


Abstract
In vivo imaging of N-methyl-d-aspartate (NMDA) glutamate receptor and γ-aminobutyric acid (GABA)-A receptor during progression of brain pathology is challenging because of the lack of imaging tracers with high affinity and specificity. Methods: We monitored changes in NMDA receptor and GABA-A receptor in a clinically relevant model of traumatic brain injury (TBI) induced by lateral fluid percussion in adult rats, using 2 new ligands for PET: 18F-GE-179 for the open/active state of the NMDA receptor ion channel and 18F-GE-194 for GABA-A receptor. Ex vivo brain autoradiography of radioligands was performed at subacute (5–6 d) and chronic (40–42 d) time points after TBI. Results: At 5–6 d after TBI, 18F-GE-179 binding was higher in the cortical lesion area, in the lesion core, and in the hippocampus than in the corresponding contralateral regions; this increase was probably related to increased permeability of the blood–brain barrier. At 40–42 d after TBI, 18F-GE-179 binding was significantly higher in the medial cortex, in the corpus callosum, and in the thalamus than in the corresponding contralateral regions. Five to 6 days after TBI, 18F-GE-194 binding was significantly higher in the lesion core and significantly lower in the ipsilateral thalamus. By 40–42 d after TBI, the reduction in 18F-GE-194 binding extended to the cortical lesion, including the perilesional cortex around the lesion core. The reduction in thalamic binding was more extensive at 40–42 d than at 5–6 d after TBI, suggesting a progressive decrease in thalamic GABA-A receptor density. Immunohistochemistry against GABA-A α1 subunit revealed a similar decrease to 18F-GE-194 binding, particularly during the chronic phase. Conclusion: Our data support the validity of novel 18F-GE-179 and 18F-GE-194 radioligands for the detection of changes in active NMDA receptor and GABA-A receptor in the injured brain. These tools are useful for follow-up evaluation of secondary postinjury pathologies.
Every 23 s, 1 person in the United States sustains a traumatic brain injury (TBI); TBI is recognized as a major cause of death and disability worldwide (1). The incidence of emergency department visits due to TBI varies depending on patient age, with the highest incidences occurring in children aged 0–4 y (1,256 per 100,000 per year) and in older adolescents aged 15–19 y (757 per 100,000 per year) (2). TBI can result in a myriad of chronic comorbidities, including behavioral and cognitive impairment as well as epileptogenesis, which significantly compromise quality of life (3). In 2010, the estimated cost of TBI, including both direct and indirect medical costs, was approximately $76.5 billion in the United States and €33 billion in Europe (4,5). Therefore, it is crucial to develop tools for understanding the pathophysiologic changes caused by TBI and for enabling personalized prediction of TBI outcome.
PET can provide spatial and temporal information about the location and type of pathologies at the molecular level (6). Previous experimental studies showed that N-methyl-d-aspartate (NMDA) glutamate receptors contribute to several post-TBI pathologies that lead to functional impairments, including hypoperfusion due to failure in cerebrovascular autoregulation (7), edema via accumulation of intracellular Na+ (8,9), glutamate-induced excitotoxicity (10), epileptogenesis (11), and cognitive decline (12). Similarly, inhibitory γ-aminobutyric acid (GABA)-A receptor–mediated functions contribute to the development and expression of post-TBI aftermath, particularly to excitability related to acute seizures and epileptogenesis (11–14). To date, no PET ligands have been available for assessment of the expression of NMDA or GABA-A receptors during the disease process.
A novel putative PET ligand, 18F-GE-179, was recently introduced for the detection of NMDA ion-channel activity (15). 18F-GE-179 has been shown to target the intrachannel phencyclidine binding site and therefore to selectively bind to the open/active state of NMDA receptor ion channels (16,17). Another novel PET ligand, 18F-GE-194, was introduced for the detection of GABA-A receptors (18). 18F-GE-194 is an 18F-labeled tracer with properties comparable to those of 11C-flumazenil, providing a longer half-life than those of 11C-labeled tracers (19).
The present study was designed to test the hypothesis that TBI triggers region-specific changes in NMDA receptor binding and in GABA-A receptor binding that can be detected using ex vivo brain autoradiography with novel PET ligands (18F-GE-179 and 18F-GE-194, respectively). We monitored the progression of brain pathology at 5–6 d and at 40–42 d after TBI in a clinically relevant rat model of TBI induced by lateral fluid percussion. Ex vivo brain autoradiographs were compared with immunohistochemistry of NMDA-R1 and GABAA-α1 receptor subunits. We found evidence of long-lasting acquired NMDA receptor and GABA-A receptor channelopathies in the thalamus after TBI that were detectable with these radioligands.
