


Abstract
Fibrillary tau aggregates in Alzheimer disease and allied neurodegenerative disorders have been visualized in vivo by PET, whereas mechanistic links between PET-detectable tau deposits and neurotoxicity remain elusive. Here, we took advantage of transgenic mouse models of tauopathies to evaluate associations between PET and postmortem measures of tau probe binding and their relation to neuronal loss. Methods: PET with a tau probe, 11C-PBB3 (2-((1E,3E)-4-(6-(11C-methylamino)pyridine-3-yl)buta-1,3-dienyl)benzo[d]thiazol-6-ol), and volumetric MRI were performed for transgenic rTg4510 mice and nontransgenic mice. Binding of 11C-PBB3 and its blockade by another tau binding compound, AV-1451 (-(6-fluoropyridine-3-yl)-5H-pyrido[4,3-b]indole), in homogenized brains of tauopathy patients and rTg4510 and PS19 mice were quantified, and 11C-PBB3–positive and phosphorylated tau lesions in sectioned brains of these mice were assessed. Results: In vivo 11C-PBB3 binding to the rTg4510 neocortex/hippocampus was increased relative to controls and correlated with local atrophy. In vitro 11C-PBB3 binding in the neocortex/hippocampus also correlated well with in vivo radioligand binding and regional atrophy in the same individual rTg4510 mice. By contrast, in vitro 11C-PBB3 binding was elevated in the brain stem but not hippocampus of PS19 mice, despite a pronounced loss of neurons in the hippocampus rather than brain stem. Finally, 11C-PBB3 and AV-1451 showed similar binding properties between mouse models and tauopathy patients. Conclusion: The present findings support the distinct utilities of 11C-PBB3 PET and MRI in rTg4510 and PS19 mice for quantitatively pursuing mechanisms connecting PET-detectable and PET-undetectable tau aggregations to neuronal death, which recapitulate 2 different modes of tau-provoked neurotoxicity.
Deposition of pathologic tau fibrils is characteristic of Alzheimer disease (AD) and related non-AD neurodegenerative disorders as exemplified by frontotemporal lobar degeneration (1). Normally functioning tau proteins are constituents of axonal cytoskeletons in neurons, and the self-assembly of tau molecules in a diseased condition gives rise to disruption of the axonal framework along with neurotoxicity induced by misfolded tau species (1,2). In line with this mechanistic view, tau abnormalities are known to be tightly associated with neurodegeneration and the emergence of clinical symptoms (3,4), providing a rationale for antitau treatments to modify the disease process (5). For the purpose of establishing etiologic, diagnostic, and therapeutic assessments of tau pathologies in living subjects, there have been growing demands for the visualization of tau fibrils in a noninvasive manner.
In vivo PET of tau lesions has recently been enabled by the development of small-molecule imaging agents selectively binding to the β-pleated sheet structure in tau filaments (6). Several tau PET probes, including 11C-PBB3 (2-((1E,3E)-4-(6-(11C-methylamino)pyridine-3-yl)buta-1,3-dienyl)benzo[d]thiazol-6-ol) (7), 18F-THK5117 (8), 18F-THK5351 (9), and 18F-AV-1451 (-(6-fluoropyridine-3-yl)-5H-pyrido[4,3-b]indole) (also known as 18F-T807 or 18F-flortaucepir) (10–12), have been applied to human subjects across the AD spectrum and have offered neuroimaging-based staging of tau pathologies. In these individuals, the distribution of PET-detectable tau deposits was reported to agree with the topography of brain atrophy and manifestation of focal symptoms (7,9,12), implicating the tau aggregation in the local neuronal deteriorations. In the meantime, it still remains elusive whether tau deposits in frontotemporal lobar degeneration are sensitively detectable by these PET probes to examine links between the localization of tau pathologies and clinical phenotypes (13–17). It is noteworthy that tau proteins comprise 6 isoforms classified into 4-repeat and 3-repeat species, and the composition of tau isoforms differs among diverse tauopathies (1,2). Indeed, AD-type tau fibrils are constituted by all 6 isoforms, in contrast to the exclusive assembly of 4-repeat tau isoforms into fibrils in a subgroup of frontotemporal lobar degeneration, such as progressive supranuclear palsy (PSP) and corticobasal degeneration (1,2). These distinct tau fibril species are ultra-structurally identified as paired helical filaments in AD and straight filaments in 4-repeat tauopathies (18,19), and such conformational variations may affect the reactivity of tau fibrils with PET probes (13). In fact, binding of 11C-PBB3 in the PSP motor cortex is not blocked by AV-1451, in contrast to a partial blockade of 11C-PBB3 binding by AV-1451 in the AD temporal cortex. The binding characteristics of these ligands can accordingly be used for discrimination between AD-like and PSP-like tau aggregates in human and mouse brain tissues.
In the nonclinical development of 11C-PBB3 (7), we used mice transgenic for a human 4-repeat tau isoform with the P301L and P301S mutations, termed the rTg4510 (Tg = transgenic) (20) and PS19 (21) lines, respectively. In vivo PET imaging and ex vivo autoradiographic imaging, and in vivo and ex vivo fluorescence imaging, of these animals provided significant information on the kinetics and detectability of tau deposits in the living brain by a candidate imaging agent (7). However, it still remains unclear whether preclinical investigation using rTg4510 and PS19 combined with 11C-PBB3 PET could provide useful information on imaging–pathologic relationships as in a clinical PET study or potentially contribute to quantitatively understanding the mechanisms connecting PET-detectable and PET-undetectable tau aggregations to neuronal death. In addition, the radioligand binding in small-animal PET assays might be underestimated in small anatomic structures because of partial-volume effects and might be overestimated in surface brain areas because of radioactivity spill-in from extracranial tissues. PET measures should accordingly be validated with reference to postmortem binding estimates in the same individuals.
