


Abstract
The hyperlipidemic mouse model HypoE/SRBI−/− has been shown to develop occlusive coronary atherosclerosis followed by myocardial infarctions and premature deaths in response to high-fat, high-cholesterol diet (HFC). However, the causal connection between myocardial infarctions and atherosclerotic plaque rupture events in the coronary arteries has not been investigated so far. The objective of this study was to assess whether diet-induced coronary plaque ruptures trigger atherothrombotic occlusions, resulting in myocardial infarctions in HFC-fed HypoE/SRBI−/− mice. Methods: HypoE/SRBI−/− mice were characterized with respect to the individual dynamics of myocardial infarctions and features of infarct-related coronary atherosclerosis by serial noninvasive molecular and functional imaging, histopathology, and a pharmaceutical intervention. Detailed histologic analysis of whole mouse hearts was performed when spontaneously occurring acute myocardial infarctions were diagnosed by imaging. Results: Using the imaging-triggered approach, we discovered thrombi in 32 (10.8%) of all 296 atherosclerotic coronary plaques in 14 HFC-fed HypoE/SRBI−/− mice. These thrombi typically were found in arteries presenting with inflammatory plaque phenotypes. Acetylsalicylic acid treatment did not attenuate the development of atherosclerotic coronary plaques but profoundly reduced the incidence of premature deaths, the number of thrombi (7 in 249 plaques), and also the degree of inflammation in the culprit lesions. Conclusion: HFC-induced ruptures of coronary plaques trigger atherothrombosis, vessel occlusions, myocardial infarctions, and sudden death in these mice. Thus, the HypoE/SRBI−/− mouse model mimics major features of human coronary heart disease and might therefore be a valuable model for the investigation of molecular and cellular parameters driving plaque rupture-related events and the development of new interventional approaches.
Cardiovascular diseases are the leading cause of deaths worldwide (1). Atherosclerosis underlies these pathologies in most cases and may trigger sudden life-threatening events such as myocardial infarction or stroke. Currently, ruptures of so-called vulnerable atherosclerotic plaques are considered a primary cause of these events. Plaque ruptures occur acutely and trigger thrombus formation and occlusion of the respective artery lumen, finally resulting in acute ischemic events.
Vulnerable plaques are advanced atherosclerotic lesions consisting of a necrotic core, an abundant accumulation of lipids and extracellular matrix, a variety of inflammatory cells, and a fibrous cap. The individual composition of plaques is widely accepted to determine the individual vulnerability and outcome rather than their size. It seems likely that multiple plaque ruptures can occur, followed by healing of the vessel wall associated with increase in lumen narrowing, whereas plaque ruptures seem to result in clinical events only when certain factors coincide (2).
For studying the pathophysiology of atherosclerosis and its clinical sequelae, a broad spectrum of animal models has been developed. As an example, advanced lesions resembling features of vulnerable plaques were found in brachiocephalic arteries of the widely used apolipoprotein E–deficient mouse (ApoE−/−) (3–5). Recently, atherothrombotic events have been reported in ApoE−/− mice on surgical carotid constriction and stress (6). However, clinical events such as myocardial infarctions or stroke were not observed in these models, questioning their clinical relevance.
In contrast, emerging mouse models such as a transgenic mouse expressing a hypomorphic mutant form of ApoE (HypoE) (7) in combination with a knockout in scavenger receptor class B type I (SRBI−/−), named HypoE/SRBI−/− (8,9), or the recently published ApoE−/−/Fbn1 (C1039G+/−) (10) carrying a mutation (C1039G+/−) in the fibrillin-1 (Fbn1) gene, present with advanced plaque phenotypes, spontaneous myocardial infarction, and sudden deaths.
However, the connection between spontaneous myocardial infarctions, plaque events, and atherothrombosis in the coronary arteries has not yet been shown. We therefore study here whether ruptures of vulnerable coronary plaques trigger occlusive thrombus formation and cause the observed myocardial infarctions in high-cholesterol diet (HFC)–fed HypoE/SRBI−/− mice using serial noninvasive imaging combined with whole-heart thin-slice histopathology and a pharmaceutical intervention.