MATERIALS AND METHODS
The study design is depicted in Supplemental Figure 1 (supplemental materials are available at http://jnm.snmjournals.org).
Synthesis of 18F-GE-179 and 18F-GE-194
18F-GE-179 and 18F-GE-194 were synthetized at Turku PET Centre Radiopharmaceutical Laboratory, as described previously (15,18). The specific activity of tracers exceeded 1 TBq/μmol, and radiochemical purity was greater than 99% in all syntheses (18F-GE-179, n = 3; 18F-GE-194, n = 4). For intravenous injection, 18F-GE-179 was dissolved in 0.9% NaCl containing 9% ethanol, and 18F-GE-194 was dissolved in Dulbecco's phosphate-buffered saline containing 11% ethanol. The chemical structures of 18F-GE-179 and 18F-GE-194 appear in Supplemental Figure 2.
Animals and Induction of TBI
Adult male Sprague–Dawley rats (Harlan Laboratories; n = 32, 325 ± 25 g at the time of the procedure) were used. Animal care complied with the guidelines of the International Council of Laboratory Animal Science. All animal procedures were approved by the Animal Ethics Committee of the Provincial Government of The Southern Finland and performed in accordance with the guidelines of the European Community Council Directives 86/609/EEC.
TBI was induced using lateral fluid percussion, as described previously (20). As expected, the mortality rate was 30% within 72 h in the TBI group. Sham animals (n = 16) underwent surgery but were not injured. The detailed protocol for TBI induction is described in the supplemental materials.
Administration of 18F-GE-179 or 18F-GE-194 Tracers
Ex vivo studies with 18F-GE-179 and 18F-GE-194 tracers were performed at 5–6 d (subacute condition) or 40–42 d (chronic condition) after TBI. In both the subacute and the chronic conditions, 4 rats were used for each tracer. Rats were anesthetized with 2.5% isoflurane/O2 and placed on a heating pad. A bolus injection of tracer was administered intravenously via the tail (maximum volume, 1 mL). In the subacute 18F-GE-179 experiment, the mean dose of tracer injected into the TBI group was 19.4 ± 14.0 MBq (n = 4); in the chronic experiment, the mean dose was 8.6 ± 1.7 MBq (n = 4). In the subacute 18F-GE-194 experiment, the mean dose of tracer injected was 30.2 ± 4.7 MBq (n = 4), and the mean dose was 26.7 ± 10.3 MBq (n = 4) in the chronic experiment.
Ex Vivo Quantification of 18F-GE-179 and 18F-GE-194 Uptake in Brain and Peripheral Organs
Thirty minutes after tracer injection, rats were anesthetized with 2.5% isoflurane/O2. Blood was collected via cardiac puncture for measurement of 18F radioactivity, the rats were killed via decapitation, and the organs were dissected.
After radioactivity was measured, brains were immediately frozen in dry ice–chilled isopentane. Coronal sections (20 μm) of the brain were cut using a cryostat starting at the level +1.8 mm rostral to the bregma and continuing caudally to the level −2.8 mm from the bregma (21). For digital autoradiography, sections were exposed to an imaging plate (Fuji BAS Imaging Plate; Fuji Photo Film Co., Ltd.) for approximately 4 h. Imaging plates were scanned using a BAS-5000 reader (Fuji Photo Film Co. Ltd.) with a resolution of 25 μm. Count densities in the autoradiographic images (photostimulated luminescence per unit area) were analyzed using Aida 2.14 (Raytest Isotopenmessgeräte GmbH).
For both 18F-GE-179 and 18F-GE-194 experiments, 14 regions of interest (ROIs) were drawn over several brain regions (Supplemental Figs. 3 and 4, respectively). ROIs were drawn around the cortical lesion area, including the immediate perilesional cortex and the lesion core; the lesion core only, defined by higher tracer uptake inside the cortical lesion area; the medial cortex, including the cortex medial to the lesion; the ipsilateral thalamus; the ipsilateral hippocampus, including the dentate gyrus and the CA3 and CA1 subfields; and the ipsilateral striatum. ROIs were also drawn around the contralateral areas corresponding to the areas listed above. For 18F-GE-179 autoradiographs, additional ROIs were drawn on the corpus callosum and the corresponding contralateral area in the chronic condition. For 18F-GE-194 autoradiographs, additional ROIs were drawn around the cortical area immediately below the cortical lesion area and the corresponding contralateral area in the chronic condition, because of a complete lack of binding in the cortical lesion area. ROIs were drawn on at least 10 sections to represent each region.
