


The network degeneration hypothesis suggests that neurodegenerative disorders are characterized by the spread of molecular neuropathologic features along specific neuronal brain networks, leading to functional impairment of these networks. Interestingly, the symptomatic phenotype of neurodegenerative disorders may be more strongly driven by the type of network affected rather than by the type of underlying neuropathology. This could explain why etiologically different disorders may be clinically confused when affecting similar networks. Plus, it helps to understand the atypical symptomatic variants, for example, of Alzheimer disease (AD), which appear to be characterized by spread of the identical neuropathology into different brain networks, associated with the particular cognitive function impaired in these patients.
Evidence for this hypothesis has been collected from the convergence of two fields whose progress is essentially driven by imaging—research on the organization of the brain into functional networks, and research on the anatomic location and topographic spread of neurodegenerative diseases.
A rising number of neuroimaging studies show that the brain contains consistent functional networks, or modular units, characterized by the synchronicity of their activation and deactivation (1,2). Although the first references to such networks emerged from PET activation studies, knowledge in this field has increased exponentially since the advent of resting-state functional MRI (3), a procedure that measures blood oxygen level–dependent brain activity under resting conditions for several minutes. Postprocessing methods then create time–activity curves for activity in the various regions of the brain, and if any show a synchronous course of activity over time, a functional interaction between those regions can be assumed.
Using this approach, several functionally connected networks of the brain have been reliably and reproducibly identified (4). Interestingly, the identified networks are between regions previously known to interact (e.g., from activation studies) in defined cognitive processes such as sensory and motor integration, visual and auditory learning, salience processing, and executive functioning and have therefore been given names reflecting these processes (3,5). Functional MRI has revealed that communication between such brain regions can be detected even when the subject is in the resting state—that is, while not performing a specific cognitive task—thus strongly promoting the view that cerebral organization is based on division into modular functional units. Among the detected functional networks, one of particular interest in neuropathologic development, known as the default-mode network, has been found to be associated with introspective cognitive functions such as the so-called internal mentation (7). This network can be robustly identified under resting conditions, even across species boundaries (e.g., monkeys and rats) (6). It contains anatomic regions known to be strongly activated when a subject is not interacting with the environment but, often, deactivated when a subject is performing externally oriented cognitive tasks. These regions include the right and left precunei/cingulate gyri, mesial prefrontal cortices, superior parietal lobuli, and hippocampi (8).
A striking overlap has become increasingly apparent between the architecture of these functional networks and the anatomic distribution of some neurodegenerative brain diseases (9). This insight forms the basis for the network degeneration hypothesis (10,11). The overlap is particularly evident between the architecture of the default-mode network and the anatomic distribution of atrophy and hypometabolism in AD. The typical distribution patterns of hypometabolism in both AD and mild cognitive impairment have long been known from 18F-FDG PET imaging. The affected brain regions include the precuneus/posterior cingulate cortex, the temporoparietal cortex, and often, in later stages, the frontal cortex (12–14). Consistently, atrophy of cortical density accompanied by pronounced hippocampal atrophy has been found in similar cortical regions among many cases of AD (15,16), and these affected regions correspond strongly to those constituting the default-mode network in healthy subjects (Fig. 1).