To address these issues, we conducted 11C-PBB3 PET and volumetric MRI on rTg4510 mice, followed by postmortem assessments of the brains from the same animals, and assessed whether in vivo PET signals reflect the amount of specific 11C-PBB3 binding and phosphorylated tau deposits. Associations of these binding estimates with cerebral atrophy determined by MRI were also analyzed to demonstrate the ability of 11C-PBB3 PET to quantify tau species tightly linked to the neurotoxicity. Similarly, in vitro binding assays were applied to brain tissues collected from PS19 mice to clarify whether toxicity-related tau assemblies can be captured by 11C-PBB3 across different tauopathy models. Finally, in vitro binding of 11C-PBB3 versus AV-1451 in rTg4510 and PS19 mouse samples was compared with those of AD and PSP tissues for examining the translatability of the probe binding data in mouse models to AD and 4-repeat tauopathies. Assessments of the competition between 11C-PBB3 and AV-1451 binding in various brain tissues would also provide information on the presence of common off-target binding sites for these 2 compounds.
MATERIALS AND METHODS
Ethics Statement
The mice studied here were maintained and handled in accordance with the National Research Council’s Guide for the Care and Use of Laboratory Animals and our institutional guidelines. Protocols for the present animal experiments were approved by the Animal Ethics Committees of the National Institute of Radiologic Sciences, National Institutes for Quantum and Radiologic Science and Technology. The research using autopsied human samples was approved by the Ethics Committee of National Institute of Radiologic Sciences.
Reagents and Antibody
PBB3 and a desmethyl precursor for radiosynthesis of 11C-PBB3 were obtained from Nard Institute. AV-1451 was in-house–synthesized according to a previous report (US 2012/0302755 A1). BTA-1 (2-(4′-methylaminophenyl)benzothiazole), clorgyline, and selegiline (also known as deprenyl) were purchased from Sigma-Aldrich. AT8, a mouse monoclonal antibody against human tau phosphorylated at Ser202 and Thr205, was purchased from Endogen/Thermo Fisher Scientific.
Animal Models
Two mouse models, PS19 and rTg4510, were used in the present study. PS19 transgenic mice heterozygous for human T34 (4-repeat tau isoform with a single N-terminal insert), with P301S mutation driven by the mouse prion protein promoter (21), were provided by the Perelman School of Medicine, University of Pennsylvania, and were bred and kept on a C57BL/6 background. rTg4510 mice, tau responder mice, and tetracycline-controlled transactivator mice were obtained from the University of Florida. A parental mutant tau responder line on an FVB/N strain (Clea Inc.) and a tetracycline-controlled transactivator activator line in a 129+ter/SV strain (Clea Inc.) were generated and maintained. To generate a tau responder line expressing human T43 (4-repeat tau isoform without N-terminal inserts) with P301L mutation, complementary DNA was placed downstream to a tetracycline-operon-responder construct. The tetracycline-controlled transactivator system was inserted downstream to the Ca2+-calmodulin kinase IIα promoter. Hemizygous mice from each parental line were cross-bred to produce rTg4510 mice possessing both responding and tetracycline-controlled transactivator constructs and nontransgenic littermates (20). A workflow of in vivo PET and MRI and in vitro binding assays with rTg4510 and nontransgenic mice is shown in Supplemental Figure 1 (supplemental materials are available at http://jnm.snmjournals.org).
Radiosynthesis and Small-Animal PET Imaging
Radiosynthesis of 11C-PBB3 was performed as described previously (7), and the resulting radiochemical purity was more than 90%. The molar activity of 11C-PBB3 was 78–93.1 GBq/μmol at the end of synthesis. PET was performed on a microPET Focus 220 animal scanner (Siemens Medical Solutions) as described previously (22). At 7–10 mo old, rTg4510 mice (n = 5; body weight, 23.6 ± 0.8 g) and age-matched nontransgenic mice (n = 7; body weight, 29.1 ± 1.5 g) were anesthetized with 1.5% (v/v) isoflurane. Emission scans were acquired for 60 min in 3-dimensional list mode with an energy window of 350–750 keV, immediately after intravenous injection of 11C-PBB3 (18.2–23.4 MBq/mouse; 20.76 ± 2.21 MBq for nontransgenic mice and 21.24 ± 1.24 MBq for transgenic mice). Images were reconstructed by either a maximum-a-posteriori method to generate single-frame average images for nonquantitative displays or filtered backprojection using a 0.5-mm Hanning filter to generate dynamic images for quantitative assays. All transgenic and 4 nontransgenic mice were scanned by T2-weighted MRI within 1 wk of the PET scans. Volumes of interest (VOIs) were placed on multiple anatomic structures, including the neocortex (coronal sections from 1.3 mm anterior to 2.7 mm posterior to the bregma), whole hippocampus, and cerebellum (6.4–7.0 mm posterior to the bregma) using PMOD image analysis software (PMOD Technologies LLC) with reference to individual T2-weighted MR images for all transgenic and 4 nontransgenic mice or a brain template generated as described previously (7) for the other 3 nontransgenic mice (Fig. 1A and Supplemental Fig. 2). The PET VOIs did not contain areas near the olfactory bulbs to circumvent radioactivity spillover from the Harderian glands. The neocortex more than 2.7 mm posterior to the bregma was excluded from the PET VOIs, since profound atrophy of this region in an aged rTg4510 mouse caused a partial-volume effect on PET data, impeding an accurate quantification of the radioligand retention. Tracer uptake in each VOI was estimated as percentage injected dose per tissue volume, which was uncorrected for body weights of animals, since changes in body weights during aging may not necessarily be proportional to changes in brain weights. Nondisplaceable binding potential for 11C-PBB3 binding in these VOIs, which is not influenced by either body or brain weights, was quantified with PMOD based on a simplified reference tissue model using the cerebellum as a reference region lacking tau deposits.