Analysis of the binding of 18F-GE-179 and 18F-GE-194 in the ipsilateral and contralateral striatum did not differ between the TBI and sham groups (Supplemental Fig. 5). Therefore, the ipsilateral region-to-striatum binding ratio was compared with the contralateral region-to-striatum ratio in the same animal.
GABA and NMDA Receptor Immunohistochemistry
Frozen sections adjacent to those used for autoradiography were processed for NMDA-NR1 and GABA-A receptor α1 subunit immunohistochemistry. Protocol details appear in the supplemental materials.
Thionin Staining
Brain slices used for ex vivo brain autoradiography were stained with thionin to detect integrity of the main neuronal layers. Protocol details appear in the supplemental materials.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism Software 5.01. Differences in the region-to-striatum uptake ratio between the ipsilateral and contralateral side in animals in the TBI group were tested using paired t tests. Differences in the region-to-striatum uptake ratios between the subacute and chronic conditions were tested using unpaired t tests. Data are presented as mean ± SD. Differences were considered significant when the P value was less than 0.05.
RESULTS
Ex Vivo Uptake of 18F-Radioactivity in Brain
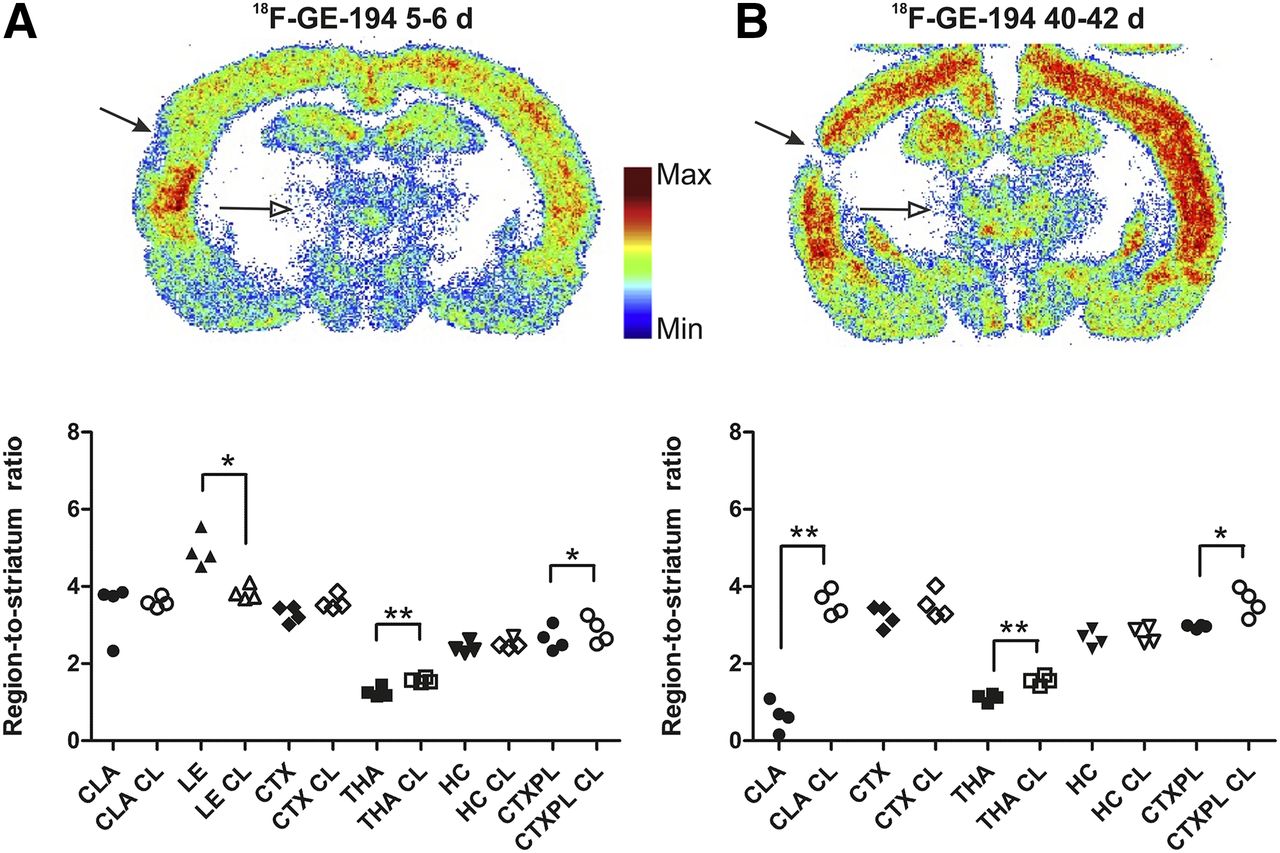
Representative autoradiographic images of 18F-GE-179 binding in the subacute and chronic TBI groups are shown in Figure 1. In the subacute TBI group, 18F-GE-179 binding was significantly higher ipsilaterally in the cortical lesion area (P < 0.01), the lesion core (P < 0.01), and the hippocampus (P < 0.05) than in the corresponding contralateral regions (Fig. 1A). In the chronic TBI group, 18F-GE-179 binding was significantly higher in the medial cortex (P < 0.01), the corpus callosum (P < 0.05), and the thalamus (P < 0.01) than in the corresponding contralateral regions (Fig. 1B). Binding in the cortical lesion area and the medial cortex was significantly lower in the chronic experiment than in the subacute experiment (P < 0.001 and 0.05, respectively) (Fig. 1).

