


Abstract
In preclinical studies, 5-fluoro-2′-deoxycytidine (FdCyd), an inhibitor of DNA methyltransferase and DNA hypermethylation, has shown treatment efficacy against multiple malignancies by suppressing epigenetic hypermethylation in tumor cells. Several ongoing clinical trials are using FdCyd, and although some patients may respond to this drug, in most patients it is ineffective. Thus, establishing a noninvasive imaging modality to evaluate the distribution of the drug may provide insight into the variable responses. A novel experimental radiopharmaceutical, 18F-labeled FdCyd, was developed as a companion imaging agent to the nonradioactive form of the drug, FdCyd. We present the first-in-humans radiation dosimetry results and biodistribution of 18F-FdCyd, administered along with tetrahydrouridine, an inhibitor of cytidine/deoxycytidine deaminase, in patients with a variety of solid tumors undergoing FdCyd therapy. Methods: This phase 0 imaging trial examined the 18F-FdCyd biodistribution and radiation dosimetry in 5 human subjects enrolled in companion therapy trials. In each subject, 4 sequential PET scans were acquired to estimate whole-body and individual organ effective dose, using OLINDA/EXM, version 1.0. Tumor-to-background ratios were also calculated for the tumor sites visualized on PET/CT imaging. Results: The average whole-body effective dose for the experimental radiopharmaceutical 18F-FdCyd administered in conjunction with tetrahydrouridine was 2.12E−02 ± 4.15E−03 mSv/MBq. This is similar to the radiation dose estimates for 18F-FDG PET. The critical organ, with the highest absorbed radiation dose, was the urinary bladder wall at 7.96E−02 mSv/MBq. Other organ doses of note were the liver (6.02E−02mSv/MBq), kidneys (5.26E−02 mSv/MBq), and gallbladder (4.05E−02 mSv/MBq). Tumor target-to-background ratios ranged from 2.4 to 1.4, which potentially enable tumor visualization in static PET images. Conclusion: This phase 0 imaging clinical trial provides evidence that 18F-FdCyd administered in conjunction with tetrahydrouridine yields acceptable individual organ and whole-body effective doses, as well as modest tumor-to-background ratios that potentially enable tumor visualization. Dose estimates for 18F-FdCyd are comparable to those for other PET radiopharmaceuticals, such as 18F-FDG. Further studies with larger study populations are warranted to assess 18F-FdCyd imaging as a predictor of FdCyd treatment effectiveness.
A relation between malignancy and DNA methylation was recognized a few decades ago (1). DNA methyltransferase expression is elevated in most cancers, resulting in epigenetic alterations in the cancer cell genome (2). One form of epigenetic modification observed in cancer cells is alterations in DNA methylation that alter regulation of gene transcription (2). Cancer cells often demonstrate global DNA hypomethylation leading to genomic instability, as well as foci of hypermethylation, particularly within CpG islands associated with tumor suppressor genes, resulting in tumor suppressor gene silencing and thus promoting tumor growth (3).
FdCyd, 5-fluoro-2′-deoxycytidine, is an antineoplastic agent that inhibits DNA methyltransferase and DNA methylation by forming a covalent bond with the enzyme after incorporation into DNA (4), resulting in reexpression of genes silenced by DNA hypermethylation. FdCyd can be phosphorylated to 5-fluoro-2′-deoxycytidylate (FdCMP) by deoxycytidine kinase and the nucleotide deaminated to 5-fluoro-2′-deoxyuridine 5′-monophosphate by deoxycytidylate (dCMP) deaminase (5). The activity of dCMP deaminase is reported to be higher in human malignancies than in normal tissues, which may result in selective cytotoxicity (6,7).
The efficacy of this developmental antineoplastic therapeutic agent, particularly in oral form, has been limited by its rapid degradation by cytidine deaminase (8). However, unlike prior versions of the drug such as 5-azacytidine and 5-aza-2′-deoxycytidine, the breakdown of FdCyd by cytidine deaminase can be largely suppressed by coadministration of 3,4,5,6-tetrahydrouridine, an inhibitor of cytidine/deoxycytidine deaminase (9,10), thus reducing enzymatic deamination, which has shown antitumor activity superior to other antimetabolites, such as 5-fluorouracil in preclinical models (11,12) and in early-phase clinical trials (13).