Correlations of PBB3-positive tau deposits with neocortical/hippocampal atrophy in rTg4510 mouse brain. (A) Representative T2-weighted MR, PET, and PET/MR images of coronal brain sections of 9-mo-old nontransgenic and rTg4510 mice showing neocortical, hippocampal, and cerebellar VOIs (black, yellow, and red outlines, respectively). PET images were generated from averaged dynamic data at 30–60 min after injection of 11C-PBB3. (B) Time–activity curves (percentage injected dose [%ID]) for 11C-PBB3 in different brain regions of rTg4510 and age-matched nontransgenic mice at 7–10 mo old. (C) 11C-PBB3 binding potential in each VOI calculated by simplified reference tissue model with cerebellum as reference tissue using data from B, and brain volume measured using structural MRI data, including calculation of nondisplaceable binding potential for neocortex and hippocampus (CTX/HIP). **Significant increase in 11C-PBB3 nondisplaceable binding potential, and significant decrease in neocortical and hippocampal volume, for rTg4510 mice compared with nontransgenic mice (P < 0.01). (D) Correlation between 11C-PBB3 nondisplaceable binding potential and volume of neocortex and hippocampus in transgenic (▪) and nontransgenic (○) mice. *P < 0.05, **P < 0.01, and ***P < 0.001 for correlations in transgenic plus nontransgenic group (Tg+nonTg, dotted lines) and transgenic group only (Tg, solid lines).
MRI of Mouse Brains
The rTg4510 mice were anesthetized with 1.5 % (v/v) isoflurane. T2-weighted 2-dimensional multislice spin-echo (rapid acquisition with relaxation enhancement [RARE]) was applied to the mouse heads using the 7.0-T MRI system (AVANCE-III; Bruker BioSpin) with a volume coil for transmission (Bruker BioSpin) and a quadrature surface coil for reception (Rapid Biomedical). The repetition time was 4,200 ms, effective echo time was 36 ms, field of view was 25.6 × 14.5 mm, slice thickness was 0.5 mm, number of slices was 28 (gapless), matrix was 256 × 256, RARE factor was 8, number of acquisitions was 8, and nominal in-plane resolution was 100 × 57 μm. VOIs across all slices of the hippocampus and cortex were manually drawn with reference to the mouse brain atlas of Paxinos and Franklin using PMOD software.
In Vitro 11C-PBB3 Binding Assays
All rTg4510 and nontransgenic mice were killed by cervical dislocation after the scans. Brains were sampled from 3 PS19 and 3 nontransgenic mice at 13 mo old. The mouse brains were removed and divided into right and left hemispheres. The right hemispheres were kept at −80°C for postmortem analyses, and the left hemispheres were fixed with 4% paraformaldehyde in phosphate buffer overnight, followed by 30% sucrose in phosphate buffer. Brain tissue from patients with AD and PSP were homogenized using MicroSmash (MS-100R; TOMY Digital Biology) in 50 mM Tris-HCl buffer (pH 7.4) containing a protease/phosphatase inhibitor cocktail (cOmplete; Sigma-Aldrich). Concentrations of proteins in the brain homogenates were measured by bicinchoninic acid assays (Thermo Fisher Scientific). Aliquots of the homogenates were stored at −80°C until the experiment.
Radioligand binding was measured by incubating 5 nM 11C-PBB3 with brain tissue homogenates from 5 rTg4510 and 7 nontransgenic mice that had undergone a PET scan, as well as from 3 PS19 and 3 nontransgenic mice. Nonspecific binding was determined in the presence of 5×10−7 M unlabeled PBB3.
Homologous and heterologous competitive binding assays were performed: 100-μL aliquots of the tissue homogenates containing 100 μg of AD brain tissue or 300 μg of mouse brain tissue were reacted with 900 μL of 50 mM Tris-HCl buffer (pH 7.4) containing 10% ethanol and 5 nM 11C-PBB3 in the presence of various unlabeled compounds, including PBB3, BTA-1, AV-1451, clorgyline, and selegiline at concentrations ranging from 10−11 to 10−6 M, for 30 min at room temperature in a dimly lit room to avoid photoisomerization of the compound. Nonspecific radioligand binding was determined in the presence of 5 × 10−7 M PBB3. Samples were run in duplicates, and the specific radioligand binding was expressed as pmol/g of tissue.