Binding of 18F-GE-179, which visualizes active (open) NMDA receptors, in rat brain after TBI. Representative, individually color-scaled, examples of autoradiography of tracer binding show increased signal in cortical lesion (filled arrows) and in ipsilateral thalamus (open arrows) versus corresponding contralateral regions, and quantifications at 5–6 d (A) and 40–42 d (B) after TBI. Values represent ratio of accumulation of 18F radioactivity in ipsilateral (filled symbols) or contralateral (open symbols) brain areas versus that in striatum at 30 min after 18F-GE-179 injection. *P < 0.05. **P < 0.01. CC = corpus callosum; CC CL = corpus callosum contralateral; CLA = cortical lesion area; CLA CL = cortical lesion area contralateral; CTX = medial cortex; CTX CL = medial cortex contralateral; HC = hippocampus; HC CL = hippocampus contralateral; LE = lesion core; LE CL = lesion core contralateral; THA = thalamus; THA CL = thalamus contralateral.
Representative autoradiography images of 18F-GE-194 binding in the subacute and chronic TBI groups are shown in Figure 2. In the subacute TBI group, 18F-GE-194 binding was significantly higher in the lesion core (P < 0.05) and significantly lower in the ipsilateral thalamus (P < 0.001) and cortical area immediately below the cortical lesion area (P < 0.05) than in the corresponding contralateral regions (Fig. 2A). In the chronic TBI group, 18F-GE-194 binding was significantly lower in the cortical lesion area (P < 0.01), ipsilateral thalamus (P < 0.01), and cortical area immediately below the cortical lesion area (P < 0.05) than in the corresponding contralateral regions (Fig. 2B).

Binding of 18F-GE-194 to GABA receptor in rat brain after TBI. Representative, individually color-scaled, examples of autoradiography of tracer binding show increased signal in cortical lesion (filled arrows) and in ipsilateral thalamus (open arrows) versus corresponding contralateral regions, and quantifications at 5–6 d (A) or 40–42 d (B) after TBI. Values represent ratio of accumulation of 18F radioactivity in ipsilateral (filled symbols) and contralateral (open symbols) brain areas versus that in striatum at 30 min after 18F-GE-194 injection. *P < 0.05. **P < 0.01. CLA = cortical lesion area; CLA CL = cortical lesion area contralateral; CTX = medial cortex; CTX CL = medial cortex contralateral; CTXPL = cortical area immediately below CLA; CTXPL CL = cortical area immediately below CLA contralateral; HC = hippocampus; HC CL = hippocampus contralateral; LE = lesion core; LE CL = lesion core contralateral; THA = thalamus; THA CL = thalamus contralateral.
The comparison of striatal binding ipsilaterally and contralaterally in the TBI and sham groups showed no differences (Supplemental Fig. 6).