MATERIALS AND METHODS
All animal experiments performed in the study were approved by the local authorizing agency of North Rhine-Westphalia. Homozygous double-transgenic HypoE/SRBI−/− mice (8) were enrolled in this study. Mice were fed a standard chow diet (4.5% fat, 0.022% cholesterol; Altromin) before the start of the experiment and were assigned to the following groups: follow-up until spontaneous death, follow-up until first defect in 18F-FDG PET, or antithrombotic intervention.
Follow-up Until Spontaneous Death
Thirty-seven mice (19 male, 18 female; median age, 18 wk) were put on an HFC diet (7.5% cocoa butter, 15.8% fat, 1.25% cholesterol, 0.5% sodium cholate; Altromin) (8), and 12 mice (3 male, 9 female) continued the chow diet. Plasma cholesterol levels were assessed at day 21 after onset of diet (chow, 253 ± 44 mg/dL; HFC, 1,300 ± 276 mg/dL). During an observation period of up to 60 d, mice were studied by serial high-resolution PET using 18F-FDG and echocardiography 3 times a week.
Follow-up Until First Defect in 18F-FDG PET
Thirty-six mice (11 male, 25 female; median age, 13 wk) were put on an HFC diet (n = 21) or left on chow (n = 15) and subjected to the identical serial imaging protocol as described above. In this cohort, 18F-FDG PET was used to immediately trigger the excision of the respective heart when the first 18F-FDG PET defect occurred. For the chow group, PET scans were performed for 8 wk. After whole-body perfusion with paraformaldehyde (4%) in phosphate-buffered saline, hearts were excised, weighed, and embedded in paraffin for detailed histology. Hearts of animals that died spontaneously before the defined endpoint (n = 7) were excluded.
Antithrombotic Intervention
Eleven HypoE/SRBI−/− mice (8 male, 3 female; median age, 15 wk) were put on a modified HFC diet for which acetylsalicylic acid (HFC-ASA) at a dose of 500 mg/kg diet was added (Altromin). Mice were studied by 18F-FDG PET at 14, 21, and 28 d on the HFC-ASA diet. Afterward, mice were sacrificed and hearts were excised and processed as detailed above.
The following sections provide a detailed description of imaging methodology, immunohistochemistry, and blood and tissue analyses.
Histology
Paraffin-embedded hearts were completely cut into 5-μm serial short-axis sections from apex to base, collecting every 4th and 5th cut (∼400 sections per heart). Alternatively, few hearts were cut in 5-μm-long axis sections perpendicular to the septum for a more gross evaluation of the myocardium. Subsequently, sections were deparaffinized, rehydrated, and subjected to hematoxylin and eosin (HE), trichrome, wheat germ agglutinin (WGA), and in situ TdT-mediated dUTP-biotin nick end labeling (TUNEL).