The inhibitory constant (Ki), which is equivalent to the dissociation constant (KD) in the homologous blocking assay, and the percentage of displacement were determined using nonlinear regression 1-site and 2-site binding models derived from the Cheng–Prusoff equation in Prism, version 5.0 (GraphPad Software), followed by an F test for model selection.
Immunohistochemical and Histochemical Analyses
Ten-micrometer-thick frozen sections from left hemispheres were generated in a cryostat (HM560; Carl Zeiss) and used for immunostaining with an antibody against phosphorylated tau (AT8; 1:250 dilution) by following a standard immunohistochemical procedure described in our previous publications (23). Fluorescence microscopic visualization of tau fibrils with PBB3 and FSB was performed as described previously (7). All stained samples were examined by an all-in-one fluorescence microscope (BZ-9000; Keyence) capable of tiling photomicrographs and merging them into a high-resolution image with a large field of view.
Statistics
Statistical analyses for comparison of mouse genotype groups were performed by unpaired t testing using Prism 5.0. The difference between groups was considered significant at a P value of less than 0.05. All error bars in the figures are expressed as SEM.
RESULTS
Correlation Between Increased In Vivo 11C-PBB3 Binding and Brain Atrophy in rTg4510 Mice
Images of 11C-PBB3 PET and structural MRI demonstrated overtly increased radioactivity retention and decreased volume in either neocortex or hippocampus of rTg4510 mice compared with age-matched nontransgenic mice (Fig. 1A). Time–activity curves also indicated increased 11C-PBB3 retention in the neocortex/hippocampus relative to the cerebellum of rTg451 mice, whereas no overt regional difference in radioligand retention was observed in nontransgenic mice (Fig. 1B). Nondisplaceable binding potential for 11C-PBB3 was significantly increased by 3- to 5-fold in the neocortex and hippocampus of rTg4510 compared with nontransgenic mice (Fig. 1C). This change was concurrent with marked reductions of the neocortical (approximately 33%) and hippocampal (approximately 38%) volumes in rTg4510 mice relative to nontransgenic mice (Fig. 1C). Indeed, intimate correlations between 11C-PBB3 nondisplaceable binding potential and the volume of either neocortex or hippocampus were found in an analysis of combined transgenic and nontransgenic mice. This approach was made by conceiving nontransgenic mice to be animals at stage 0 of the tau pathology. In an assessment of the transgenics only, nondisplaceable binding potential values were still significantly correlated with local volumes in the hippocampus and showed a tendency to correlate with each other in the neocortex, despite a small sample size (Fig. 1D).
Postmortem Assays of Tau Pathologies in rTg4510 and PS19 Mice
Abundant neuronal inclusions fluorescently stained with PBB3 and immunolabeled with AT8 were observed in the neocortex and hippocampus but not in the cerebellum (data not shown) of 7- to 10-mo-old rTg4510 mice. Similarly, accumulation of PBB3- and AT8-positive tau deposits was detected in the brain stem of 13-mo-old PS19 mice. There was a lack of intense fluorescence labeling with PBB3 in the hippocampus of these PS19 mice despite abundant immunostaining with AT8 (Fig. 2A). In vitro 11C-PBB3 binding was also increased in the neocortex/hippocampus of 7- to 10-mo-old rTg4510 mice and mixed brain stem/spinal cord tissues of 13-mo-old PS19 mice compared with corresponding nontransgenic mice, whereas 11C-PBB3 binding in the mixed neocortex/hippocampus tissues of these PS19 mice was unchanged relative to age-matched nontransgenic mice (Fig. 2B). In vitro 11C-PBB3 binding showed a good correlation with both in vivo 11C-PBB3 binding and volume reduction in the neocortex/hippocampus of rTg4510 and nontransgenic mice (Figs. 2C and 2D).
