


Abstract
Cell death is an important target for imaging the early response of tumors to treatment. We describe here the validation of a phosphatidylserine-binding agent for detecting tumor cell death in vivo based on the C2A domain of synaptotagmin-I. Methods: The capability of near-infrared fluorophore-labeled and 99mTc- and 111In-labeled derivatives of C2Am for imaging tumor cell death, using planar near-infrared fluorescence imaging and SPECT, respectively, was evaluated in implanted and genetically engineered mouse models of lymphoma and in a human colorectal xenograft. Results: The fluorophore-labeled C2Am derivative showed predominantly renal clearance and high specificity and sensitivity for detecting low levels of tumor cell death (2%–5%). There was a significant correlation (R > 0.9, P < 0.05) between fluorescently labeled C2Am binding and histologic markers of cell death, including cleaved caspase-3, whereas there was no such correlation with a site-directed mutant of C2Am (iC2Am) that does not bind phosphatidylserine. 99mTc-C2Am and 111In-C2Am also showed favorable biodistribution profiles, with predominantly renal clearance and low nonspecific retention in the liver and spleen at 24 h after probe administration. 99mTc-C2Am and 111In-C2Am generated tumor-to-muscle ratios in drug-treated tumors of 4.3× and 2.2×, respectively, at 2 h and 7.3× and 4.1×, respectively, at 24 h after administration. Conclusion: Given the favorable biodistribution profile of 99mTc- and 111In-labeled C2Am, and their ability to produce rapid and cell death–specific image contrast, these agents have potential for clinical translation.
Detection of the early responses of tumors to therapy would allow rapid selection of the most effective treatment. Currently, clinical assessment of treatment response is based on RECIST, which define partial response as a reduction of at least 30% in the sum of the diameters of the target lesion (1). The problem for early detection of response is that these morphologic changes may occur only weeks after the start of treatment and fail to detect response to cytostatic therapies (2).
Cell death is an important target for imaging early treatment response (3), because most treatments induce tumor apoptosis or necrosis. However, there is as yet no reliable technique for routine imaging of cell death in the clinic (4). Phosphatidylserine, which is externalized on the outer leaflet of the plasma membrane bilayer in apoptosis and is exposed through permeabilization of the plasma membrane in necrotic cells, can be detected using the 36-kDa phosphatidylserine-binding protein annexin-V (5). 99mTc-labeled annexin-V has been used to image drug-induced cell death in human tumors, including breast, lymphoma, lung (6), and head and neck squamous cell carcinoma (7). However, despite early promise, and the development of novel site-directed mutants of annexin-V with improved biodistribution (8), problems with this agent persisted (9), including suboptimal pharmacokinetics and nonspecific binding (10).
We have developed a phosphatidylserine-targeted agent based on the C2A domain of synaptotagmin-I. This was first used, in animal models, in the form of a dimeric glutathione S-transferase–tagged construct (84 kDa) for imaging tumor cell death using MRI (11,12) as well as in 99mTc-labeled form, for imaging tumor cell death (13) and cardiac ischemia using SPECT (14), and in 18F-labeled form, for imaging cell death in a rabbit lung cancer model using PET (15).
More recently, we have used the isolated C2A domain (16), which is much smaller (16 kDa), giving better tissue access and clearance, in which we have introduced a site-directed mutation (S78C; C2Am) that allows site-specific modification with an imaging label. Studies in vitro demonstrated that this probe showed a 4-fold-lower binding to viable cells and consequently improved specificity for detecting apoptosis and necrosis, when compared with annexin-V (16). Moreover, removal of the glutathione S-transferase tag, which is likely to be immunogenic, should facilitate translation of this agent to the clinic.
We have evaluated here the speed of detection, specificity, sensitivity, and biodistribution profiles of AlexaFluor-750– and 99mTc- and 111In-labeled C2Am, for near-infrared fluorescence and SPECT imaging, respectively, of cell death in implanted models of lymphoma (EL4) (11) and colorectal cancer (Colo-205) (17), and in a spontaneous Myc-driven model of Burkitt's lymphoma (Eμ-myc) (18). We also describe a site-directed mutant of C2Am (iC2Am, D108N) that is inactive in phosphatidylserine binding, which we used to demonstrate that there were only low levels of nonspecific probe retention.
MATERIALS AND METHODS
Probe Expression and Chemical Labeling
C2Am and iC2Am were expressed, purified, and labeled using AlexaFluor-750 C5-maleimide, maleimide-hydrazinonicotinamide (HYNIC) (for 99mTc), and maleimide-DOTA (for 111In) as described previously (16). Successful conjugation was confirmed using electrospray ionization mass spectrometry and radio–high-performance liquid chromatography (Supplemental Figs. 1 and 2; supplemental materials are available at http://jnm.snmjournals.org). Phosphatidylserine-binding activity of the conjugates was confirmed by surface plasmon resonance measurements (Supplemental Table 1).
Cell Culture
EL4, murine lymphoma, and Colo-205, human colorectal cells (American Type Culture Collection) were cultured in RPMI medium (Life Technologies) containing 300 mg/L L-glutamine and 10% (v/v) fetal bovine serum (PAA Laboratories). Cell death was induced in EL4 cells by treatment with 15 μM etoposide (Pharmachemie BV) for 24 h at 37°C. An automated analyzer (Vi-Cell; Beckman Coulter) was used to monitor cell number and viability.
Animals and Tumor Models
EL4 or Colo-205 cells (>95% viability) were washed, resuspended in chilled phosphate-buffered saline, and implanted (5 × 106 cells) subcutaneously under isoflurane anesthesia, in the flank of C57BL/6 (BL6) or between the shoulder blades of BALB/c nude mice (8- to 12-wk-old females, from Charles Rivers Laboratories), respectively. Tumors were allowed to develop for between 8 and 13 d. Animals bearing EL4 tumors were treated with etoposide (67 mg/kg of body weight; intraperitoneally) or solvent vehicle, at 24 h before imaging. BALB/c nude mice bearing Colo-205 tumors were treated with 5-fluorouracil (5-FU; Sigma-Aldrich) (250 mg/kg of body weight; intraperitoneally) or solvent vehicle, at 24 h before imaging. Eμ-myc animals (Jackson Laboratories) were monitored daily and enrolled into the study when palpable lymph node masses were detectable (18). The mice were treated with cyclophosphamide (Sigma-Aldrich) (200 mg/kg of body weight; intraperitoneally), at 24−48 h before imaging.
Biodistribution and Near-Infrared Fluorescence Imaging Studies
C2Am-AF750, iC2Am-AF750, or annexin-V-750 (Annexin-Vivo; Perkin Elmer) was administered at 0.10 μmol/kg (10 mL/kg; intravenously) to tumor-bearing BALB/c nude mice. Imaging in vivo or ex vivo was performed using Li-Cor Pearl-Impulse or Xenogen IVIS200 (Perkin Elmer) small-animal imaging systems. Li-Cor Imaging Studio software (version 3.1.4) or Perkin Elmer Living Image (version 3.2) was used for image analysis.