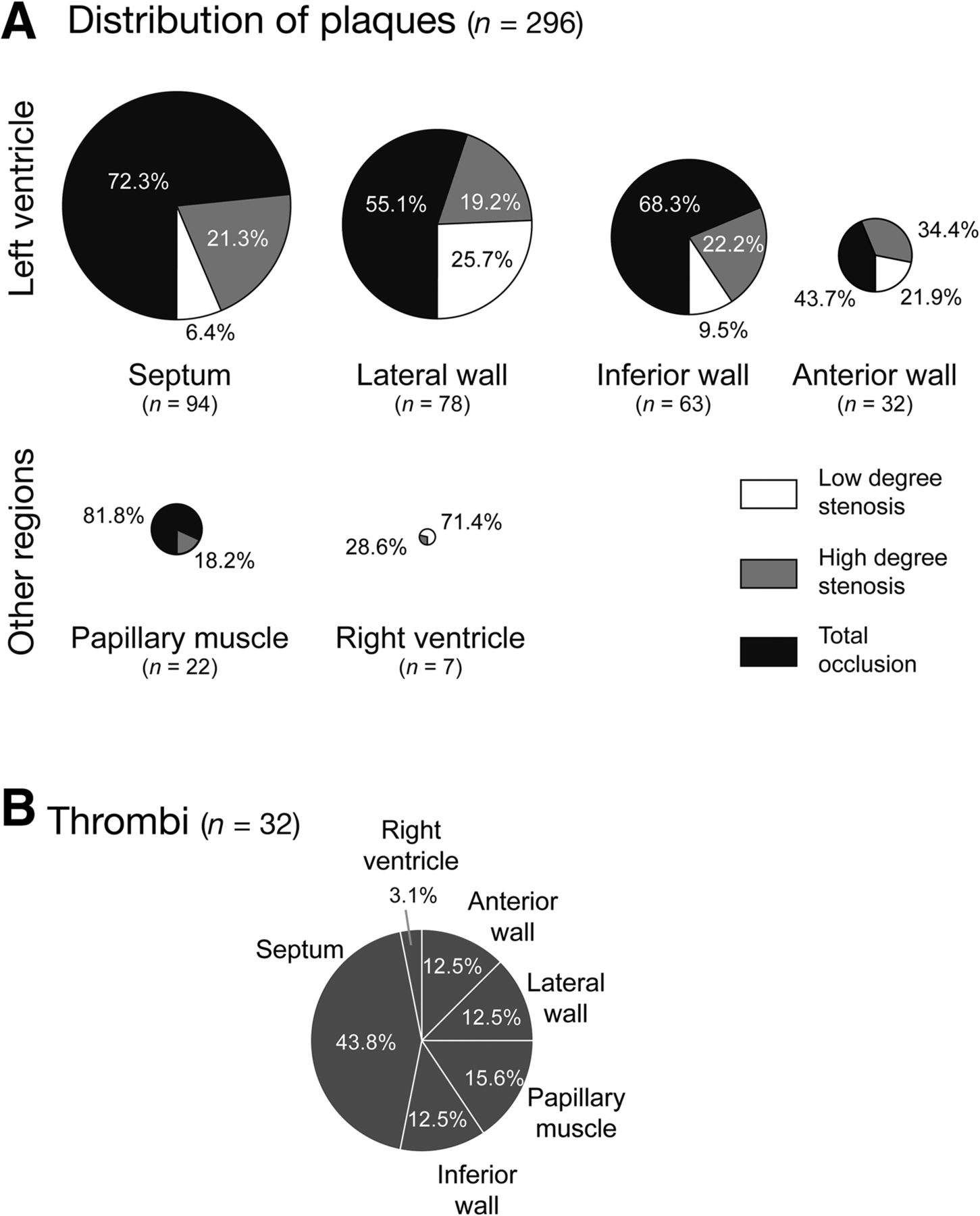
HE standard staining was performed on every 19th and 20th section of each heart (∼100 sections per heart) to evaluate atherosclerotic lesions throughout the coronary arteries. Atherosclerotic plaques observed in the HE-stained heart section series were allocated to 3 categories according to the degree of obstruction of the respective coronary artery: total occlusion, high-degree stenosis (>50% reduction of original luminal area), and low-degree stenosis (<50% reduction of original luminal area). HE staining also served to study the presence of plaque-associated thrombi. In addition, plaques were assigned to their respective location in the myocardium, with 6 myocardial regions being distinguished (anterior, lateral, inferior wall and septum; papillary muscle; right ventricle).
Masson-Goldner trichrome staining was performed to study the presence, extent, and composition of areas of postischemic myocardial damage and fibrotic remodeling. This staining allows the differentiation between viable myocardium (orange-red), postischemic (low-saturated green), and fibrotic (light green) myocardium. In addition, Shoobridge polychrome staining was applied to allow evaluation of atherothrombosis-related fibrin clots (from noncolored fresh fibrin through yellow-orange, orange-red, dark red to final blue-red and blue collagen).