Ex Vivo Uptake of 18F-GE-179 and 18F-GE-194 in Tissues
Biodistribution of 18F-GE-179– and 18F-GE-194–derived radioactivity 30 min after injection at 40–42 d after TBI is presented in the supplemental materials and in Supplemental Figure 7.
NMDA-NR1 Glutamate Receptor Immunohistochemistry
At 5–6 d after TBI (subacute condition), NMDA-NR1 receptor subunit immunoreactivity was lower in the cortical lesion area and absent from the lesion core (Fig. 3A). Immunoreactivity was lower ipsilaterally in the CA3 pyramidal cell layer of the hippocampus and in the thalamus. Furthermore, using thionin staining, we detected neuronal loss at the impact site, thalamus, and the CA3 subfield of the hippocampus (Fig. 3B). White matter areas, including the external capsule and corpus callosum, displayed increased gliosis (Fig. 3B).
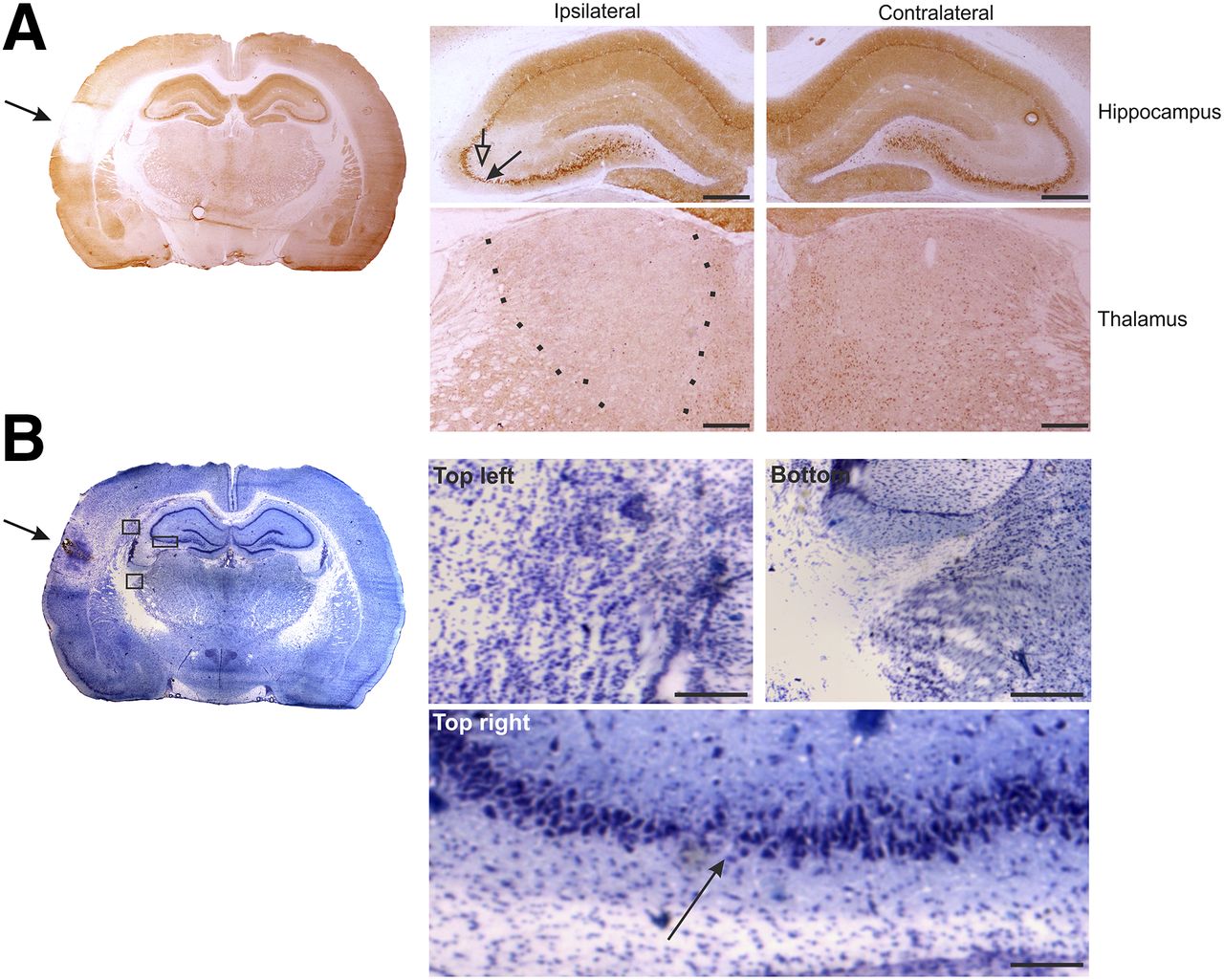
NMDA-NR1 immunohistochemistry at 5 d after TBI. (A) Whole-brain slice section stained with antibody against NMDA-NR1. In high magnification of ipsilateral hippocampus, reduced neuronal staining in CA3a region (filled arrow) that associates with reduced dendritic labeling in stratum radiatum (open arrow) can be observed, compared with contralateral side. In high magnification of ipsilateral thalamus, reduction in NMDA-NR1 immunoreactivity can be observed (between dotted lines), compared with contralateral side. (B) Adjacent thionin-stained section including lesion site (arrow). Photomicrographs in insets were taken from indicated areas in B and show gliosis ipsilaterally in external capsule ventral to perilesional cortex (top left), in dorsal aspect of reticular nucleus of thalamus (bottom), and thinning of pyramidal cell layer of CA3b/a (arrow) (top right). Scale bars: 500 μm (A); 100 μm (B).