Biodistribution and SPECT Studies
Twenty-four hours after chemotherapy treatment, tumor-bearing mice received an injection (10 mL/kg; intravenously) of either 99mTc-C2Am (EL4 and Eμ-myc models) or 111In-C2Am (Colo-205 model) at 7.5 nmol/kg (0.12 mg/kg, or ∼1.5–2 MBq per mouse). SPECT studies were conducted in separate cohorts of mice 2 h after administration of radiolabeled C2Am at 75 nmol/kg (1.2 mg/kg or ∼15–20 MBq per mouse; intravenously). Specific activities were 10–11.5 MBq/nmol for both 99mTc- and 111In-labeled C2Am. Injected doses were determined using a dose calibrator (ISO-MED 2000; MED Nuklear-Medizintechnik GmbH) and corrected for decay.
Study Approval
All animal experiments were performed in compliance with a project license issued under the Animals (Scientific Procedures) Act of 1986 and were designed with reference to guidelines of the U.K. Coordinating Committee on Cancer Research for the welfare of animals in experimental neoplasia (19). Protocols were approved by the Cancer Research U.K., Cambridge Institute Animal Welfare and Ethical Review Body.
Additional information is available in the supplemental materials.
RESULTS
Preparation and Characterization of Imaging Probes
iC2Am-AF750 was produced by site-directed mutagenesis (D108N) of C2Am. This mutation abolishes binding to phosphatidylserine (20). C2Am and iC2Am were prepared and labeled with AlexaFluor-750 (C2Am-AF750, iC2Am-AF750), as described previously (16). The proteins were fully modified, yielding a single molecular species on electrospray ionization mass spectrometry (Supplemental Figs. 1A and 1B). C2Am-AF750, C2Am-HYNIC, and C2Am-DOTA bound phosphatidylserine with similar affinities (Kd, ∼60–90 nM; Supplemental Table 1), as determined using surface plasmon resonance measurements (16). As expected, iC2Am-AF750 showed no binding. C2Am was also modified stoichiometrically with maleimide-HYNIC or maleimide-DOTA, generating a single species of molecular weights 16,426.5 and 16,749.8 Da, respectively, as determined by electrospray ionization mass spectrometry (Supplemental Figs. 1C and 1D). C2Am-HYNIC was loaded with 99mTc using methods described previously (21). C2Am-DOTA was loaded with 111InCl3 as described in the supplemental materials. The conjugates were analyzed by HPLC (Supplemental Fig. 2). Radiolabeling efficiency was greater than 94%.
Fluorescence Imaging of Tumor Cell Death In Vivo
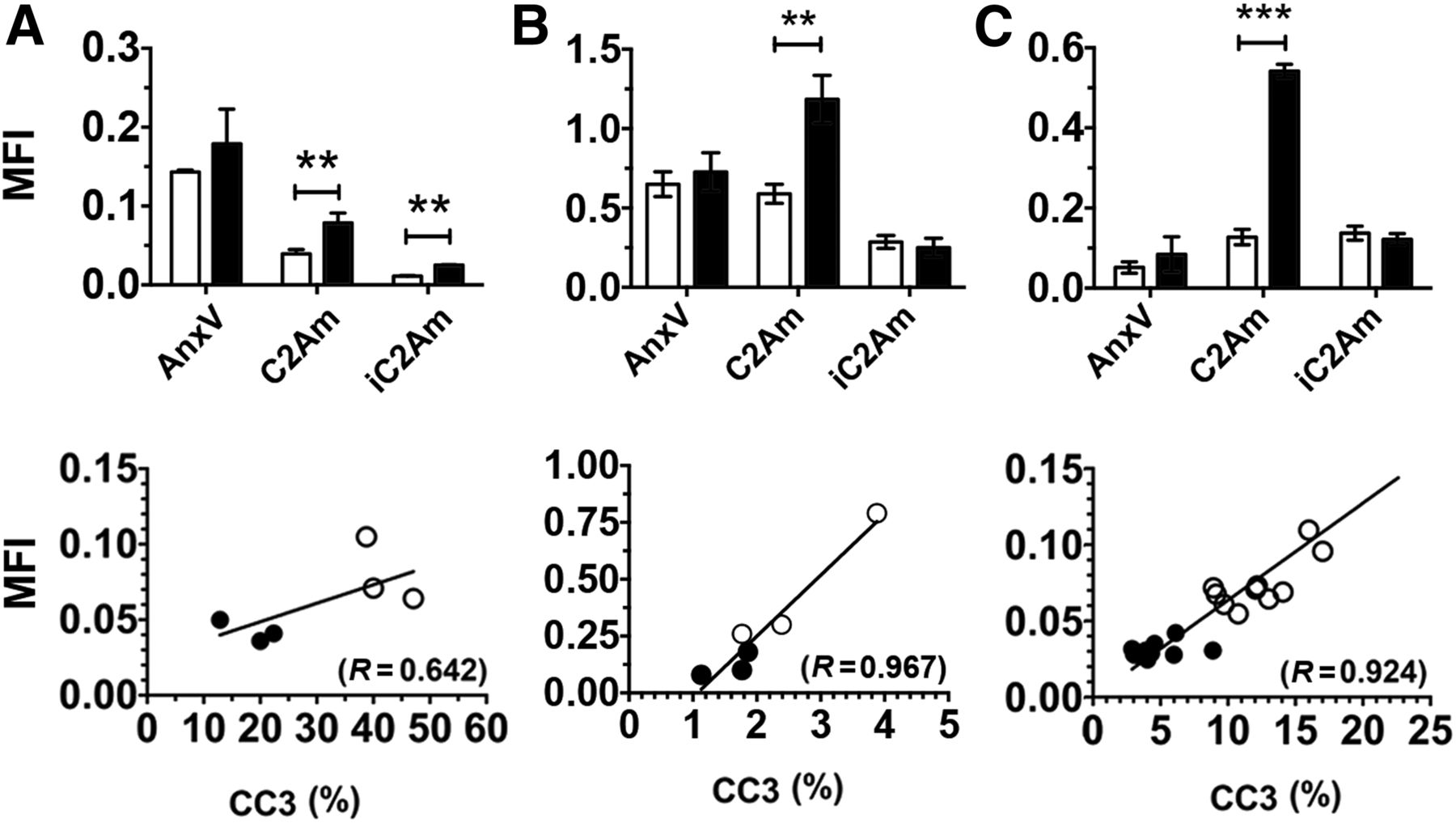
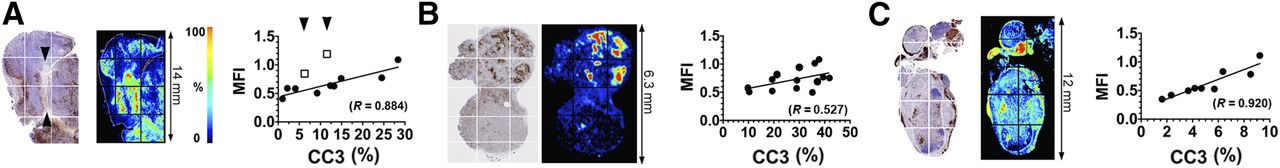