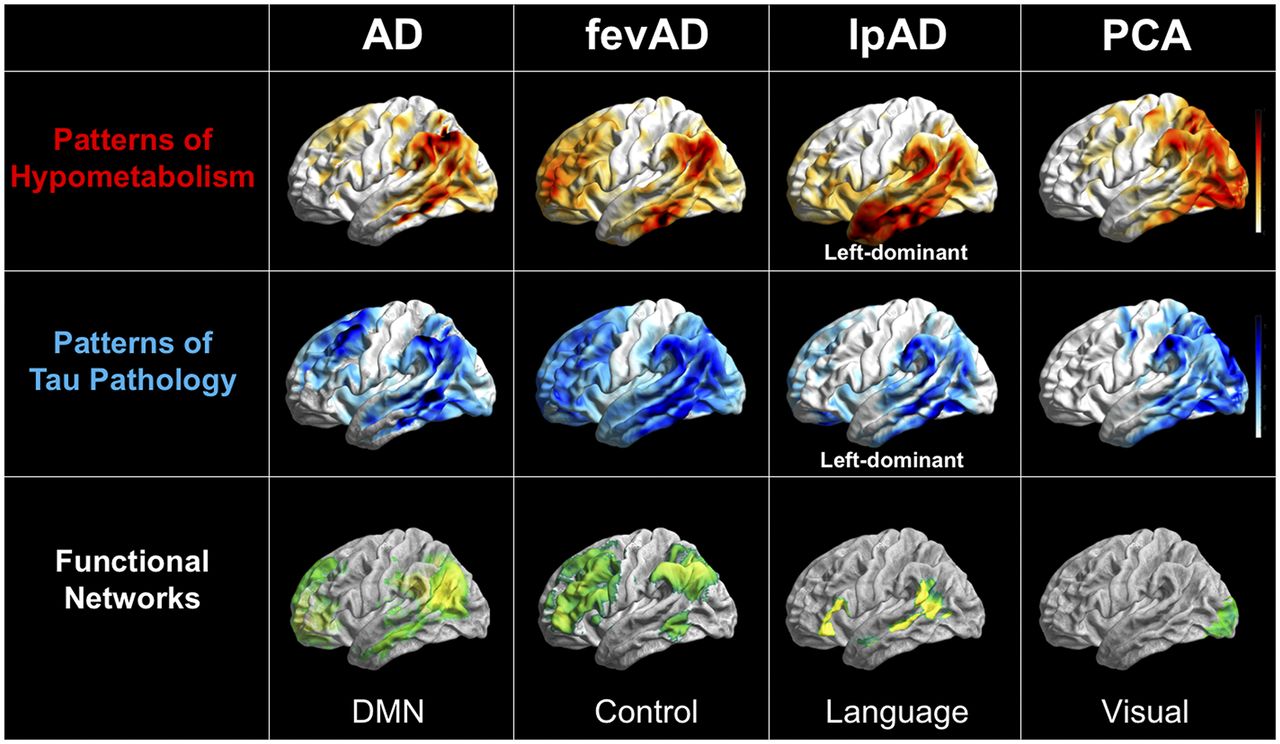
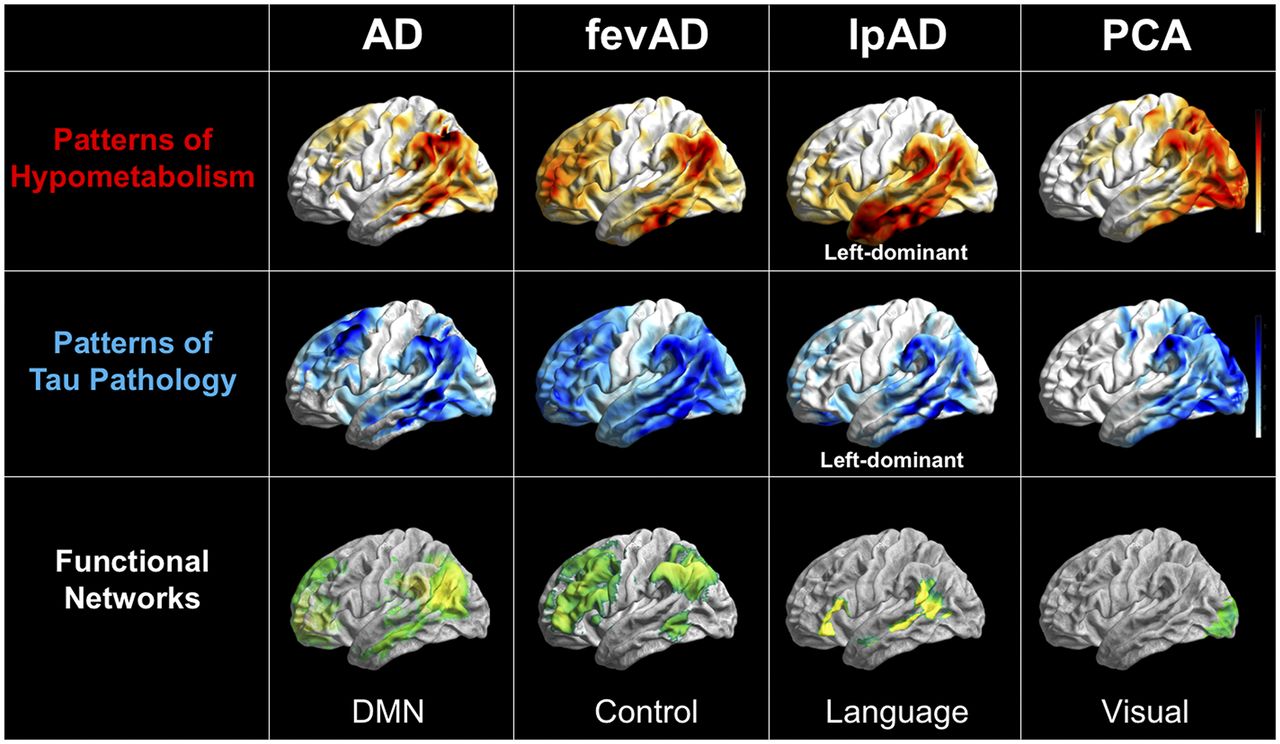
Overlap between patterns of hypometabolism on 18F-FDG PET, patterns of tau aggregation on 18F-T807 PET, and neuronal networks on resting-state functional MRI in different forms of AD. All images are in left lateral surface projection. AD = typical AD; fevAD = executive variant; lpAD = logopenic variant; PCA = posterior-cortical-atrophy variant. (Courtesy of Merle Hönig, University of Cologne.)
With the arrival of amyloid imaging, regional associations between the distribution of this molecular neuropathologic feature (amyloid plaque load) and the network architecture of the brain have also been documented. Buckner et al. were able to show that the maxima of amyloid plaque deposition in AD patients are in the hub regions (regions functionally connected to many other regions) of the default-mode network and that the intensity of the deposition in patients correlates with the intensity of the functional network connectivity in healthy subjects (17).
In addition to the mere anatomic overlap between brain networks and neuropathologic distributions, a significant and progressive loss of functional connectivity within these very networks has also been observed in patients affected by neurodegeneration. This observation serves as a strong argument for the network degeneration hypothesis. In early and even prodromal disease stages—patients with mild cognitive impairment and even asymptomatic amyloid-positive controls—a loss of functional connectivity within the default-mode network could be documented (18,19).
In general, however, amyloid plaques are deposited in a more ubiquitous and less regional pattern, especially in later stages of the disease. Thus, topographic overlap with neuronal networks and with patterns of neuronal dysfunction are less apparent (20). In several studies, a less direct relationship has been proposed—a relationship between global, rather than local, levels of amyloid plaque deposition and neurodegeneration (19,21,22). The less immediate link between amyloid plaque deposition and neuronal dysfunction/network architecture corresponds well to the fact that amyloid plaque deposition spreads extraneuronally (i.e., occurs in the interstitial space rather than within neurons or along networks), is not closely related to disease severity (i.e., appears decades before disease is apparent), and is not considered a sufficient biomarker of ongoing neuronal injury (23–26).