FdCyd and tetrahydrouridine have shown preliminary evidence of activity against solid tumors in early-phase trials, although not all patients showed clinical response (13). The establishment of a radiolabeled form to image the biodistribution in vivo during therapy may provide insight into the distribution of the therapeutic drug (3,14). Thus, to optimize the likelihood of FdCyd’s clinical success against heterogeneous solid tumors, a companion diagnostic imaging agent, chemically identical to FdCyd and labeled with a PET tracer, could be useful in identifying tumors in which the drug is likely to accumulate and, therefore, where it could potentially be clinically more effective (15). Similar to other radiolabeled pyrimidine molecules, the limiting factor for imaging is the high background activity resulting from byproducts of pyrimidine catabolism, which in the case of radiolabeled FdCyd can be minimized with simultaneous administration of tetrahydrouridine (16). Thus, the present study was undertaken to better understand the biodistribution and radiation dosimetry of 18F-labeled FdCyd with coadministration of tetrahydrouridine and to describe tumor uptake in a small cohort of patients with solid tumors who were concurrently participating in either a phase I or a phase II clinical trial of FdCyd and tetrahydrouridine
MATERIALS AND METHODS
Patients and Study Design
This study was approved by the local Institutional Review Board (Cancer Therapy Evaluation Program [CTEP] 8865; National Cancer Institute [NCI] protocol 12-C-0014) in compliance with all applicable Federal regulations governing the protection of human subjects, and informed consent was obtained from all patients before imaging. Biodistribution data from 5 subjects (3 men and 2 women; age range, 30–65 y old) who underwent 18F-FdCyd PET were used for dosimetry estimates. The patients who were recruited were simultaneously participating in 1 of the 2 ongoing clinical therapeutic trials investigating FdCyd: a phase I trial (NCI protocol 12-C-0066; CTEP 9127; advanced solid tumors) and a phase II trial (NCI protocol 09-C-0214; CTEP 8351; head/neck, lung, breast, and urinary bladder neoplasms).
Radiopharmaceutical Production
Radioactive labeling of FdCyd with 18F was performed at the University of Pennsylvania, in Philadelphia. The FdCyd was produced using 25 ± 3 mg of precursor, 2′-deoxycytidine, which was dissolved in 8 mL of acetic acid, and 25 μL of the boron trifluoride acetic acid solution were added to the solution. Then, 18F2 was bubbled through it, with unreacted 18F being captured by the KI solution. The activity was transferred to the evaporation vessel. The acetic acid was evaporated, and a second aliquot, 2 mL, of acetic acid was added and evaporated. The reaction product was transferred from the evaporation vial to the integrated high-pressure liquid chromatography injector port by the addition of 2 mL of the mobile-phase buffer (92:8 acetonitrile:deionized water). The high-pressure liquid chromatography was started, and the 5-formyl-2′-deoxycytidine was collected after approximately 45 min. After collection, the high-pressure liquid chromatography solvent was evaporated, and the final product was rinsed off the walls of the evaporation vessel using isotonic saline and transferred into a laminar flow or biosafety cabinet for final compounding. The drug substance was collected directly through a 0.2-μm sterilizing filter into a vented sterile vial. Samples were aseptically removed for analysis of product quality. The amount of injected drug was no more than 30 mg of 5-formyl-2′-deoxycytidine, providing a minimum specific activity of 80 mCi/mmol. A semiautomated synthesis unit was used so that each step in the procedure could be performed under computer control, minimizing the variance in the chemical reaction.
18F-FdCyd PET Imaging
FdCyd and tetrahydrouridine were administered according to the specifications of the parent protocol, NCI clinical protocol 09-C-0214 (CTEP 8351) or NCI protocol 12-C-0066 (CTEP 9127). Briefly, an intravenous bolus of 20% of the total dose of tetrahydrouridine (total dose of tetrahydrouridine, 350 mg/m2) was administered, followed by the remaining 80% of the dose, diluted in 250 mL of D5W, infused for 3 h or until the end of imaging. Twenty minutes after the start of intravenous tetrahydrouridine, and concurrent with or within 1 h after the start of the nonradioactive FdCyd therapeutic dose, the experimental PET radiopharmaceutical, 18F-FdCyd (average dose, 140.6 ± 36.8 MBq), was administered intravenously over approximately 1 min. Three sequential static whole-body PET emission scans were performed from the top of the head to the upper thigh for up to 1 h. A fourth additional PET emission scan was acquired at about 2 h after injection. Low-dose CT scans (120 keV, 60 mAs) were acquired for attenuation correction and anatomic correlation.