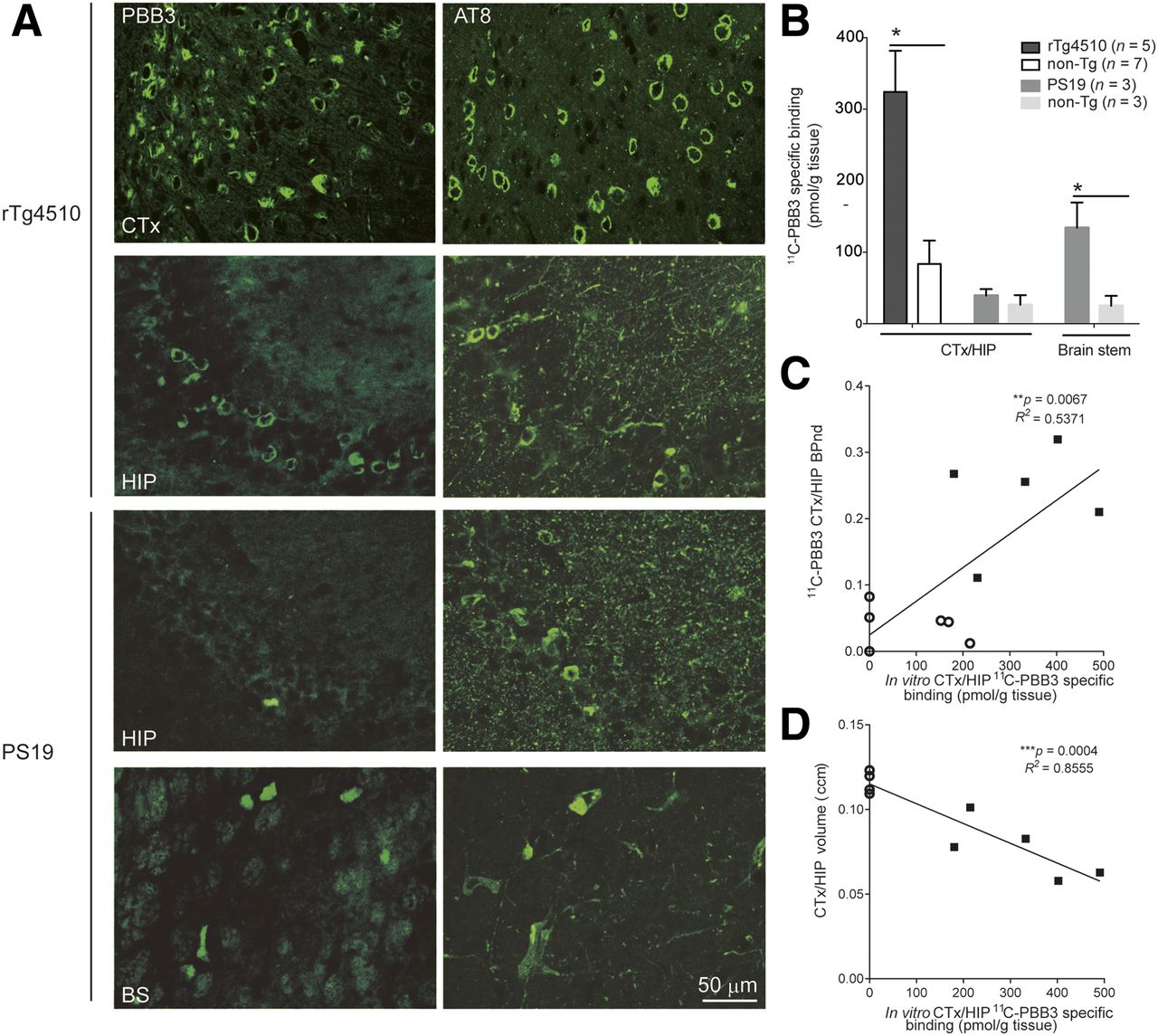
Comparisons of in vitro 11C-PBB3 binding and histochemical and immunohistochemical findings in brains of rTg4510 and PS19 mice. (A) Representative images of PBB3-positive tau deposits and phosphorylated tau (AT8) in cortex (CTx), hippocampus (HIP), and brain stem (BS) of 9-mo-old rTg4510 mice and 13-mo-old PS19 mice. (B) Increased specific binding of 11C-PBB3 (5 nM) in CTX/HIP of rTg4510 mice and in brain stem of PS19 mice compared with nontransgenic mice (*P < 0.05). No significant difference in 11C-PBB3 binding in CTX/HIP was observed between PS19 and nontransgenic mice. (C and D) Significant correlation of in vitro 11C-PBB3 binding with in vivo nondisplaceable binding potential (C) and regional volume (D) in CTX/HIP of rTg4510 (▪) and nontransgenic (○) mice.
Characteristics of Tau Fibrils in Mouse Models Versus AD and PSP Based on Binding of PBB3 and AV-1451
We assumed that conformational features of tau fibrils in the brain could be probed by their reactivity with different ligands. Following this rationale, 11C-PBB3 binding in homogenized frontal cortical tissues of an AD patient with abundant amyloid plaques and neurofibrillary tangles in the frontal cortex (Supplemental Fig. 3) was measured under a homologous blockade by nonlabeled PBB3 and a heterologous blockade by AV-1451 or BTA-1. The homologous blocking of 11C-PBB3 binding by nonlabeled PBB3 indicated the presence of high- and low-affinity binding components for this ligand, with Ki (=KD) values of 3.9 and 246.6 nM, respectively (Fig. 3A). The heterologous blockade of 11C-PBB3 binding by BTA-1, which is more selective for Aβ versus tau filaments, was well described by a 1-site model, with a Ki value of 79.0 nM (Fig. 3A). According to this inhibition curve, BTA-1 at a concentration below 50 nM in the AD brain, which could reflect its high-affinity binding to Aβ aggregates, minimally blocked the specific 11C-PBB3 binding. AV-1451 displaced 30%–40% of 11C-PBB3 binding at maximum in the AD tissue (Fig. 3A). This blockade was described by a 1-site model with a Ki value of 89.6 nM, which was approximately 50-fold weaker than the homologous blockade by nonlabeled PBB3. In the PSP basal ganglia tissue, a 1-site model indicated high-affinity binding of 11C-PBB3 to tau deposits, with a Ki (=KD) value of 2 nM, and this was barely blocked by AV-1451 even at a high concentration (Fig. 3B).