At 40–42 d after TBI (chronic condition), immunohistochemistry revealed lower NMDA-NR1 glutamate receptor staining in the CA3 pyramidal layer of the ipsilateral hippocampus and fewer immunopositive cells in the ventroposterior thalamus (Fig. 4A). Thionin staining indicated an enlargement of the lateral ventricle and a reduction in the volume of the hippocampus ipsilaterally (Fig. 4B). Staining was also higher in the cortical lesion area, in the cortex medial to the lesion, and in the underlying external capsule (Fig. 4B). Similar to the subacute condition, neurodegeneration was evident in the thalamus and in the hippocampus (Fig. 4B).
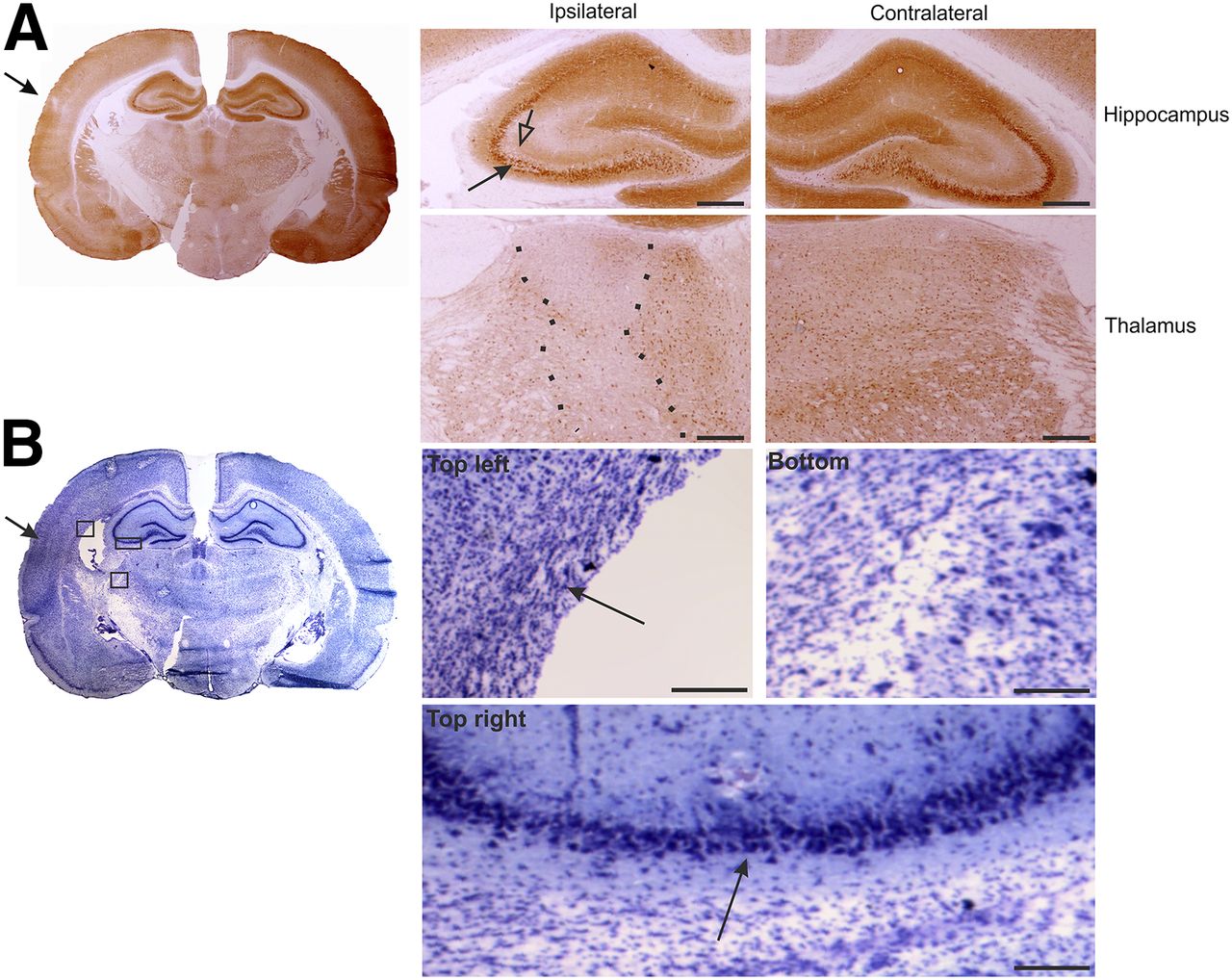
NMDA-NR1 immunohistochemistry at 42 d after TBI. (A) Whole-brain slice section stained with antibody against NMDA-NR1. In high magnification of ipsilateral hippocampus, reduced neuronal staining in CA3a region (filled arrow) that associates with reduced dendritic labeling in stratum radiatum (open arrow) can be observed, compared with contralateral side. In high magnification of ipsilateral thalamus, reduction in NMDA-NR1 immunoreactivity can be observed (between dotted lines), compared with contralateral side. (B) Adjacent thionin-stained section of lesion site. Note enlarged lateral ventricle ipsilaterally. Photomicrographs in insets were taken from indicated areas in B and show gliosis ipsilaterally in external capsule ventral to perilesional cortex (top left), in dorsal aspect of reticular nucleus of thalamus (bottom), and thinning of pyramidal cell layer of CA3b/a (top right). Scale bars: 500 μm (A); 100 μm (B).
GABA-A Receptor Immunohistochemistry
At 5–6 d after TBI, reduction of GABA-A receptor α1 subunit staining was not observed either in the cortex or in the hippocampus. In contrast, a clear decrease in immunoreactivity in the ipsilateral thalamus was detected (Fig. 5). This observation was in agreement with the reduction in 18F-GE-194 binding in the same area.