Image Analysis
For each PET/CT examination, volumes of interest were manually drawn over all organs using MIM version 5.6.7 (MIM Software). When the entire organ volume could not be separated from the adjacent structures or the whole organ volume could not be delineated (e.g., heart wall, bone marrow, and muscle), a smaller volume of interest was drawn to estimate the organ activity concentration. For each PET examination, the activity concentration extracted from the volumes of interest was decay-corrected and normalized to the time of the initial dose injection. Tumor volumes of interest were manually drawn, as identified from prior diagnostic anatomic imaging studies and review of patient electronic medical records. Tumor SUVmax was measured at 4 time points to assess biodistribution. Tumor-to-background ratios (TBRs) were also calculated.
Radiation Dosimetry
Time–activity curves for each organ region of interest were generated using Excel (Microsoft). An exponential decay function based on the half-life of 18F was used to estimate the remainder of the time–activity curve after the final time point. Normal organ radioactivity residence times were obtained by integrating the area under each time–activity curve. The residence times were entered into the kinetic input field in OLINDA/EXM version 1.0 (Vanderbilt University), which enabled calculation of individual-organ and whole-body effective doses. In the OLINDA model input form, the appropriate sex model was selected for each patient. No bladder-voiding or gastrointestinal models were used because of the short half-life of 18F.