Wheat germ agglutinin–tetramethylrhodamine conjugate (Molecular Probes/Life Technologies GmbH; #W849, 10 μg/mL) staining was performed for quantitative analysis of cardiomyocyte diameters. Staining was performed in at least 100 cells/heart (left ventricle) in 10 mice for chow and HFC groups.
In situ TUNEL staining was applied using a commercial kit assay (In Situ Cell Death Detection Kit, TMR red; Roche Diagnostics GmbH) to localize apoptotic and late necrotic cells. Nuclei were counterstained by DAPI-containing mounting medium.
Immunohistochemistry
Immunohistologic stainings were used to further characterize the myocardium and coronary plaque phenotypes in terms of inflammatory activity and plaque vulnerability. Applied primary antibodies were monoclonal rabbit anti-α-smooth muscle actin (clone E184 [Novus Biological], #NB110-55432; 1:500), rat monoclonal anti Mac-3 (clone M3/84, catalog # 550292 [BD Pharmingen]; 1:50), rabbit polyclonal anti-myeloperoxidase (ab65871 [Abcam]; 1:500), and affinity-purified rabbit polyclonal S100 calcium-binding protein A9 (S100A9 [also known as migration inhibitory factor–related protein 14; kindly provided by Thomas Vogl, Institute of Immunology, University of Münster, Germany; 1:600) (11). Corresponding biotinylated secondary antibodies were labeled using a diaminobenzidine chromogenic staining kit.
S100A9 immunolocalization as a matter of local inflammatory activity in coronary arteries was quantified by calculating the percentage of immunopositive area to the overall vessel crosssectional area (excluding the free vessel lumen). Quantitative histologic analysis was restricted to plaques from septal coronary arteries. The reason for this limitation was to achieve equivalent plaque subpopulations for each experimental group. Selected plaques were 9 from 9 animals in the chow-fed group, 17 from 14 animals in the HFC-diet group, and 13 from 9 animals in the HFC group treated with ASA. For each plaque to be analyzed, 1–2 slides were taken from the site of maximal stenosis. All analyses were performed by a masked experienced reader.
All histologic/immunohistochemistry slides were scanned with a Nikon Eclipse Ni digital microscope, equipped with digital cameras for brightfield (DS-Fi1) and fluorescence (DS-Qi1) microscopy (Nikon GmbH) and evaluated by the software packages Nikon NIS Elements AR or the free available ImageJ distribution Fiji (12).
Blood Analysis
Mice 14 days under the respective diet/treatment were anesthetized with isoflurane, and blood samples were collected from the abdominal vena cava to perform a whole-blood analysis in a ScilVet ABC Animal Blood Counter (Scil Animal Care Company GmbH). In addition, analysis of platelet aggregation was performed with the PAP-8E Aggregometer (Bio/Data Corporation, USA). For this, blood was immediately transferred to a tube precoated with ACD-buffer (citric acid 78 mM, sodium citrate 117mM, glucose 282 mM) in a blood-to-anticoagulant ratio of 1 to 6 and centrifuged at 200g for 10 min at room temperature. The resultant platelet-rich plasma was collected and again centrifuged (1,000g, 10 min, room temperature) to pellet the platelets. The pellet was washed twice and resuspended in Tyrode buffer (2.7 mM KCl, 3 mM NaH2PO4, 10 mM HEPES, 5.6 mM dextrose, 10 mM NaHCO3) for analysis. Samples with 100,000 platelets each were placed under stirring in the aggregometer rack and monitored for 10 min before different agonists were added. Used agonists were ADP (10 μM), collagen (20 μL), and thrombin (1 u/mol).
PET Imaging
Baseline PET scans were obtained before the onset of diet intervention. After start of the HFC diet, serial PET measurements were performed 3 times a week until the endpoint of the respective study group. 18F-FDG (10 MBq) in 100 μL of 0.9% saline was injected intravenously 1 h before each PET scan. Mice were anesthetized with isoflurane/oxygen inhalation, and body temperature was maintained using a heating pad during the experimental procedure. PET list-mode data were acquired for 15 min using the 32-module quadHIDAC scanner (Oxford Positron Systems+) dedicated to small-animal imaging. The scanner has an effective resolution of 0.7 mm (full width at half maximum) in the transaxial and axial directions when using an iterative resolution recovery reconstruction algorithm.
