target using fibroblast-activating protein inhibitors (42). Whether high uptake of fibroblast-activating protein inhibitor might predict failure of ICI therapy remains to be assessed.
Other PET tracers that have already been used in humans may also be repurposed for specific interrogation of tumor biology. For example, proliferative markers such as 3′-18F-fluoro-3′-deoxythymidine might find a role in differentiating hyperprogression, which would have a high signal in tumor sites, from pseudoprogression, for which active proliferation might be expected to be indicated by T-cell expansion in draining lymph nodes and the spleen (Fig. 3). A small study on patients being treated for metastatic melanoma has compared 18F-FDG and 3′-18F-fluoro-3′-deoxythymidine PET/CT with an anti-CTLA-4 agent, tremelimumab, demonstrating significantly increased splenic uptake after treatment (43).
Serial imaging with 3′-18F-fluoro-3′-deoxythymidine PET/CT of non–small cell lung cancer before and after chemoradiotherapy. (A) At baseline, there is low uptake in spleen (arrow), which is primarily a repository of immune cells, but high uptake in proliferating bone marrow (bracket), as well as in tumor in right hilum (arrowhead). (B) Early in treatment, reduction in bone marrow activity is observed (bracket), particularly within radiation treatment volume, but persistence of low uptake in spleen (arrow) and higher uptake in tumor (arrowhead). (C) At 3 mo after radiotherapy, there has been recovery in marrow proliferation beyond high-dose radiation field (brackets) but development of increased uptake in spleen (arrow) and lower uptake in tumor (arrowhead). This potentially reflects proliferation of immune cells representing immune response to release of tumor-associated antigens.
INTEGRATING NOVEL TRACERS INTO CLINICAL IMAGING PARADIGMS
In selecting the appropriate therapy, the simplest potential approach is to label that therapeutic agent to assess expression of the putative target. Because most current ICIs are intact monoclonal antibodies, the kinetics of blood clearance mandate use of a relatively long-lived radionuclide. Accordingly, several have been labeled with 89Zr or 64Cu (44). However, these agents require imaging at late time-points, which is inconvenient for patients, and dosimetry considerations limit the administered activity, potentially compromising image quality. Accordingly, efforts to develop small-molecule tracers have accelerated. Significant promise has been demonstrated for a PD-L1–targeting adnectin that has been labeled with 18F (26).
How to integrate these new tracers into clinical practice remains unclear. It is unlikely, given the complexity of the immune environment, that any one tracer will soon replace 18F-FDG as a diagnostic or treatment-monitoring investigation, especially with the regulatory hurdles that confront new diagnostic tests. Nevertheless, the cost and potential toxicity of ICIs suggest that more efficient selection and monitoring of treatment could both improve patient outcomes and reduce health-care costs. On the basis of the key management decisions that confront oncologists with the current generation of ICIs, and recognizing that new combination therapies are emerging and that definitive data on the utility of these new agents are lacking, we propose a possible diagnostic algorithm incorporating both 18F-FDG PET/CT and immune-PET to aid key management decisions (Fig. 4). This speculative algorithm assumes that PET is complementary to other established tissue-based biomarkers, which might include techniques such as the “Immunoscore,” a pathologic scoring system that assesses the type and density of immune cell infiltrates (45). The algorithm also suggests that 18F-FDG PET/CT will remain the primary technique for evaluating the extent of disease and monitoring response to treatment. However, for more sophisticated characterization of immune context, specific tracers will be used to select treatments and to resolve diagnostic dilemmas that currently require serial imaging follow-up but that potentially delay cessation of inactive treatment or recognition of toxicity. We have selected agents that are already in human trials, but these may well be replaced by any of a myriad of other agents being tested in preclinical models or being developed as companion diagnostics for new immunotherapy agents. Clinical validation of these agents will likely prove challenging given the rapidly evolving clinical paradigms and increasing use of combination therapies, but we recommend integrating prospective molecular imaging into baseline evaluation and at response assessment to increase our understanding of both the utility of the diagnostic agent and the mechanisms of resistance to immunotherapy, since treatment failure is still the norm.

Speculative diagnostic algorithm using PET agents to interrogate immune therapy. Through combination of baseline 18F-FDG PET/CT and biopsy, prognostic factors for response to ICIs, including whole-body metabolic tumor volume and TILs, could be assessed. For disease with low or absent TILs on biopsy, imaging with CD8+ agents could provide baseline for assessment of interventions to enhance immune cell infiltration, particularly including anti-CTLA-4 therapy or use of radiotherapy to induce T-cell priming by release of tumor-associated antigens. For those with high TILs, radiolabeled anti-PD-1/PD-L1 or 18F-adnectin PD-L1 agents could be used to select patients for ICI monotherapy against these targets. Early (6–8 wk) during given therapy, 18F-FDG PET/CT could be repeated. Conventional 18F-FDG PET/CT response would enable ongoing treatment, whereas increase in 18F-FDG uptake or development of new lesions could be further assessed by 18F-Ara-G or granzyme B imaging to detect T-cell activation as manifestation of pseudoprogression. Although not shown in this algorithm, 3′-18F-fluoro-3′-deoxythymidine PET/CT may also be helpful for demonstrating enhanced proliferation in tumor in case of hyperprogression or draining nodes and spleen in case of pseudoprogression. In patients receiving anti-CTLA-4 monotherapy agents but not responding on 18F-FDG PET/CT, reimaging with CD8+ T-cell agents to confirm efficacy of T-cell recruitment or empiric addition of anti-PD-1/PD-L1 agents may be appropriate, whereas for patients already on such treatment, withdrawal of treatment might be contemplated. Therapeutic agents that target immunosuppressive factors might also be considered in such cases. CMR = complete metabolic response; PMD = progressive metabolic disease; PMR = partial metabolic response; SMD = stable metabolic disease; wbMTV = whole-body metabolic tumor volume.