RESULTS
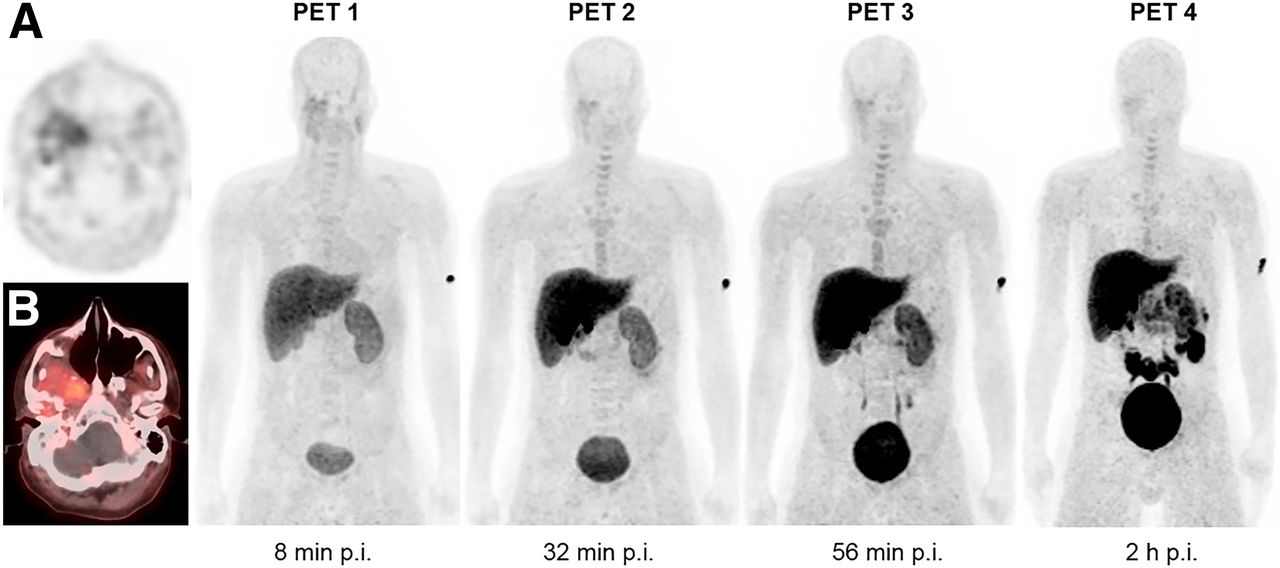
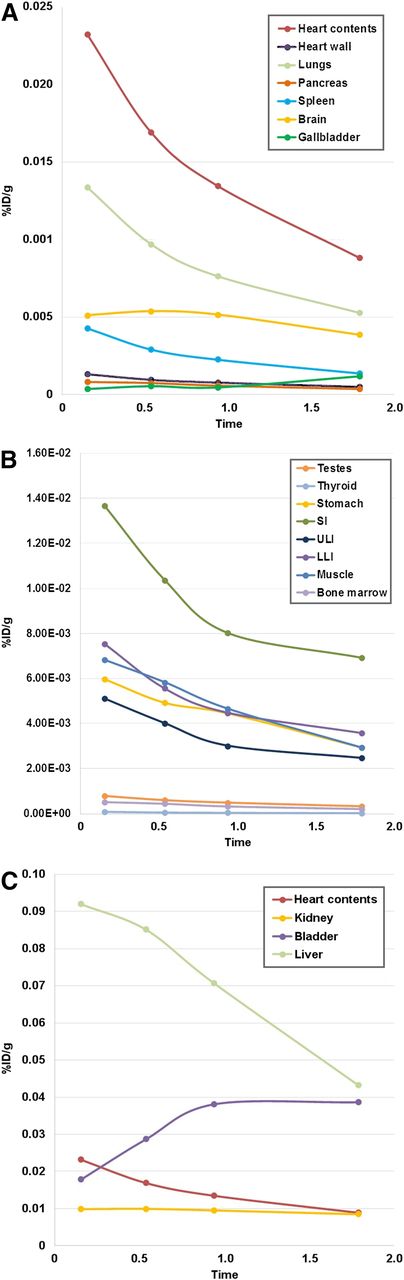
Tumor types represented in this study included head and neck cancer (n = 2), non–small cell lung cancer (n = 2), and hepatocellular carcinoma (n = 1). Patients had at least 1 target lesion measuring 1 cm or larger. Approximately 140.6 ± 36.8 MBq (∼3.8 ± 0.9 mCi; range, 2.1–5 mCi) of 18F-FdCyd was infused intravenously. Figure 1 shows maximum-intensity-projection 18F-FDCyd PET images over time in a representative patient. The activity from the blood pool cleared rapidly, with evidence of hepatic and renal routes of excretion and radiotracer accumulation within the bladder. Physiologic radiotracer uptake was seen in the liver and small intestine. Time–activity curves for individual normal organs are shown in Figure 2.
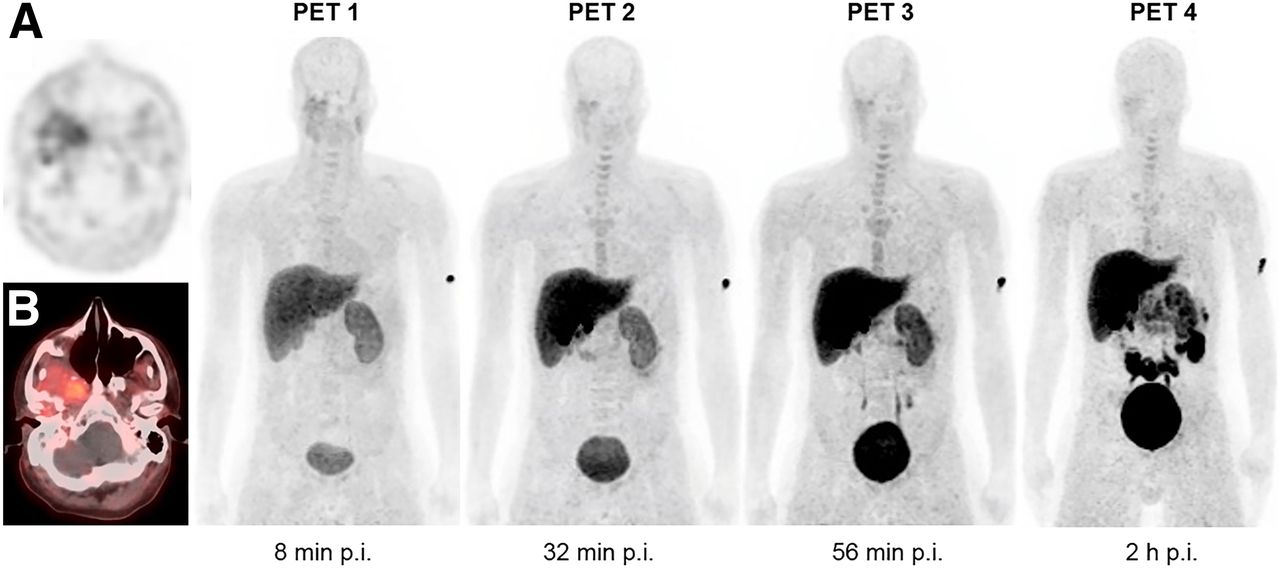
Maximum-intensity-projection PET imaging sequence after 18F-FdCyd injection in patient 2. Images demonstrate physiologic uptake in liver, gallbladder, kidneys, urinary bladder, small intestine, and mildly in bone marrow. There is modest tumor uptake in histologically confirmed spindle cell carcinoma at right parapharyngeal and masticator spaces, best seen on axial PET (A) and PET/CT (B). p.i. = after injection.