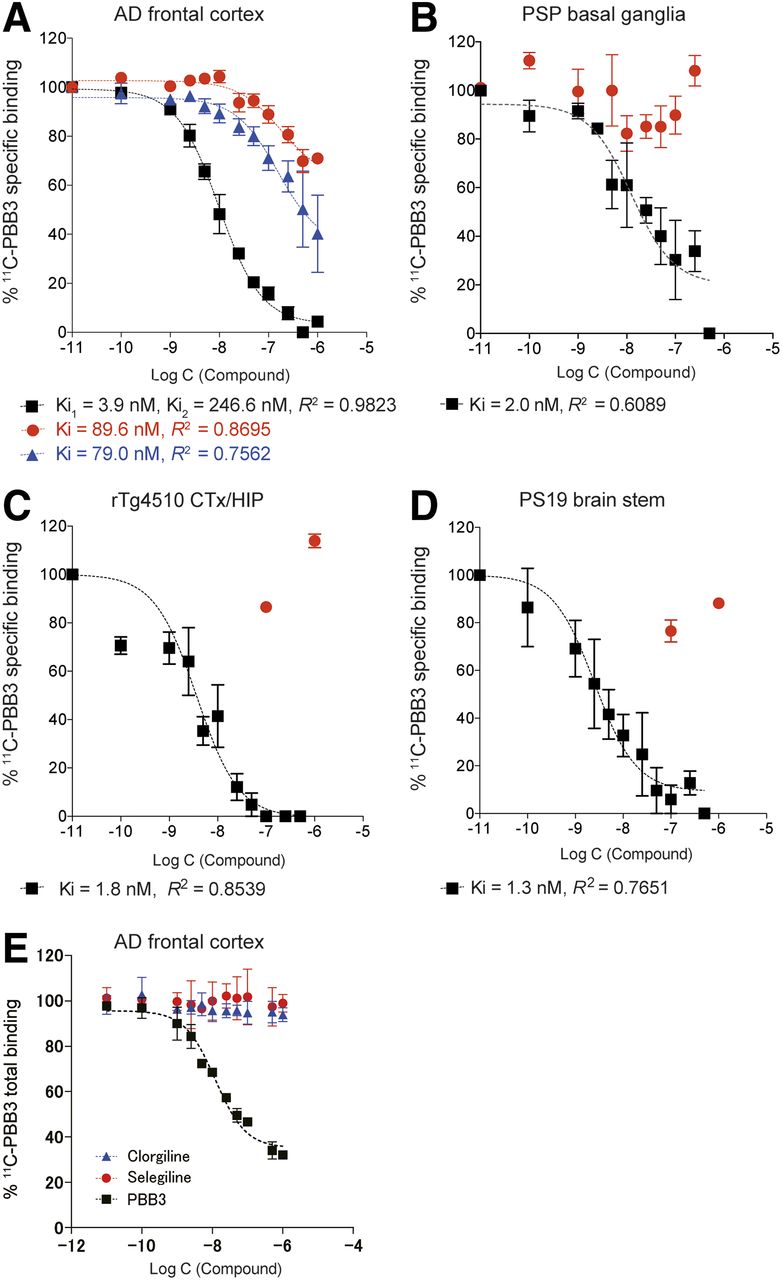
Assays of 11C-PBB3 binding in brain tissues derived from mouse models and AD patient. Graphs show specific binding of 11C-PBB3 (5 nM) in frontal cortex obtained from AD patient (A), basal ganglia from PSP patient (B), neocortex (CTx)/hippocampus (HIP) from rTg4510 mice (C, samples from 5 mice undergoing PET), and brain stem/spinal cord from PS19 mice (D, samples from three 13-mo-old mice). 11C-PBB3 binding was blocked in homologous fashion by nonlabeled PBB3 (black symbols) and in heterologous fashion by nonlabeled AV-1451 (red symbols) and BTA-1 (blue symbols). (E) No notable displacement of 11C-PBB3 binding by clorgyline or selegiline was observed in frontal cortex of AD, in contrast to significant blockade of radioligand binding by unlabeled PBB3. Data were generated from more than 3 independent experiments.
Likewise, 11C-PBB3 exhibited high-affinity binding to tau deposits in the neocortex/hippocampus of rTg4510 mice (Fig. 3C) and the brain stem/spinal cord of PS19 mice (Fig. 3D), with Ki (=KD) values of 1.8 and 1.3 nM, respectively, and this radioligand binding was only minimally inhibited by AV-1451 at high concentrations (Figs. 3C and 3D).
The lack of competition between 11C-PBB3 and AV-1451 binding in PSP and transgenic mouse brain samples supports the view that 11C-PBB3 does not react with probable off-target binding components for AV-1451, including monoamine oxidase A and iron-containing deposits (24). Finally, minimal displacement of 11C-PBB3 binding was observed in the presence of the monoamine oxidase A inhibitor clorgyline or the monoamine oxidase B inhibitor selegiline in homogenates from the AD frontal cortex even at high concentrations (Fig. 3E), suggesting that 11C-PBB3 can barely react with off-target binding sites on monoamine oxidases.
DISCUSSION
The current findings provide in vitro and in vivo evidence for the conformational homology between pathologic tau assemblies in human tauopathies and mouse models as probed by 11C-PBB3. Binding assays of 11C-PBB3 in comparison with AV-1451 have demonstrated that tau fibrils in transgenic mice overexpressing a single 4-repeat tau isoform particularly resemble straight filaments in 4-repeat tau disorders represented by PSP. These indications support the notion that PET imaging of the model animals along with postmortem investigations with 11C-PBB3 offers assay systems seamlessly linking nonclinical and clinical evaluations of candidates for therapeutic and diagnostic agents targeting the tau depositions. To further justify the use of the nonclinical 11C-PBB3 PET system with rTg4510 and PS19 mice, a more extensive in vitro analysis of the relevance between radioligand binding profiles in transgenic mouse and human tissues will be required with a larger sample size and different brain areas. There might be regional and interindividual differences in the conformational properties and neurotoxicity of pathologic tau assemblies within a single disease, although the present data using the AD frontal cortex and PSP basal ganglia (Figs. 3C and 3D) agreed with previous findings in different tangle-rich brain regions of patients with these illnesses (13).