Quantification of segmental tracer uptake and volumes of the left ventricle were performed using a contour-detection algorithm developed in-house and validated against MRI (13). Here, the left ventricle was segmented into 8 segments per short-axis plane, with 7 planes per heart, resulting in 56 segments. To allow for parametric analyses, segmental values were normalized to the maximum uptake in the left ventricle. The mean segmental normalized 18F-FDG uptake and standard deviations were determined in a reference group (49 HypoE mice under chow diet, before onset of HFC diet). Finally, for each individual 18F-FDG scan, at follow-up the deviation from the reference group was calculated and expressed in z scores (difference between individual segments and mean reference segments divided by the SD of the reference segments). Segments with z score values below −3 were considered as 18F-FDG defects.
Echocardiography
Mice were anesthetized with isoflurane (1.5% in oxygen). The chest was shaved, and 2-dimensional–guided M-mode echocardiography was performed using a HDI 5000 (Philips Medical Systems) equipped with a 15-MHz linear array transducer to derive left ventricular (LV) functional and dimensional parameters. Long- and short-axis views were obtained as described previously (14).
Statistics
Statistical analysis was performed using Systat Sigmaplot 11. Depending on the existence of equal variances and normal distribution of data, datasets were compared using an unpaired or paired group comparison test or the equivalent nonparametric rank-sum test, respectively. For the comparison of frequencies, the Pearson χ2 test was applied. P values of less than 0.05 were considered statistically significant.
RESULTS
Molecular and Functional Imaging Discovers Multiple Spontaneously Occurring Myocardial Infarctions
As expected, all HFC-fed mice prematurely died within 28 d, whereas no death was observed in the chow group (Fig. 1A).

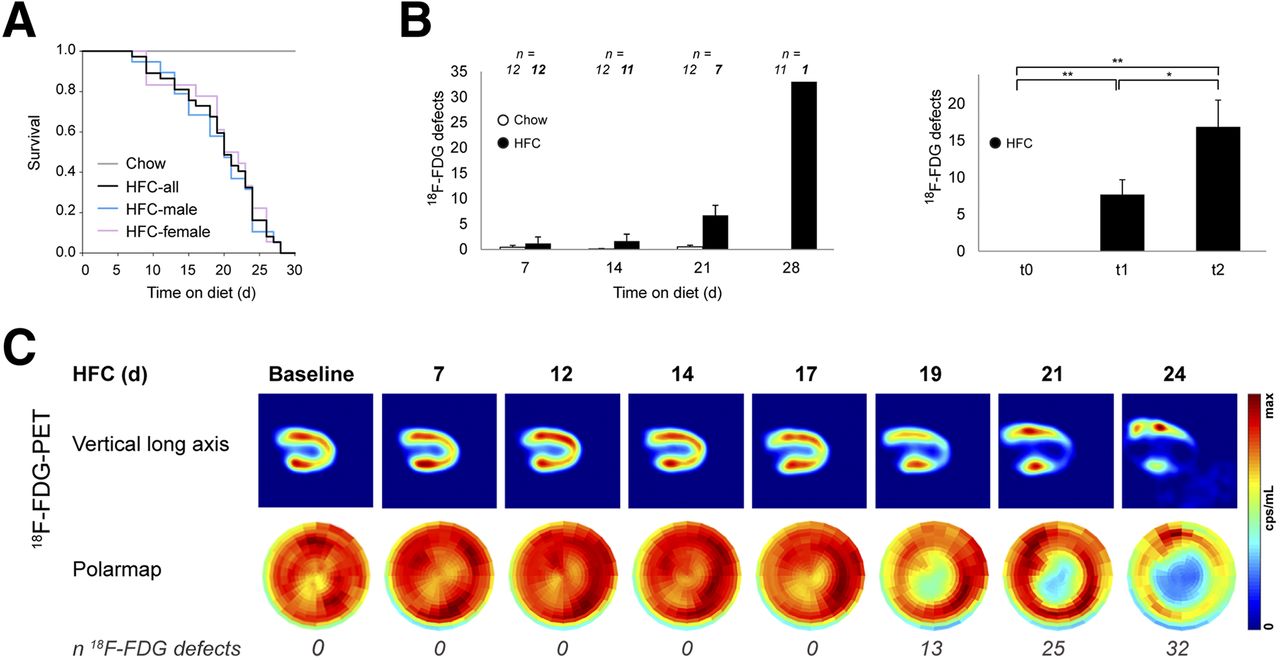
Premature deaths and serial PET imaging in HypoE/SRBI−/− mice under HFC diet. (A) HFC-fed mice (n = 37, 19 male, 18 female) died spontaneously within 28 d whereas all chow-fed mice (n = 12) survived (Mantel–Cox test, P < 0.001). Mortality curves are similar for both genders. (B) Animals were imaged by 18F-FDG PET 3 times a week until spontaneous death (HFC, n = 12) or until day 30 (chow, n = 12). (Left) HFC-fed group showed exponential increase in 18F-FDG defects over time in contrast to chow group. (Right) At baseline, no 18F-FDG defects were detected (t0). First 18F-FDG defects (t1) considerably vary in time (days on HFC-diet: median, 19; range, 7–26) and significantly grew toward last 18F-FDG PET (t2) before death (days on diet: median, 21; range, 7–28). Data are mean + SEM. *P < 0.05, **P < 0.01 by paired t test and Wilcoxon signed-rank test. (C) Sample case showing physiologic uptake of 18F-FDG throughout left ventricle at baseline. PET reveals suddenly occurring 18F-FDG defects indicative of myocardial infarctions in apex and inferoseptal wall. First defect was observed at day 19 of HFC diet, with considerable increase in defect size over time.