There was increased retention of C2Am-AF750 in drug-treated as compared with untreated EL4 (Figs. 1A and 2A), Colo-205 (Figs. 1B and 2B), and Eμ-myc tumors (Figs. 1C and 2C) at 24 h after probe injection. Anx-AF750 showed no increase in retention after drug treatment in any of the tumor models. Although Anx-AF750 fluorescence was greater than that of C2Am-AF750 in the EL4 tumor model, this was not increased by drug treatment. The retention of iC2Am-AF750 in drug-treated tumors was less than that of C2Am-AF750 in all of the tumor models, and there was no significant difference in the retention of iC2Am-AF750 in untreated or treated Colo-205 (Fig. 2B) and Eμ-myc tumors (Fig. 2C). In drug-treated EL4 tumors, there was an increase in tumor fluorescence in animals injected with iC2Am-AF750 (Fig. 2A); however, the levels were 3 times lower than with C2Am-AF750, possibly due to a treatment-induced increase in the enhanced permeability and retention effect in this tumor model (22). The heterogeneous skin pigmentation of Eμ-myc mice prevented accurate optical imaging in vivo, and therefore for this tumor model fluorescence was measured ex vivo (Fig. 2C), which may explain the better contrast observed. All 3 tumor models showed a correlation between whole-tumor mean C2Am-AF750 fluorescence intensities and cleaved and activated caspase-3 (CC3) staining of histologic sections obtained postmortem (Figs. 2A–2C, lower panels). This correlation was confirmed microscopically (Fig. 3). In Eμ-myc tumors, C2Am-AF750 fluorescence was detected from regions in which CC3 staining occupied as little as 1%–2% of the approximately 1 mm2 regions of interest (Fig. 3C). Differences in the relationship between fluorescence intensity and the levels of cleaved caspase-3 in the different tumors (Fig. 3) may reflect differences in phosphatidylserine exposure.

Near-infrared fluorescence imaging of cell death. Imaging in vivo of untreated and treated EL4 (A), Colo-205 (B), and Eμ-myc (C) tumors. Images are overlays of bright-field images and 800 nm-channel fluorescence signals, acquired 24 h after C2Am-AF750 administration. AnxV = annexin-V.
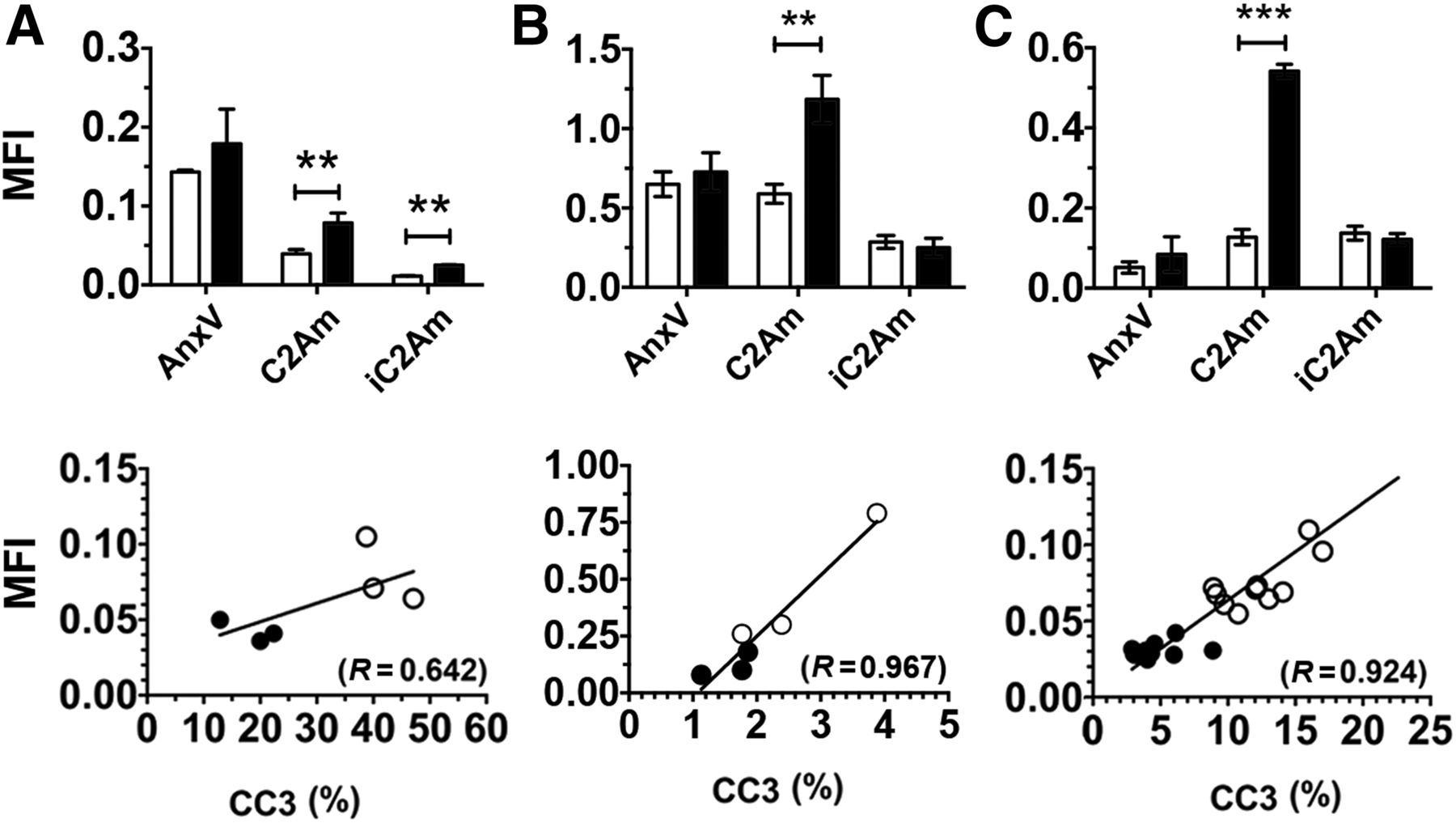
Quantification of tumor mean fluorescence intensity (MFI) for untreated (open bars) and treated (filled bars) EL4 (A), Colo-205 (B), and Eμ-myc (C) tumors. Data for Eμ-myc model was acquired ex vivo, due to skin pigmentation artifacts. (Bottom) Correlation of C2Am whole-tumor MFIs with corresponding CC3 staining, measured in sections of excised EL4 (A), Colo-205 (B), and Eμ-myc (C) tumors. ○ = drug-treated (48 h after treatment); ● = untreated. **P < 0.01, ***P < 0.001, n = 3/group, values are mean ± SD. AnxV = annexin-V.