The situation is somewhat different for the newer PET tracers, which depict deposition of tau protein in the brain. Tau aggregation in the form of neurofibrillary bundles occurs intraneuronally. Accordingly, the first studies on these newer tracers found that tau protein is deposited in an anatomically circumscribed pattern that is similar to the AD pattern of hypometabolism and, consequently, to the default-mode network (Fig. 1). Several studies have found overlap between the distribution of tau tracers in the diseased brain and the architecture of neural networks. For example, a recent study found that the brains of AD patients showed several independent components of tau tracer accumulation that overlapped with known functional networks of the brain, especially the default-mode network (27). Another study found that AD patients had a higher accumulation of tau tracer in brain network nodes that are typically more strongly connected (28). Interestingly, tau deposits have also been found to correlate locally with reduced metabolism (20) and network function in AD (28). These findings support the pathophysiologic concept that the intraneuronal deposition of tau protein may have a more immediate impact on neuronal function and, thus, on network damage (20). Plus, these findings agree with previous data implicating a stronger link between tau deposition and cognitive decline (29).
Of the known functional networks of the brain, it is obvious that the default-mode network is particularly affected in AD. Clear overlap between the patterns of dysfunction and the default-mode network is recognizable, and malfunction of the network can be detected early and consistently. However, not all patients are affected equally or in the same chronologic order, and the disease can also involve other networks. Of particular interest are the atypical symptomatic manifestations of AD, as supported by recent data from multimodal neuroimaging studies. Such manifestations include the executive variant, which is characterized by deficits in executive function and behavior; the logopenic variant, which is characterized by progressive aphasia and the posterior-cortical-atrophy variant, which is characterized primarily by visual symptoms (30–32). These manifestations show a relatively ubiquitous increase in amyloid plaque deposits, comparable to typical AD, without any regional reference to cognitive deficits. However, characteristic reductions in metabolism and synchronous deposition of tau protein within the networks responsible for these cognitive functions can differentiate atypical from typical AD (30,31)—that is, within the frontal network for the executive variant; unilaterally and asymmetrically within the temporoparietal language-associated network for the logopenic variant; and occipitally, within the visual network, for the posterior-cortical-atrophy variant (Fig. 1) (33). These findings indicate that the clinical appearance of AD is driven by the affected functional network. Even if the default-mode network is that most prominently affected in typical AD, the atypical manifestations seem to be caused by spread of disease into other networks or by a chronologically atypical propagation of disease within the default-mode network.
In general, however, the phenomenon of network degeneration is not limited to AD but can also be observed in specific networks for other neurodegenerative diseases (Fig. 2). In a groundbreaking study, Seeley et al. identified the typical regions of peak cortical atrophy for several different neurodegenerative diseases, such as semantic dementia, progressive nonfluent aphasia, and the behavioral variant of frontotemporal dementia (10). The investigators assumed that the reason these regions are severely affected by atrophy is because they represent crystallization points where neurodegeneration begins before later spreading successively to other parts of the brain. Supporting this hypothesis, resting-state functional MRI data from healthy subjects showed that specific networks could be induced from these regions of peak atrophy and that these networks in turn overlapped strongly with the typical pattern of atrophy for each type of disease. This study revealed that the anatomic distribution of the various diseases coincides with specific networks at whose center is the point of maximum neurodegeneration. A subsequent study found the potential for differential diagnosis between neurodegenerative diseases through combined examination of atrophy and neuronal dysfunction in the respective disease-typical network cores (34). Together, these results support the notion that degenerative spread along functional networks from a central starting point is a universal phenomenon in neurodegenerative disorders. Consequently, such disorders can be considered to represent nexopathies, or network disorders, of different origins, clinically characterized by the network affected (35).