Serial 18F-FDG PET found the first myocardial infarctions already 4 d after onset of the HFC diet, with an increase in defect size starting after 2 wk of HFC (Figs. 1B and 1C). In clear contrast, no myocardial infarction occurred in the 12 chow-fed animals (Fig. 1B, left), of which 5 were even followed up until day 60.
In recognition of the broad temporal variability of the occurrence of myocardial infarctions and the time point of individual death, we defined 3 distinct time points for all mice on HFC diet: t0 = baseline, t1 = first 18F-FDG defect, and t2 = final 18F-FDG PET scan before spontaneous death (Supplemental Figure 1; supplemental materials are available at http://jnm.snmjournals.org). The right panel of Figure 1B illustrates that 18F-FDG defect sizes increase toward the time of the last PET scan in HFC-fed mice. Echocardiography showed a significant decrease of ejection fractions by the time of the first 18F-FDG defect, with progressive deterioration in HFC-fed animals, accompanied by an increase of end-systolic volumes. LV myocardial mass increased over time. No changes in ventricular function or myocardial mass were observed in the chow group (Supplemental Fig. 2). In summary, myocardial infarctions occurring in HFC-fed HypoE/SRBI−/− mice gradually led to an impairment of cardiac function.
To study whether coronary plaque ruptures and atherothrombosis underlie myocardial infarctions, a separate group of HypoE/SRBI−/− mice was sacrificed as soon as the first myocardial infarction occurred. Here, characteristic signs of subacute infarctions were found: HFC hearts were bigger and showed inhomogeneous white-colored patches in the LV myocardium especially at the apex as compared with the chow group (Supplemental Fig. 3A). In line with the increase in myocardial mass, relative heart weights of HFC-fed mice were higher than those of chow-fed mice (Supplemental Fig. 3B). However, mean cardiomyocyte diameters were similar between the HFC and chow groups (Supplemental Fig. 3C), suggesting structural changes rather than myocardial hypertrophy causing the increased heart weights in HFC-fed mice. Moreover, areas of myocardial infarction on histologic slices matched the respective 18F-FDG defects in coregistered slices (Fig. 2). Distinct regions of tissue demise, inflammation, and remodeling characterizing postischemic myocardial tissue were identified in situ by immunohistochemistry (Supplemental Fig. 4).