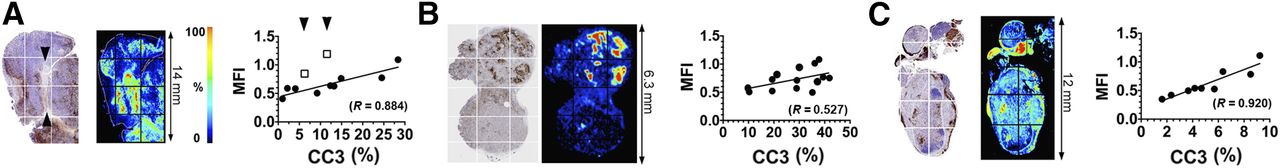
Maps of CC3 staining (left), C2Am-AF750 fluorescence (middle), and correlation (right) of fluorescence intensities of regions of interest (grids indicated on the left), with staining for CC3 in same regions of interest. Tumors were excised 24 h after C2Am-AF750 administration and 48 h after drug treatment. The correlation coefficients (R) of the linear fits to the data (A–C, right) are shown. Arrows in A (left) indicate decellularized regions of tissue. Sections are from EL4 (A), Colo-205 (B), and Eμ-myc (C) tumors. MFI = mean fluorescence intensity.
Cell Binding Experiments with 99mTc-C2Am
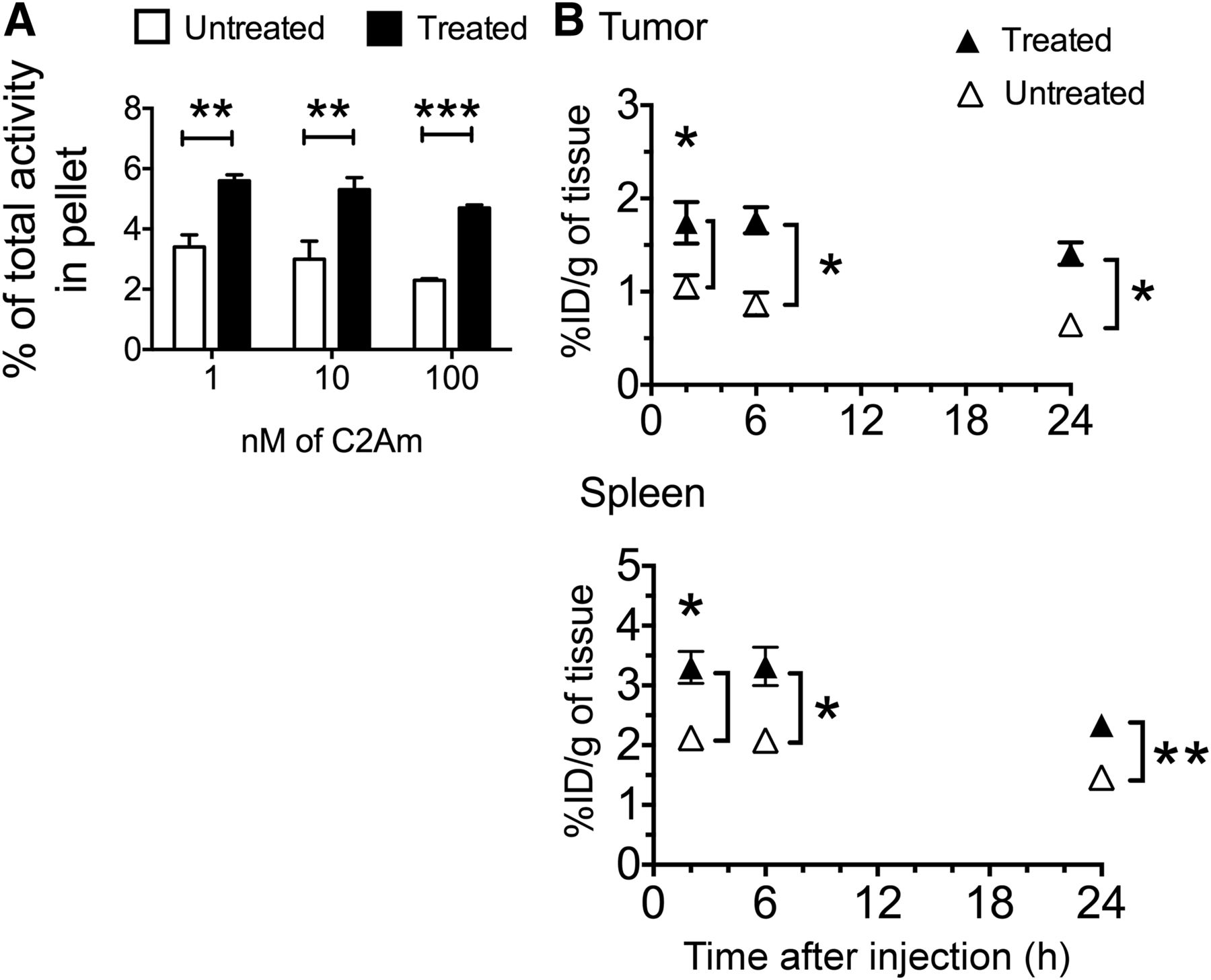
Because we were unable to detect treatment response with annexin-V-AF750, despite using the most recent agent and according to the manufacturer’s instructions, radionuclide imaging studies were performed solely with C2Am labeled with either 99mTc or 111In. There was a significant increase in the radioactivity retained by washed cell pellets prepared from suspensions of EL4 lymphoma cells that had been treated with etoposide (15 μM, for 24 h) and then incubated with 99mTc-C2Am in the concentration range of 1–100 nM (Fig. 4A). Cell viability decreased from 95% to 75% in these drug-treated cells, as assessed by trypan blue dye exclusion.