GABA-A receptor α1 subunit immunohistochemistry at 5 d after TBI. Whole-brain slice section stained with antibody against GABA-A receptor α1 subunit. No changes were observed in ipsilateral hippocampus, compared with contralateral side. In high magnification of ipsilateral thalamus, reduction in GABA-A receptor α1 subunit immunoreactivity can be observed, compared with contralateral side. Scale bars: 500 μm.
At 40–42 d after TBI (chronic condition), we observed decreased GABA-A receptor α1 subunit staining in the cortical lesion area and thalamus (Fig. 6), which matched with the evident reduction in signal in ex vivo autoradiography with 18F-GE-194 (Fig. 2B). Hippocampal immunostaining in the ipsilateral area was comparable to that in the contralateral area.

GABA-A receptor α1 subunit immunohistochemistry at 41 d after TBI. Whole-brain slice section stained with antibody against GABA-A receptor α1 subunit. No changes were observed in ipsilateral hippocampus, compared with contralateral side. In high magnification of ipsilateral thalamus, reduction in GABA-A receptor α1 subunit immunoreactivity can be observed (marked with dotted lines), compared with contralateral side. Scale bars: 500 μm.
DISCUSSION
The present study tested the hypothesis that intravenous administration of 18F-GE-179 and 18F-GE-194 after TBI can be used to detect TBI-induced molecular pathology. As first steps, signal was detected using ex vivo autoradiography, and immunohistochemistry was performed for NMDA receptor and GABA-A receptor. Our data indicate that both radioligands were synthetized with good specific activity; no uptake of 18F radioactivity in bone was observed (Supplemental Fig. 6), underscoring the stability of the 18F label. 18F-GE-179 binding displayed time-dependent changes in the cortical lesion, in the cortex located medial to the lesion, the corpus callosum, and in the thalamus. 18F-GE-179 binding showed a limited association with immunostaining for NMDA-R1 glutamate receptor. In the case of 18F-GE-194 after an initial increase in binding in the core of the cortical lesion, binding in the lesion core as well as in the ipsilateral thalamus dramatically decreased. Less 18F-GE-194 signal was associated with lower levels of immunostaining for the GABA-A α1 receptor subunit.