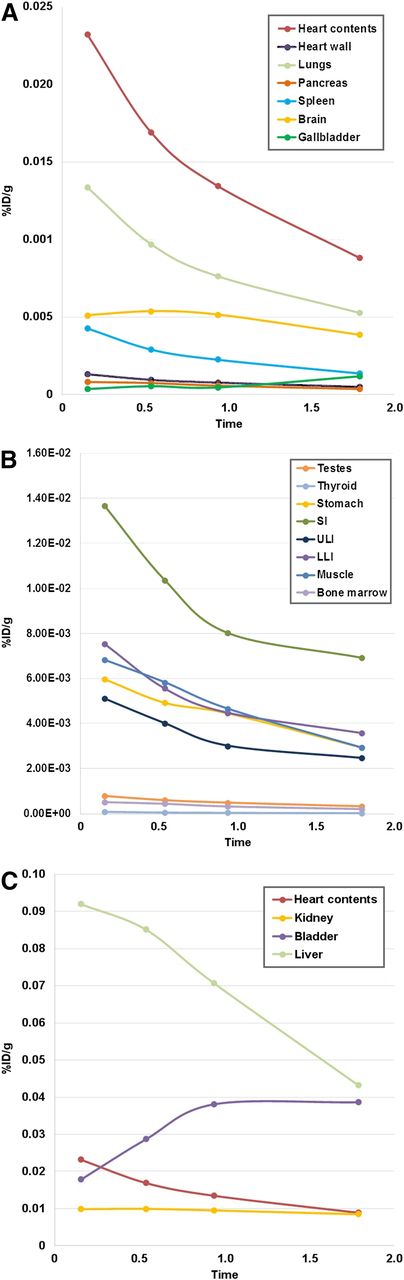
Mean biodistribution curves plotted for percentage injected dose per gram of organ mass vs. time. (A) Organs with higher uptake. (B) Organs with lower uptake. (C) Increasing urinary bladder activity. LLI = lower large intestine; SI = small intestine; ULI = upper large intestine; %ID/g = percentage injected dose per gram of organ mass.
Estimates of whole-body effective dose ranged from 1.86E−02 to 2.80E−02 mSv/MBq, with an average of 2.12E−02 ± 4.15E−03. The urinary bladder wall received the highest individual-organ dose, at 7.96E−02 mSv/MBq. Other organ doses of note were the liver (6.02E−02 mSv/MBq), kidneys (5.26E−02 mSv/MBq), and gallbladder (4.05E−02 mSv/MBq). Individual-organ and whole-body effective doses are shown in Table 1.
Dosimetry Estimates in mSv/MBq
Four of the 5 patients showed visual tumor uptake, except for patient 5, whose target tumor was within the liver and obscured by the high background uptake of the liver. The average tumor SUVmax was 3.3 (range, 2.6–3.9) for the first scan (average time, 9 min; range, 6–15 min), 2.4 (range, 2.0–3.1) for the second scan (average time, 32 min; range, 29–37 min), 2.1 (range, 1.9–2.1) for the third scan (average time, 56 min; range, 52–64 min), and 1.8 (range, 1.7–1.8) for the delayed scan at about 2 h after injection (average, 107 min; range, 86 min to 2 h), with calculated TBRs of 1.8, 1.7, 1.5, and 1.7, respectively. The patient-specific clinical information and tumor TBRs are detailed in Table 2.
Patient Demographics and TBR of Target Lesions on PET at 4 Time Points After Injection
DISCUSSION
The cytidine analog FdCyd has shown treatment efficacy in preclinical studies of solid and hematologic tumors (11). In the clinical setting, FdCyd was studied in combination with a fixed dose of tetrahydrouridine in a multicenter phase I study including 58 patients with advanced solid tumors (13); response data from 40 of those patients showed partial response for about 15 mo in 1 of the patients, and stable disease in 20 patients (13). Multiple factors have hampered the clinical effectiveness of FdCyd. The principal limiting factor in FdCyd activity is its rapid degradation by cytidine deaminase (8), leading to a very short half-life in circulation; however, this enzyme can be largely suppressed by coadministration of 3,4,5,6-tetrahydrouridine, which greatly prolongs the clearance of the FdCyd (16). This is the first-in-humans study using 18F-FdCyd and tetrahydrouridine PET/CT imaging presenting biodistribution and dosimetry data from 5 patients with solid tumors undergoing therapeutic trials with FdCyd. The intent of imaging 18F-FdCyd in combination with tetrahydrouridine was to determine whether it would result in decreased background activity, and thus achieve acceptable TBRs, sufficient for tumor imaging. Similarly, other groups have used enzymatic inhibitor coadministration with developmental imaging agents as a rational approach to minimize undesirable background activity from radiopharmaceutical catabolism to improve TBR, that is, disulfiram mitigating defluorination in 18F-FCWAY PET imaging (17). Other potential factors limiting drug effectiveness include heterogeneity of methylation patterns and limitations in the ability to deliver the drug in sufficient concentrations. 18F-FdCyd was developed as a companion imaging agent to therapeutic FdCyd to provide a method for better patient selection. The aim of the study was to obtain preliminary safety data, human dosimetry, and biodistribution before deploying the agent on a wider basis.