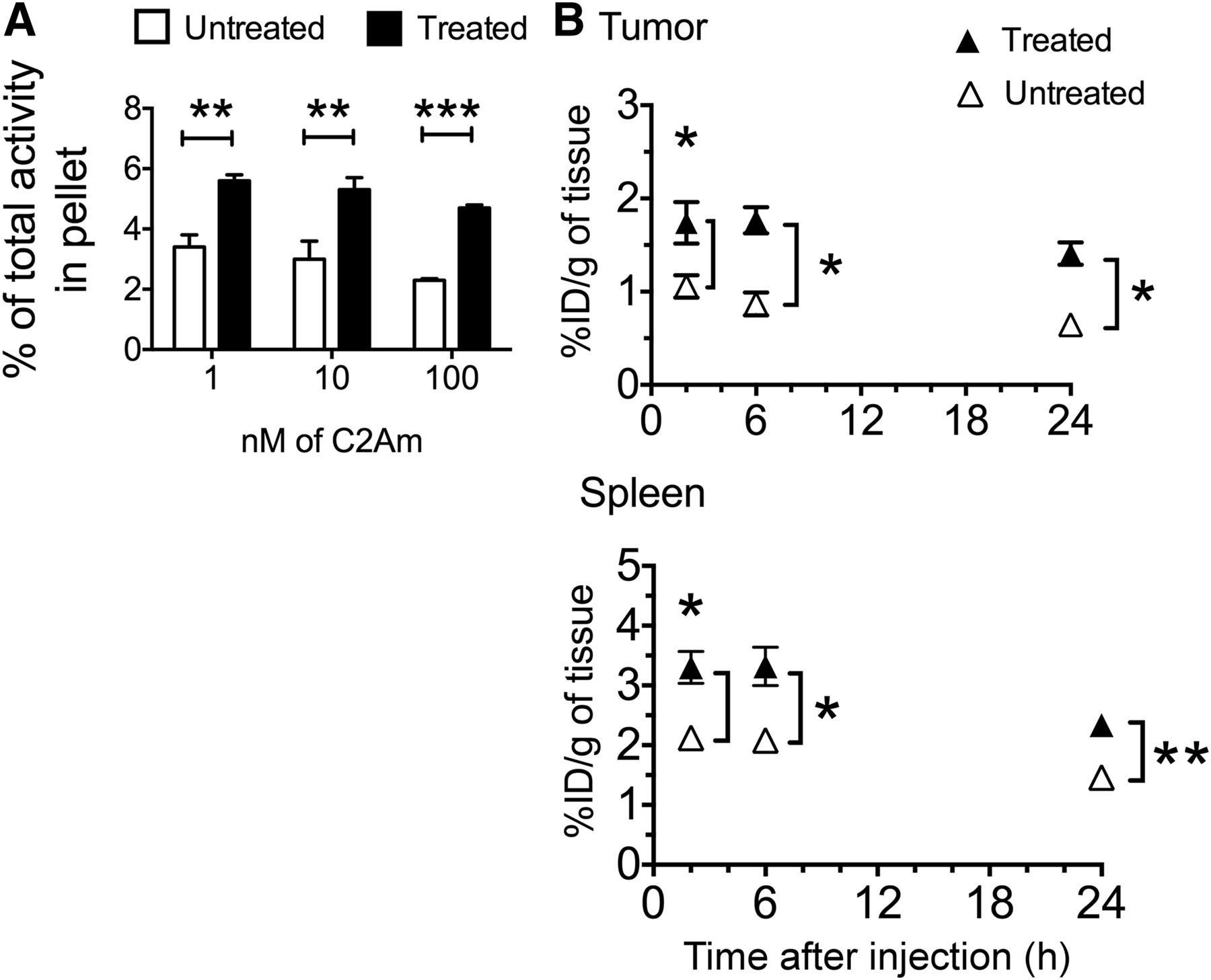
(A) Binding of 99mTc-C2Am to EL4 cells. Labeling of drug-treated and untreated cells is expressed as percentage of total 99mTc activity retained by cell pellets. **P < 0.01, ***P < 0.001, n = 3/group, values are mean ± SD. (B) Retention of 99mTc-C2Am in tumors (top) and spleens (bottom) from EL4 tumor–bearing mice, in untreated (△) and drug-treated (▲) animals, at indicated times after probe administration. Tissue retention is expressed as percentage injected dose per gram (%ID/g) of tissue. *P < 0.05, **P < 0.01, n = 3/group, 2-way ANOVA, with Bonferroni posttest correction, was used for group comparisons. Values are mean ± SD.
Biodistribution
99mTc-C2Am and 111In-C2Am showed favorable biodistribution profiles, with predominantly renal clearance (Table 1). 99mTc-C2Am had blood half-lives in EL4 tumor–bearing animals of 7.2 ± 0.6 and 9.7 ± 0.1 h in etoposide-treated and untreated animals, respectively. 111In-C2Am showed similar half-lives in Colo-205 tumor–bearing animals of 11.4 ± 1.2 and 8.0 ± 0.8 h in 5-FU–treated and untreated animals, respectively. Tumor-to-blood ratios increased progressively in chemotherapy-treated EL4 tumor–bearing animals, from 1.71 ± 0.40 at 2 h to 6.96 ± 1.08 at 24 h for 99mTc-C2Am and from 1.38 ± 0.65 at 2 h to 5.43 ± 0.96 at 24 h for 111In-C2Am in Colo-205 tumor–bearing animals. There was significantly greater retention in tumors and spleens (Fig. 4B, P < 0.05) from etoposide-treated EL4 tumor–bearing animals, from as early as 2 h after drug administration. The spleens showed significant increases in the levels of cell death after drug treatment (Supplemental Fig. 3).
Biodistribution of 99mTc-C2Am in Tumor-Bearing EL4 Mice and 111In-C2Am in Tumor-Bearing Colo-205 Mice, 24 Hours After Etoposide and 5-FU Treatment, Respectively
The biodistribution profiles of C2Am-AF750 and iC2Am-AF750 (Supplemental Table 2) were similar to those of 99mTc-C2Am and 111In-C2Am (Table 1). As with 99mTc-C2Am and 111In-C2Am, C2Am-AF750 generated EL4 tumor-to-muscle contrast from as early as 2 h (2.2 ± 0.49) and up to 24 h (3.63 ± 0.4) after injection of the imaging agent (Supplemental Table 2).