18F-GE-179 Binding Remains Chronically Elevated in Thalamocortical Region
Our data revealed a remarkable elevation in ex vivo 18F-GE-179 binding at 5–6 d after TBI in the cortical lesion area and lesion core. This observation is in contrast with previous messenger RNA or immunohistochemical studies that reported no changes or transient reduction in the levels of NMDA-R1 transcript or protein during the first week after lateral fluid percussion injury in rats (22,23); these transient changes could mask reductions in receptor density (24). The high 18F-GE-179 binding in the lesion core and cortical lesion area could be explained by a mix of the damage to the blood–brain barrier and a subsequent leakage of tracer into the tissue as well as by the increased NMDA binding due to perilesional gliosis. The decrease in the signal ratios in the damaged cortical area from the subacute to the chronic time point can be due to the closure or repair of the blood–brain barrier and the resolution of gliosis.
NMDA-R1 immunohistochemistry showed reduced receptor subunit binding in the ipsilateral pyramidal cell layer and in the stratum radiatum of the CA3a subfield of the hippocampus; these changes were not evident on ex vivo 18F-GE-179 brain autoradiography because of the small size of the area and the limited resolution.
Interestingly, we observed substantially more 18F-GE-179 binding in the ventroposterior nuclei of the thalamus at 40–42 d after TBI. This increase in binding extended along the internal capsule to the deep layers of the cortex. These data suggest increased NMDA receptor activity in the thalamocortical pathway.
There is an apparent contradiction between increased 18F-GE-179 thalamic binding and decreased NMDA-R1 immunohistochemical staining. However, this can be explained by the loss of some NMDA-R1–expressing neurons while there is a large increase in the activity of the NMDA receptors due to gliosis. The compatibility of the increase in total NMDA receptor activity with a decrease in the number of NMDA receptors due to partial neuronal damage in the same area has been shown in epileptic syndromes (25).
18F-GE-194 Binding Remains Chronically Reduced in Thalamocortical Region
We observed a remarkable increase in 18F-GE-194 binding in the lesion core at 5–6 d after TBI that likely relates to damage to the blood–brain barrier. Interestingly, at 5–6 d after TBI 18F-GE-194 binding was reduced in the thalamus. This difference was greater at 40–42 d after TBI, and the decrease in 18F-GE-194 binding extended to the cortical lesion area, including the perilesional cortex around the lesion core. A clear decrease in GABA-A receptor α1 was evident by 40–42 d after TBI. These observations suggest a progressive decrease in thalamic GABA-A receptor density that is consistent with our current histologic analysis of the progression of GABAergic neuronal pathology as well as with our recent real-time polymerase chain reaction, gene array, and immunohistochemical analyses of GABA-A receptor subunits in the same model of TBI (26,27). The decrease in GABA receptors in the thalamus detected with 18F-GE-194 may also underlie the increase in NMDA activity in the same area revealed by 18F-GE-179.
CONCLUSION
The current data support the validity of novel 18F-GE-179 and 18F-GE-194 radioligands for the detection of changes in NMDA receptor and GABA-A receptor in the injured brain. Post-TBI changes in NMDA receptor and GABA-A receptor binding were detected with 18F-GE-179 and 18F-GE-194 ligands, respectively, at subacute and chronic time points in areas outside the vicinity of the primary impact, providing insight into the evolution of secondary postinjury pathologies. In particular, the regional development of acquired NMDA and GABA-A channelopathies may yield insight into the mechanisms that underlie the development of TBI-associated comorbidities such as cognitive decline and epileptogenesis.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This study was supported by The Academy of Finland projects 272249, 273909, and 271683; project 266891; the EUROCORES EuroEPINOMICS Program (EpiGENet); Strategic Funding of the University of Eastern Finland (UEF-Brain); and GE Healthcare Ltd., which also provided the chemicals for 18F-GE-179 and 18F-GE-194 synthesis. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Jarmo Hartikainen, Elisa Riuttala, and Marko Vehmanen for technical help.
Footnotes
Published online May 19, 2016.
- © 2016 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication September 28, 2015.
- Accepted for publication April 7, 2016.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.