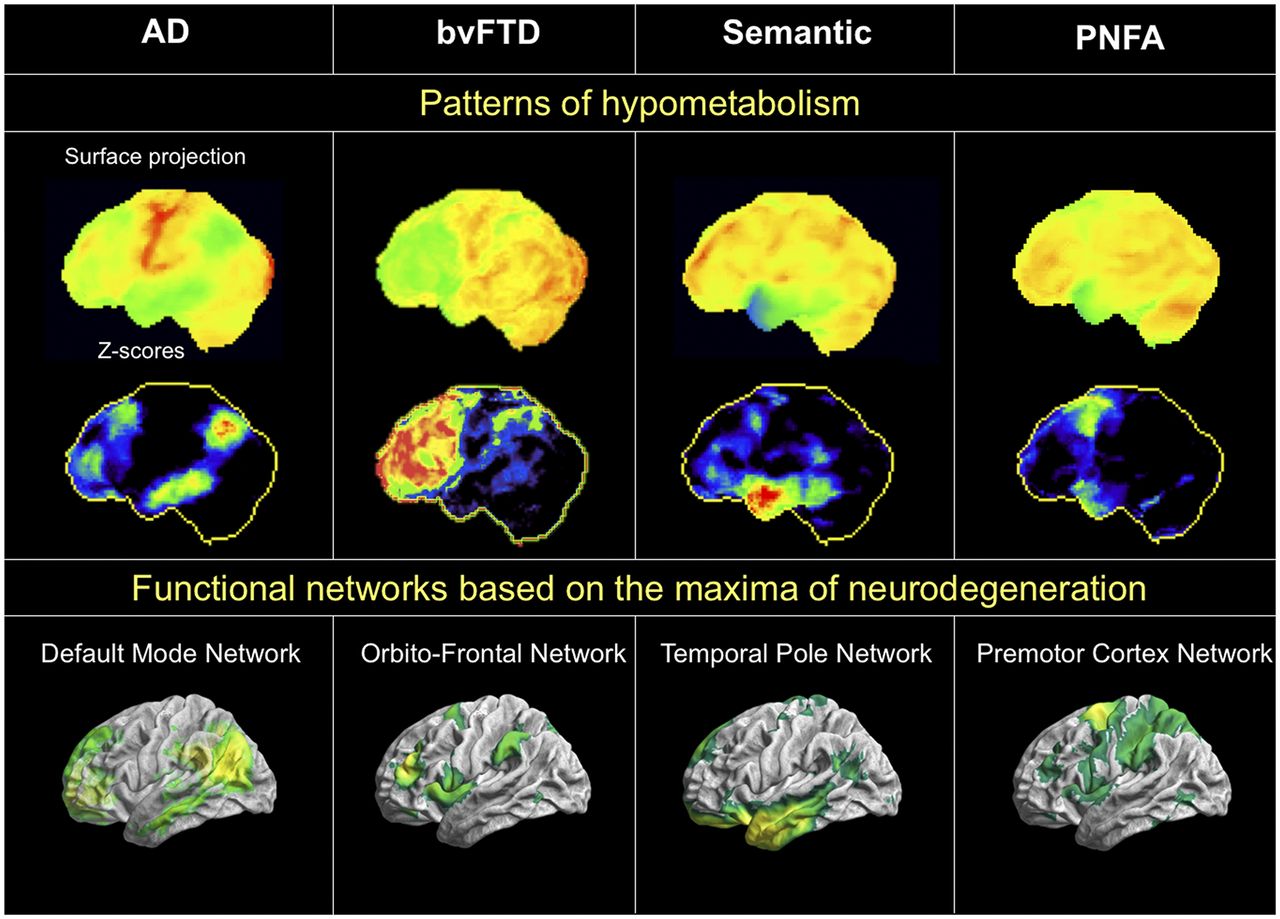
Overlap between individual patterns of hypometabolism on 18F-FDG PET (top row), corresponding z score images (middle row), and neuronal networks on resting-state functional MRI (bottom row) in different forms of neurodegeneration. All images are in left lateral surface projection. AD = typical AD; bvFTD = behavioral variant of frontotemporal dementia; svPPA = semantic variant of primary progressive aphasia; nfvPPA = nonfluent variant of primary progressive aphasia. (Courtesy of Merle Hönig, University of Cologne.)