Atherothrombosis induces myocardial infarctions in HFC-fed HypoE/SRBI−/−. HFC-fed mouse heart explanted after 14 d on diet on occurrence of first 18F-FDG defect. (A) Masson Goldner–stained cross section taken from basal region of heart reveals atherothrombosis in septal coronary artery (yellow box) but no sign of myocardial infarction. Corresponding PET image shows normal 18F-FDG uptake. (B) Downstream of intraluminal thrombus, subacute myocardial infarction was identified in the septum characterized by massive postischemic myocardial fibrosis (black arrows) corresponding to septal 18F-FDG PET defect (white arrows). (C) HE-stained serial cross sections of septal coronary artery reveal occluding thrombus associated with perivascular inflammation and inflammatory infiltrates (§). LV = left ventricle; MV = mitral valve; P = papillary muscle; RV = right ventricle.
Imaging-Triggered Histology Reveals Vulnerable Plaque Phenotypes Associated with Coronary Atherothrombosis
Analysis of hearts from HFC-fed mice revealed not only regions of subacute infarctions, but also a clear correspondence of these regions with atherothrombosis in the supplying coronary artery (Fig. 2). We found a striking difference in the number and phenotype of coronary plaques between the HFC-fed and the chow-fed HypoE/SRBI−/− group: in 14 HFC-fed mice enrolled, 296 coronary atherosclerotic plaques were observed (Fig. 3A). This is approximately 10 times more plaques per mouse than in the chow group (34 plaques in 15 hearts, Supplemental Fig. 5).
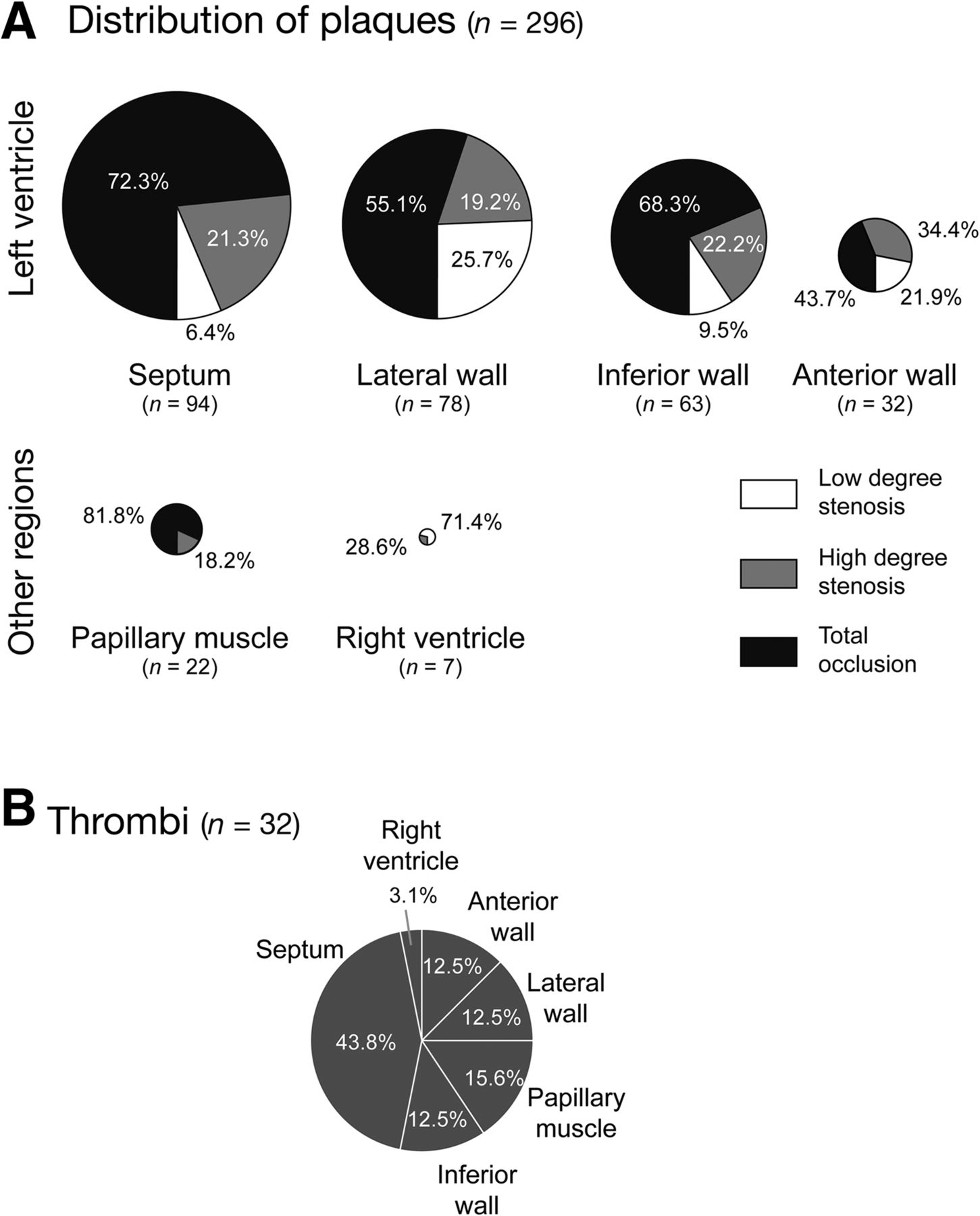
Histologic evaluation of coronary plaques and thrombi in HFC-fed HypoE/SRBI−/−. (A) Degree of stenosis and location of coronary atherosclerotic plaques (low-degree stenosis equals < 50% luminal narrowing, high-degree stenosis equals 50%–99% luminal narrowing, and total occlusion). In HFC-fed mice (n = 14), a total of 296 plaques were found, predominantly in left ventricle and with a high rate of occluded arteries. (B) Thirty-two of 296 plaques (10.8%) showed thrombus formation, with a higher incidence in arteries supplying the septum.