SPECT Imaging of Tumor Cell Death In Vivo Using 99mTc-C2Am and 111In-C2Am
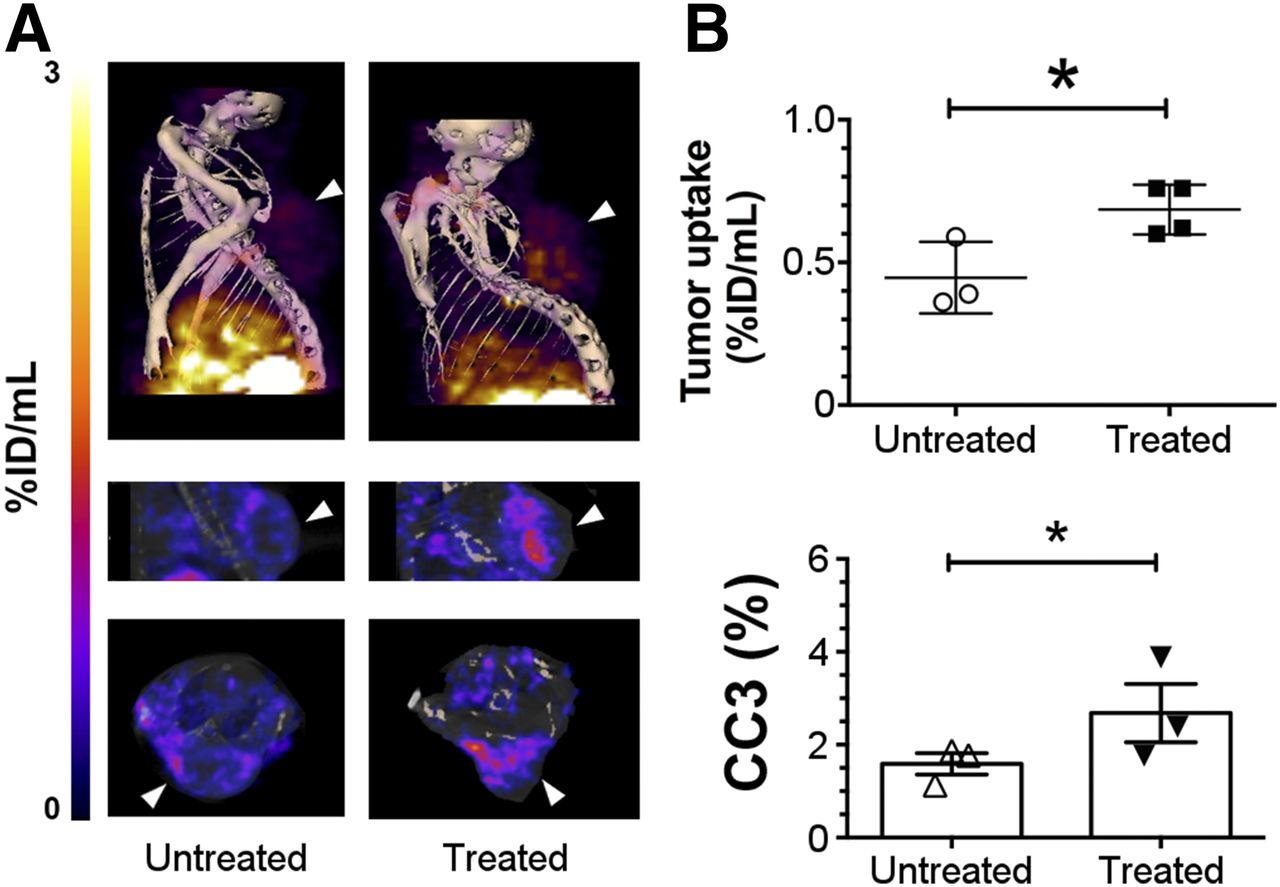
Images were acquired from animals with implanted Colo-205 (Fig. 5A) tumors and from tumor-bearing Eμ-myc mice (Fig. 6A) before and 24 h after treatment with 5-FU or cyclophosphamide, respectively, and 2 h after injection of 111In-C2Am or 99mTc-C2Am, respectively. Corresponding studies with 99mTc-C2Am in etoposide-treated EL4 tumors are shown in Supplemental Figure 4. Maximum activity was observed in the kidneys and bladder, consistent with a predominantly renal excretion route (Table 1). Renal retention was mostly cortical (Supplemental Fig. 4A). In 5-FU–treated Colo-205 tumors (Fig. 5), 111In-C2Am uptake was increased by approximately 1.6 times (Fig. 5B, upper panel, P < 0.05), reflecting a similar increase (1.65×) in the percentage of dead cells determined histologically in tumor sections (Fig. 5B, lower panel, P < 0.05). In Eμ-myc mice, 99mTc-C2Am detected baseline levels of cell death in cervical tumors (Fig. 6A, left), and there was an approximately 1.9 times increase (P < 0.0001) in tumor signal after treatment (Fig. 6B, upper panel). Both cervical (Fig. 6A, 1) and axillary (Fig. 6A, 2) node tumors were clearly visible (Fig. 6A, arrowheads) after treatment (Supplemental Video 1). Histologic analysis of tumor sections showed a similar increase in tumor cell death after treatment (∼3×; P < 0.05) (Fig. 6B, lower panel). In EL4 tumors 99mTc-C2Am uptake increased after treatment by approximately 1.3 times (P < 0.05), paralleling the modest increase and reflecting the wide variation in the levels of cell death in this tumor model (∼1.6×; P < 0.05). Treatment response was also observed in the spleen and salivary glands of these etoposide-treated animals (Supplemental Fig. 4; Supplemental Video 1).
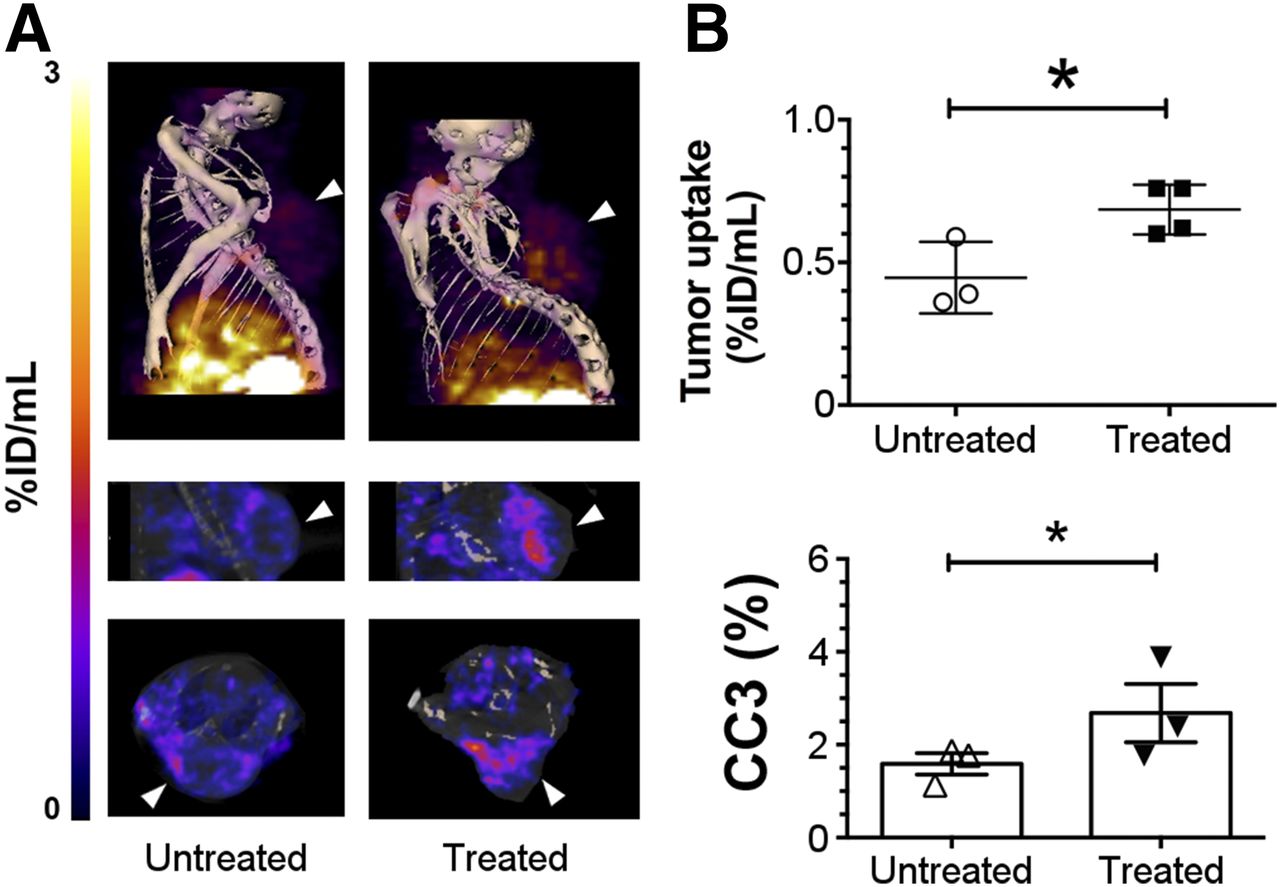
SPECT imaging of cell death in vivo in Colo-205 tumors. Imaging of 111In-labeled C2Am was performed 2 h after probe administration and 24 h after drug treatment. (A) SPECT/CT fusion images of representative untreated Colo-205 tumor–bearing mouse (A, left) and 5-FU–treated animal (A, right), 2 h after administration of 111In-C2Am. Tumor location is indicated by arrowheads. (B) Tumor retention (percentage injected dose [%ID]/mL) (upper) and CC3 staining (lower) in untreated and 5-FU–treated tumors. *P < 0.05, n = 3−4 tumors/group.