Although the overlap between neurodegenerative patterns and functional networks suggests a causal relationship, little is known about the underlying pathogenic mechanisms. One hypothetical mechanism is a greater a priori susceptibility (neuroanatomically, genetically, or developmentally) of particular networks to neurodegenerative processes (35,36). Another possibility is the long-established theory of wear and tear, by which exhaustion from strain (e.g., from oxidative stress) on certain heavy-duty networks over an entire lifetime may predispose them to later neurodegeneration (37). In fact, the AD-related default-mode network is characterized by continuously high baseline activity (17). Also supporting this hypothesis are studies showing that synaptically active regions produce increased amounts of β-amyloid peptides (38), which simultaneously exert a synaptotoxic effect (39). Other theories focus on the potentially noxious metabolic phenomena that are seen, particularly, in highly active brain regions (e.g., aerobic glycolysis, or excessive glycolytic metabolism despite an existing oxygen supply) (40,41). However, a recent study by Cope et al. instead speaks against primarily metabolic causes, having found increased tau deposits in highly cross-linked brain regions without regard to metabolic demand (28).
Perhaps the most intriguing theory on the cause of network degeneration is that tau protein aggregation directly disrupts the function of the network (resulting in molecular nexopathies) (35). This hypothesis is strongly fueled by recent evidence of a prion-like spread of tau aggregation across the brain. Studies have shown that, for tau aggregates, a neuron-to-neuron or even transsnyaptic spread seems possible (42–44). If so, an affected neuron could pass noxious tau aggregates across a synapse to the next neuron and thus contribute to migration of the degeneration along the network. This theory could well explain the network-dependent spread of neurodegeneration and is therefore the subject of active research, being relevant to staging, prognosis, therapy, and follow-up with imaging biomarkers. Such pathologic mechanisms may hold true not only for the tau-based neurofibrillary tangles in AD but also for other types of protein aggregation, particularly those that develop intraneuronally.
Together, these theories suggest that neurodegeneration occurs in a specific sequence. First, protein aggregation propagates from central points of crystallization to the rest of a brain network. Function is then lost in consecutive regions of that network as the aggregation reduces synaptic function and connectivity. A reduction in metabolism follows, and neuronal loss finally results in network atrophy. Such a concept might explain why different diseases may manifest with similar symptoms (the diseases spread along the same network) or why the same disease may manifest differently between patients (the disease spreads along different networks). Apparently, the symptoms of a disease allow conclusions to be drawn not about the underlying neuropathology but about the affected network. Knowledge of these mechanisms is of high clinical relevance. For example, in a large study on AD treatment, many patients diagnosed with typical AD by expert clinical raters had no AD-typical neuropathologic findings on imaging (45). The spread of non-AD diseases along typical AD networks might have led to this misdiagnosis. Conversely, clinically atypical forms of AD are to be expected when the disease spreads along networks other than the usual one. Therefore, it is only the combination of suitable neurodegeneration biomarkers (for information on functionally affected networks, e.g., through 18F-FDG PET) along with molecular disease biomarkers (for information on amyloid deposition, e.g., in amyloid imaging) that can be conclusive in the diagnosis of neurodegenerative disease, and what is most important is that nuclear medicine offers methods covering both aspects (24–26).
Several questions remain about the network degeneration hypothesis. First, because distinct functional connectivity networks can be generated starting from almost every region in the brain, the overlap between a given disease and a network seeded from the peak region of that disease may be coincidental. Also, it is yet unclear why the same molecular disease may propagate along different network pathways. Furthermore, the concept of neuron-to-neuron propagation is not consistent with the knowledge that some neurodegenerative disorders have glial rather than neuronal protein aggregation. Finally, a recent longitudinal study reported a rather uniform accumulation of tau protein across brain regions, a finding that is not consistent with spread only from one region to another. The investigators mentioned, however, that their findings are still consistent with the hypothesis of a prion-like propagation of tau aggregation (46).
Altogether, cumulating evidence supports the hypothesis that neurodegenerative disorders are characterized by the spread of specific molecular neuropathologic features along specific neuronal brain networks. Molecular and functional imaging with nuclear medicine modalities may allow reliable classification of such disorders by providing information both on the type of neurodegeneration and on the type of network affected.
DISCLOSURE
No potential conflict of interest relevant to this article was reported.
- © 2018 by the Society of Nuclear Medicine and Molecular Imaging.





{kind=link}
{kind=link}