The estimated whole-body effective dose for 18F-FdCyd was acceptable, with a mean of 2.1E−02 mSv/MBq, similar to 18F-FDG (18), which has an estimated whole-body effective dose of 1.9E−02 mSv/MBq (19). The effective dose was also similar to another 18F-labeled developmental radiopharmaceutical, 18F-fluorodihydrotestosterone, with an estimated whole-body effective dose of 1.8E−02 mSv/MBq (20). The largest estimated target organ dose for 18F-FdCyd and tetrahydrouridine was the urinary bladder wall (7.96E−02 mSv/MBq), which was slightly less than the estimated dose to the urinary bladder wall in 18F-FDG (8.65E−02 mSv/MBq) (21). With 18F-FdCyd plus tetrahydrouridine, other high absorbed doses were found in the liver (6.02E−02mSv/MBq), the kidneys (5.26E−02 mSv/MBq), and the gallbladder (4.05E−02 mSv/MBq). These values modestly exceed those observed for corresponding organs with 18F-FDG: 1.57E−02, 2.00E−02, and 1.32E−02 mSv/MBq, respectively (21). Although slightly higher, these doses do not approach regulatory limits.
The 18F-FdCyd TBRs ranged from 2.4 to 1.4 (in n = 4), depending of type of tumor and scan time (Table 2). In 1 subject with a hepatocellular carcinoma, the tumor was not visualized because of intense liver background activity; thus, the TBR was not calculated. The minimal PET TBR necessary for detection depends on many factors such as imaging system and scan time; however, prior studies have demonstrated that a TBR of 3.0 can potentially detect a lesion as small as 0.4 mL and that a TBR of 1.85 can potentially detect a lesion as small as 0.5 mL (21,22). Our preliminary data show that 18F-FdCyd PET imaging with tetrahydrouridine provides sufficient TBRs for detection of multiple types and sizes of tumors.
The biodistribution and dosimetry estimates of 18F-FdCyd revealed that the mechanism of radiopharmaceutical excretion is both renal and hepatic, thus limiting use for tumor detection in these 2 organs.
CONCLUSION
This phase 0, first-in-humans, clinical study provides evidence that PET imaging with 18F-FdCyd and tetrahydrouridine is feasible and safe, with acceptable target organ and whole-body effective radiation doses, and produces modest TBRs sufficient for tumor localization. This study also provides dose estimates for 18F-FdCyd that are comparable to those of other PET radiopharmaceuticals, such as 18F-FDG. Further studies using 18F-FdCyd in larger trials may be helpful to investigate its ability to enable selection of suitable candidates for FdCyd treatment and monitor therapy effectiveness.
DISCLOSURE
This research was supported by the Intramural Research Program of the NIH, NCI, Center for Cancer Research, and by the Division of Cancer Diagnosis and Treatment, NCI. No other potential conflict of interest relevant to this article was reported. Esther Mena is a military service member (or employee of the U.S. Government). This work was prepared as part of her official duties. Title 17 U.S.C. §105 provides that “Copyright protection under this title is not available for any work of the United States Government.” Title 17 U.S.C. §101 defines a U.S. Government work as a work prepared by a military service member or employee of the U.S.
Acknowledgments
We thank Richard H. Freifelder, PhD, for the synthesis of the tracer at the University of Pennsylvania, Philadelphia, PA. The opinions or assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the Department of the Navy, the Department of the Army, the Department of Defense, or the U.S. Government. Any citations of commercial organizations and trade names in this report do not constitute an official endorsement of the products or services of these organizations by the Department of the Navy, Department of the Army, or Department of Defense.
Footnotes
Published online Nov. 2, 2018.
- © 2019 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication July 5, 2018.
- Accepted for publication September 12, 2018.





{kind=link}
{kind=link}