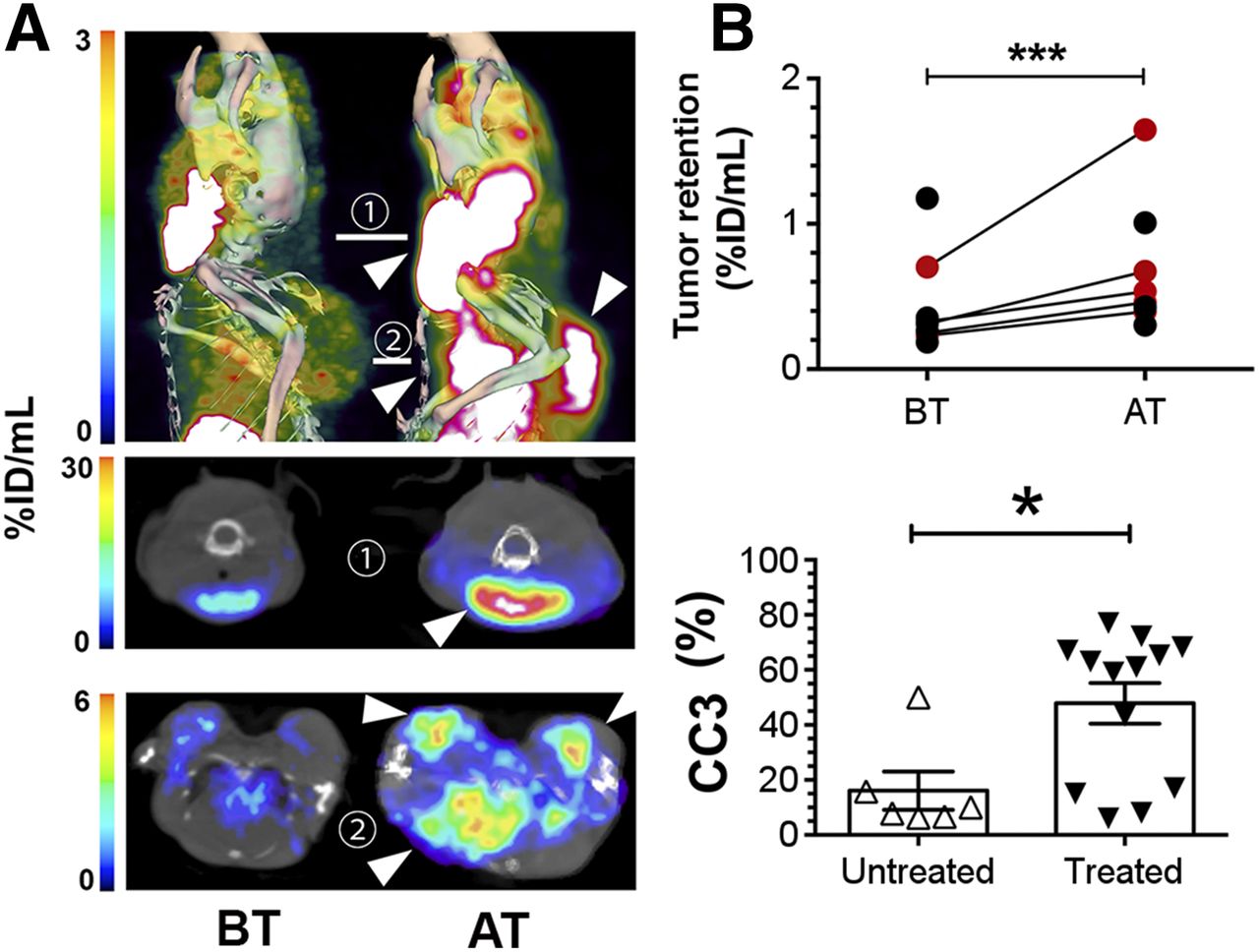
SPECT imaging of cell death in vivo in Eμ-myc tumors. Imaging of 99mTc-labeled C2Am was performed 2 h after probe administration, and 24 h after drug treatment. (A) SPECT/CT fusion images of representative Eμ-myc mice before (left) and after (right) cyclophosphamide treatment. Tumors in neck, axillary region, and chest cavity were visible (arrowheads, top) and in axial sections across cervical (1, middle) and axillary (2, bottom) planes. Red circles in B correspond to percentage injected dose [%ID]/mL retention values for tumors of animals shown in A. CC3 staining (B, bottom) in untreated and drug-treated tumors. *P < 0.05, ***P < 0.0001, n = 6−7 tumors/group (A); n = 6−13 tumors/group (B). AT = after treatment; BT = before treatment. Supplemental materials provide statistical analysis. Three-dimensional rendering of SPECT data are shown in Supplemental Video 1.