We have not considered the evolving field of adoptive cell therapies, including chimeric-antigen T-cell therapies (46). There have been significant efforts to develop robust methods of tracking these cells, including the use of reporter genes, which complement the vast arrays of probes that comprise the immunoimaging toolbox, as recently reviewed by Mayer and Gambhir (47).
CONCLUSION
The simultaneous strength and weakness of molecular imaging is the plethora of imaging targets identified through our increasing understanding of the extremely complex regulators of the immune environment and increasingly sophisticated techniques for producing tracers directed toward these regulators. In view of the regulatory hurdles that face approval and reimbursement of novel radiopharmaceuticals, as well as the costs of comparative studies to determine which option for imaging the immune environment will most effectively guide treatment choices, it seems unlikely that the clinical community will soon have access to the evolving tools for precision medicine, outside clinical trials. The nuclear medicine community may therefore need to initially focus on developing a limited number of agents, guided by the first principles and promising leads that have already entered clinical studies. These agents might, for example, be based on patient-care pathways and the therapeutic options currently available and be integrated with more conventional imaging paradigms.
Specific imaging of the immune system during immune therapy may not be necessary if an early and complete metabolic response is seen on 18F-FDG PET/CT, but early differentiation of pseudoprogression from hyperprogression may have significant benefits for further treatment choices. An increase in infiltrating CD8+ cells or evidence of increased granzyme B release may be instructive in this setting.
Academia needs to partner with industry to develop relevant companion diagnostic agents that rationalize the use of treatment with the simultaneous goals of increasing efficacy, reducing unnecessary toxicity, and minimizing costs both directly from the therapy and indirectly from managing complications. In this regard, 18F-FDG PET/CT will remain an important diagnostic tool in the selection of patients for ICI therapy and monitoring of response but will be, in our opinion, complemented by specific imaging of components of the immune system (Table 1).
Learning Points and Speculation on Future Directions for Imaging Immune System with 18F-FDG and New Tracers
Acknowledgments
We are particularly indebted to our medical oncology colleagues for their ongoing interest in the use of molecular imaging to characterize cancer and monitor response to novel therapies, and we thank them for their tolerance while we learned the nuances and complications of immune-oncology regimens. We are also grateful to our radiochemistry team, led by Dr. Peter Roselt, for indulging our interest in novel tracers and bringing them to the clinic, and we thank our nuclear medicine technologists who fit research studies seamlessly into a busy clinical program. Finally, we are grateful for a longstanding collaboration with Dr. Paul Donnelly and his team in developing peptide and antibody-based radiopharmaceuticals for the benefit of our patients.
Footnotes
Published online Sep. 4, 2020.
Learning Objectives: On successful completion of this activity, participants should be able to describe (1) the potential complementary role of immune-PET to FDG PET/CT for selecting and monitoring of immune checkpoint inhibitor therapy; (2) the aspects of CD8+ T cell biology that can currently be imaged; and (3) a framework for future development and advancement of immune-PET in the field of immune-oncology.
Financial Disclosure: Professor Hicks is a scientific advisor to and holds shares in Telix Pharmaceuticals, with proceeds donated to his institution. Telix Pharmaceuticals is undertaking a clinical trial using 89Zr-girentuximab, which is mentioned in this review. Professor Hicks is also a member of the Independent Data Management Committee for AAA/Novartis. The authors of this article have indicated no other relevant relationships that could be perceived as a real or apparent conflict of interest.
CME Credit: SNMMI is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to sponsor continuing education for physicians. SNMMI designates each JNM continuing education article for a maximum of 2.0 AMA PRA Category 1 Credits. Physicians should claim only credit commensurate with the extent of their participation in the activity. For CE credit, SAM, and other credit types, participants can access this activity through the SNMMI website (http://www.snmmilearningcenter.org) through November 2023.
- © 2020 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication May 18, 2020.
- Accepted for publication August 18, 2020.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Approaches to Imaging Immune Activation Using PET
- Preclinical Evaluation and Pilot Clinical Study of CD137 PET Radiotracer for Noninvasive Monitoring Early Responses of Immunotherapy
- ICAM-1 orchestrates the abscopal effect of tumor radiotherapy
- FAPI PET/CT: Will It End the Hegemony of 18F-FDG in Oncology?