As illustrated in Figure 3, 11C-PBB3 is capable of binding to tau deposits in the PSP and transgenic mouse brain tissues with a high affinity, and this binding was not blocked by AV-1451 even at a high concentration above 100 nM. This observation demonstrates the resemblance of straight filaments composed of 4-repeat taus in PSP and transgenic models, raising the possibility that PET scans of these mice will facilitate the development of a probe for high-contrast imaging of tau lesions in PSP and allied 4-repeat tauopathies. Such new imaging agents would be generated on the basis of the PBB3 backbone structure, in light of the current data.
In the AD brain, Aβ and tau aggregates share a common β-pleated sheet structure that is in principle reactive with diverse β-sheet ligands, but our present and previous results (7,13) suggest that 11C-PBB3 at a concentration below 10 nM, which is comparable to the radioligand concentration in the brain achievable in a clinical PET study, primarily reacts with tau versus Aβ deposits in the brains of AD patients. Indeed, the peak concentration of 11C-PBB3 is conceived to be 10 nM or below in human and mouse PET studies, according to the current and previous (7) data, and is therefore considered to be close to the concentration of 11C-PBB3 (i.e., 5 nM) used for the current homogenate binding assays. Because BTA-1 binds to Aβ aggregates with high affinity, the blockade of specific 11C-PBB3 binding in AD tissues by 1–50 nM BTA-1 should reflect the binding of 11C-PBB3 to Aβ deposits, which was found to be minimal (Fig. 3A). Meanwhile, the heterologous blockade by BTA-1 at high concentrations above 100 nM is likely to stem primarily from the low-affinity binding of BTA-1 to tau fibrils. This finding agrees with previous autoradiographic binding measurements indicating that 11C-PBB3 at a concentration below 10 nM reacts with mostly high-affinity components on tau fibrils, relative to low-affinity elements on Aβ fibrils, in AD brain sections. Moreover, the affinities of 11C-PBB3 for tau in the brains of AD patient and transgenic mouse were close to each other, validating the use of these animals for nonclinical characterization of 11C-PBB3 and in vivo screening of PBB derivatives for imaging AD-type tau pathologies. Notwithstanding this supportive evidence, the properties of 11C-PBB3 and resemblance between AD and transgenic mouse tau fibrils will need to be further examined by increasing the sample size of AD tissues.
It is noteworthy that 11C-PBB3 binding in AD brain homogenates was only partially blocked by AV-1451, with a Ki value of 89.6 nM (Fig. 3A), which is consistent with our previous report (13). Because specific binding of 5 nM 11C-PBB3 was barely inhibited by 1–50 nM AV-1451, PBB3 and AV-1451 may not share high-affinity binding components in AD tau filaments. In addition, the high-affinity binding of 5 nM 11C-PBB3 was only modestly blocked by AV-1451 at high concentrations (Fig. 3A). Hence, 11C-PBB3 and 18F-AV-1451 in living AD patients may detect different tau species that differentially contribute to neurotoxicity as implied by a previous in vitro assessment (13), and this probability will be examined by a head-to-head comparison of these 2 radioligands in the same human subjects.
The present work also indicates an intimate correlation between in vivo and in vitro binding of 11C-PBB3 in the same rTg4510 mice at 7–10 mo old. We defined PET VOIs in a subportion of the neocortex to circumvent technical issues of radioactivity spill-in and partial-volume effects stemming from the small volumes of the mouse brain regions, enabling quantification of the radioligand binding reflecting the number of binding components. Because we observed a marked colocalization of PBB3 fluorescence labeling and AT8 immunostaining in the brain sections derived from these mice, in vivo 11C-PBB3 PET is presumed to be able to robustly quantify insoluble tau aggregates constituted of phosphorylated tau molecules. Meanwhile, there still remains a possibility of inaccurate measurements of the radioligand binding caused by insufficient volumes of target VOIs, particularly in mice with severe pathologies, highlighting the need for further technical considerations in small-animal PET imaging.
According to the correlation between local brain atrophy and in vivo 11C-PBB3 binding in rTg4510 mice, neurons in the brains of these animals may die mainly at a pathologic stage characterized by the accumulation of PBB3-labeled inclusions, and our previous data also demonstrate that neuronal loss is less prominent in the absence of PBB3 positivity notwithstanding the high abundance of tau oligomers (25), suggesting a potential role of PBB3-positive inclusions in the advancement of the neurotoxicity. In the meantime, extracellular tau deposits dubbed ghost tangles were not found in our histochemical and immunohistochemical analyses of the brain sections collected from rTg4510 mice. This implies death of tangle-bearing neurons followed by immediate elimination of tau aggregates or loss of tangle-negative neurons due to neurotoxicity of relatively immature tau assemblies. To examine these possibilities, longitudinal PET and intravital microscopic imaging experiments need to be conducted and are currently under way. In PET scans of patients with AD or mild cognitive impairment (7,26), an increase in 11C-PBB3 binding has been reported to be correlated with either local atrophy or cognitive performance, suggesting that the neuronal loss in these diseases is mediated by molecular mechanisms linked to PBB3-positive tau fibrils similar to those in rTg4510 mice.