DISCUSSION
Phosphatidylserine exposed by dying cells constitutes a temporally stable and abundant biomarker for detection of cell death (23). We have been developing the C2Am domain of synaptotagmin-I, which binds phosphatidylserine with nanomolar affinity, as a cell death imaging agent (16). Previous work with mouse lymphoma (EL4) and human triple-negative breast cancer (MDA-MB-231) cells demonstrated that C2Am has a higher specificity for binding to dead and dying cells in vitro, when compared with annexin-V (16). We have demonstrated here, in vivo, that 99mTc-C2Am and 111In-C2Am have favorable biodistribution profiles, with predominantly renal clearance (Table 1; Supplemental Fig. 4), and that C2Am was capable of detecting cell death in vivo with high sensitivity and specificity, in 3 mouse models, from as early as 2 h after administration.
The binding of C2Am-AF750 was closely correlated with tumor cell death (Figs. 1−3), detecting cell death in regions with levels of CC3 staining as low as approximately 2% (Fig. 2B, lower panel; Figs. 3A and 3C, right panels). Although iC2Am-AF750 showed a small increase in uptake in EL4 tumors after treatment (Fig. 2A, upper panel), this likely reflects an increased enhanced permeability and retention effect after treatment (24). This effect was not observed in the other 2 tumor models, Eμ-myc and Colo-205 (Figs. 2B and 2C, upper panels), possibly due to lower levels of cell death in these models leading to better clearance of the probe from the tumor.
111In-C2Am detected a treatment response in Colo-205 tumors within 2 h of probe administration, where the 60% increase in probe retention in treated tumors paralleled the increase in cell death determined histologically in tumor sections obtained postmortem, from 1.59% ± 0.17% to 2.62% ± 0.48% (Fig. 5B, P < 0.05). In Eμ-myc tumors, treatment resulted in a greater proportional increase in cell death (3×) and consequently a greater increase in 99mTc-C2Am retention (1.9×) (Fig. 6). In EL4 tumors, despite high levels of preexisting cell death and a modest increase after treatment (1.6×) (Supplemental Fig. 4B), 99mTc-C2Am was nevertheless able to detect a significant treatment response (1.3× increase in probe retention, P < 0.05, Supplemental Fig. 4B). The capability of C2Am to detect small percentage increases in cell death suggests that C2Am should be capable of detecting treatment response in the clinic. For example, levels of cell death can range from less than 2% before treatment to 5%–15% after treatment (25). These data also suggest that C2Am would be effective clinically in those tumors that show high levels of preexisting cell death, such as non-Hodgkin lymphoma, in which high levels of spontaneous cell death (necrosis) have been reported in 25% of patients, frequently correlating with advanced disease and poor prognosis (26). Increased uptake of 99mTc-C2Am was also observed in the spleen and salivary glands of etoposide-treated animals (Fig. 4B; Supplemental Fig. 4A) and can be explained by etoposide-induced cell death in these tissues (27,28).
Several imaging agents have been developed to detect tumor cell death, some of which have progressed to the clinic. Annexin-V binds phosphatidylserine with high affinity (5) and has been used widely as a preclinical tool for detecting cell death, both in vitro and in vivo. However, binding of annexin-V to tumor tissue after therapy is thought not to be entirely phosphatidylserine-specific, possibly explaining the limited success of annexin-V in the clinic, which has been hampered by high levels of nonspecific binding to viable tissues, including the liver, gut, and kidneys (29). Modifications to annexin-V have shown limited improvements in biodistribution or contrast agent performance in vivo (8). Duramycin is a 19-amino-acid peptide that binds with high affinity and specificity to the phosphatidylethanolamine externalized by dying cells (30). 99mTc-labeled duramycin has been used recently to detect response to chemotherapy in a mouse model of colorectal cancer, in which accumulation of the agent was found to correlate with markers of cell death (31). However, the clinical utility of duramycin has yet to be demonstrated. Imaging agents that target CC3, an executioner caspase in the apoptosis pathway, have also been developed for detection of cell death in vivo (25), and a probe capable of detecting CC3 has progressed to clinical trials (32). However, because CC3 is a transient biomarker of cell death, the choice of temporal imaging window after treatment is critical (33). Furthermore, CC3-targeted imaging agents are unable to identify caspase-independent modes of cell death, such as necrosis (34), which is often present after chemotherapy or radiotherapy. 18F-ML-10, which appears to bind to dead cells by an unknown mechanism, has shown favorable safety and biodistribution profiles in humans (35). In a clinical study in 10 patients with brain metastasis, there was enhanced retention (up to 2-fold) of 18F-ML-10 after radiotherapy and a good correlation between probe uptake and tumor size reduction (36). However, there was no histologic demonstration of increased cell death and the average reported decrease in tumor size after therapy was substantial (∼60% reduction). Therefore, the utility of 18F-ML-10 for detecting cell death in the most common clinical scenarios, in which therapy induces low levels of tumor cell death, has yet to be demonstrated.
CONCLUSION
We have demonstrated the capability of C2Am to detect tumor cell death in vivo as early as 2 h after administration. Radiolabeled C2Am derivatives showed favorable biodistribution profiles, with predominantly renal clearance, and there was a close correlation between C2Am binding and histologic markers of cell death. The capability of C2Am to detect relatively modest increases in cell death suggests that these agents will have sufficient sensitivity to detect tumor cell death in the clinic.
DISCLOSURE
This work was supported by a Cancer Research U.K. programme grant to Kevin M. Brindle (17242) and by the CRUK-EPSRC Imaging Centre in Cambridge and Manchester (16465). Sarah Fawcett was the recipient of a PhD studentship from the Cambridge Biomedical Research Centre of the National Institute of Health Research with financial support from GlaxoSmithKline U.K. Tiago B. Rodrigues received Intra-European Marie Curie (FP7-PEOPLE-2009-IEF, Imaging Lymphoma) and Long-term EMBO (EMBO-ALT-1145-2009) fellowships. C2Am is under a licensing agreement with Cambridge Enterprise, and has been patented (US2011/0038798). Some of the authors of this study (André A. Neves, Israt S. Alam, Maaike M. de Backer, and Kevin M. Brindle) are coinventors on this patent. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Peter Sharat and Len Packman at the Protein and Nucleic Acid Facility (PNAC) of the Department of Biochemistry, University of Cambridge, and Chandra Solanki and Kishor Solanki, of the Nuclear Medicine Department of Addenbrooke’s Hospital Cambridge, for their advice and support as well as Sarah Dawson for data analysis. We are grateful to the CRUK Cambridge Institute Histopathology, Research Instrumentation, Imaging, Bioresources, Bioinformatics Core Units, for their support. We thank Li-Cor Inc. for the lease of a Pearl Impulse small-animal imaging system and Jane Gray for her support in the use of a Li-Cor Odyssey scanner. We are grateful to Sandra Fulton for her guidance on site-directed mutagenesis.
Footnotes
Published online Feb. 16, 2017.
- © 2017 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication August 24, 2016.
- Accepted for publication January 17, 2017.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}