In contrast to the pathologies in rTg4510 mice, AT8-immunoreactive tau deposits in the neocortex/hippocampus of PS19 mice barely reacted with PBB3. Meanwhile, a pronounced increase of tau lesions doubly positive for AT8 and PBB3 occurred in the brain stem and spinal cord of the PS19 strain. Because regional atrophy was severe in the neocortex/hippocampus and modest in the brain stem and spinal cord of these animals (7,21), the concurrence of PBB3-negative, AT8-positive inclusions with loss of neurons and absence of ghost tangles in the hippocampus of PS19 mice might indicate the neurotoxicity of PBB3-negative tau assemblies, or immediate elimination of neurons bearing PBB3-positive tau tangles. Furthermore, the PBB3 negativity in the PS19 hippocampus may be attributable to the formation of region-specific conformers and posttranslational modifications of tau molecules, leading to the loss of their reactivity with PBB3. The reactivity of different PET probes with tauopathy mouse models in addition to rTg4510 and PS19 mice is also of significant interest. A previous PET study documented that another tau PET ligand, 18F-THK5117, enabled quantification of tau deposits in 2 different tau transgenic mouse models (27), whereas the relationships between tau PET signals and regional atrophy in these strains remain undetermined. Furthermore, it should be noted that THK5117 and related quinoline derivatives may cross-react with monoamine oxidase B (28), which is abundantly present in activated astrocytes, whereas 18F-AV-1451 may bind to monoamine oxidase A (24). Our in vitro assays indicate that 11C-PBB3 binding in AD brain homogenates does not compete with inhibitors of either monoamine oxidase A or monoamine oxidase B, and therefore 11C-PBB3-PET signals in human and mouse brains are considered to reflect tau lesions, not other neuropathologic events such as astrogliosis enriched with monoamine oxidase B.
CONCLUSION
Our PET and MRI analyses of tau transgenic mice in conjunction with postmortem radiochemical, histochemical, and immunohistochemical assays have proven the applicability of 11C-PBB3 to the research on neurodegenerative tauopathies reciprocally linking nonclinical and clinical evidence. The findings obtained here have also revealed the distinct utility of rTg4510 and PS19 models for assessing putative etiologic pathways triggered by either PBB3-positive or PBB3-negative tau multimers.
DISCLOSURE
This work was supported by grants-in-aid for Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS; 15653129) to Tetsuya Suhara and Makoto Higuchi; Research and Development Grants for Dementia (16768966) to Makoto Higuchi and the COI STREAM award for MRI devices to Ichio Aoki from the Japan Agency for Medical Research and Development; 2 grants-in-aid for Scientific Research to Bin Ji (15K09979) and Makoto Higuchi (16678815) and a postdoctoral Fellowship to Ruiqing Ni (PE15760) from the Japan Society for the Promotion of Science. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Prof. John Q. Trojanowski and Prof. Virginia M.-Y. Lee (Center for Neurodegenerative Disease Research, University of Pennsylvania) for kindly providing human brain tissues and PS19 mice; Prof. Jada Lewis of the University of Florida for supporting analyses of rTg4510 mice; and Takeharu Minamihisamatsu, Shoko Uchida, Sayuri Sasaki, Sayaka Shibata, Nobuhiro Nitta, and Yoshikazu Ozawa for technical assistance.
Footnotes
Published online Feb. 1, 2018.
- © 2018 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication September 5, 2017.
- Accepted for publication January 9, 2018.





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Reduced levels of synaptic vesicle protein 2A in the extracellular vesicles and brain of Alzheimers disease- associations with A{beta}, tau and synaptophysin
- Increased regional P2X7R uptake detected by [18F]GSK1482160 PET in a tauopathy mouse model
- Reduced SV2A and GABAA receptor levels in the brains of type 2 diabetic rats revealed by [18F]SDM-8 and [18F]flumazenil PET
- Relationship between reactive astrocytes, by [18F]SMBT-1 imaging, with amyloid-beta, tau, glucose metabolism, and microgliosis in mouse models of Alzheimers disease
- Imaging {alpha}-synuclein pathologies in animal models and patients with Parkinsons and related diseases
- Efficient characterization of multiple binding sites of small molecule imaging ligands on amyloid-beta, 4-repeat/full-length tau and alpha-synuclein
- Aquaporin 4 is differentially increased and depolarized in association with tau and amyloid-beta
- Rapamycin attenuated zinc-induced tau phosphorylation and oxidative stress in animal model: Involvement of dual mTOR/p70S6K and Nrf2/HO-1 pathways
- High-contrast in-vivo imaging of tau pathologies in Alzheimers and non-Alzheimers disease tauopathies
- Tracking tau fibrillogenesis and consequent primary phagocytosis of neurons mediated by microglia in a living tauopathy model
- DTI reveals whole-brain microstructural changes in the P301L mouse model of tauopathy
- Tau deposition is associated with imaging patterns of tissue calcification in the P301L mouse model of human tauopathy
- Tau imaging detects distinctive distribution of tau pathology in ALS/PDC on the Kii Peninsula