The most striking finding was the occurrence of intraluminal thrombi in 32 (10.8%) of the 296 atherosclerotic plaques in 12 of 14 HFC-fed mice (Fig. 3B). Detailed analysis of the atherosclerotic lesions associated with local atherothrombosis demonstrated a high inflammatory activity, as indicated by local accumulation of macrophages, MPO-expressing cells (e.g., activated neutrophils), and strong expression of the phagocyte activity marker S100A9 (Fig. 4A). Most interestingly, inflammatory activity in atherosclerotic lesions was higher in those associated with thrombus formation than nonthrombotic lesions as indicated by S100A9 (Fig. 4B).

Atherothrombosis in HFC-fed HypoE/SRBI−/− is related to local inflammatory activity. (A) Cross sections of atherosclerotic plaque with thrombotic vessel occlusion. Shoobridge polychrome staining allows for discriminating between cardiomyocytes (red-orange) and connective tissue (blue) but also serves to identify fibrin clots (*). Smooth muscle cells (α-SMA+) in arterial vessel wall demonstrates medium degradation. Immunohistochemistry shows macrophages (Mac-3), neutrophils (MPO), and phagocyte/neutrophil activity (S100A9) reflecting inflammatory activity in the lesion. (B) Inflammatory activity assessed by S100A9 staining was significantly higher in HFC-fed than chow-fed mice and further increased in thrombus-associated plaques (Mann–Whitney rank-sum test).
Intervention with ASA Reduces Incidence of Atherothrombosis and Premature Deaths
To further study the causal relationship between coronary atherothrombosis and myocardial infarctions, we fed HypoE/SRBI−/− mice with HFC and ASA (n = 13, HFC-ASA) as published before (15). Remarkably, all HFC-ASA mice survived the observation period of 28 d (Fig. 5A). Although 18F-FDG PET proved the occurrence of myocardial infarctions, the defects were significantly smaller than in HFC-fed mice and typically occurred later (Fig. 5B).
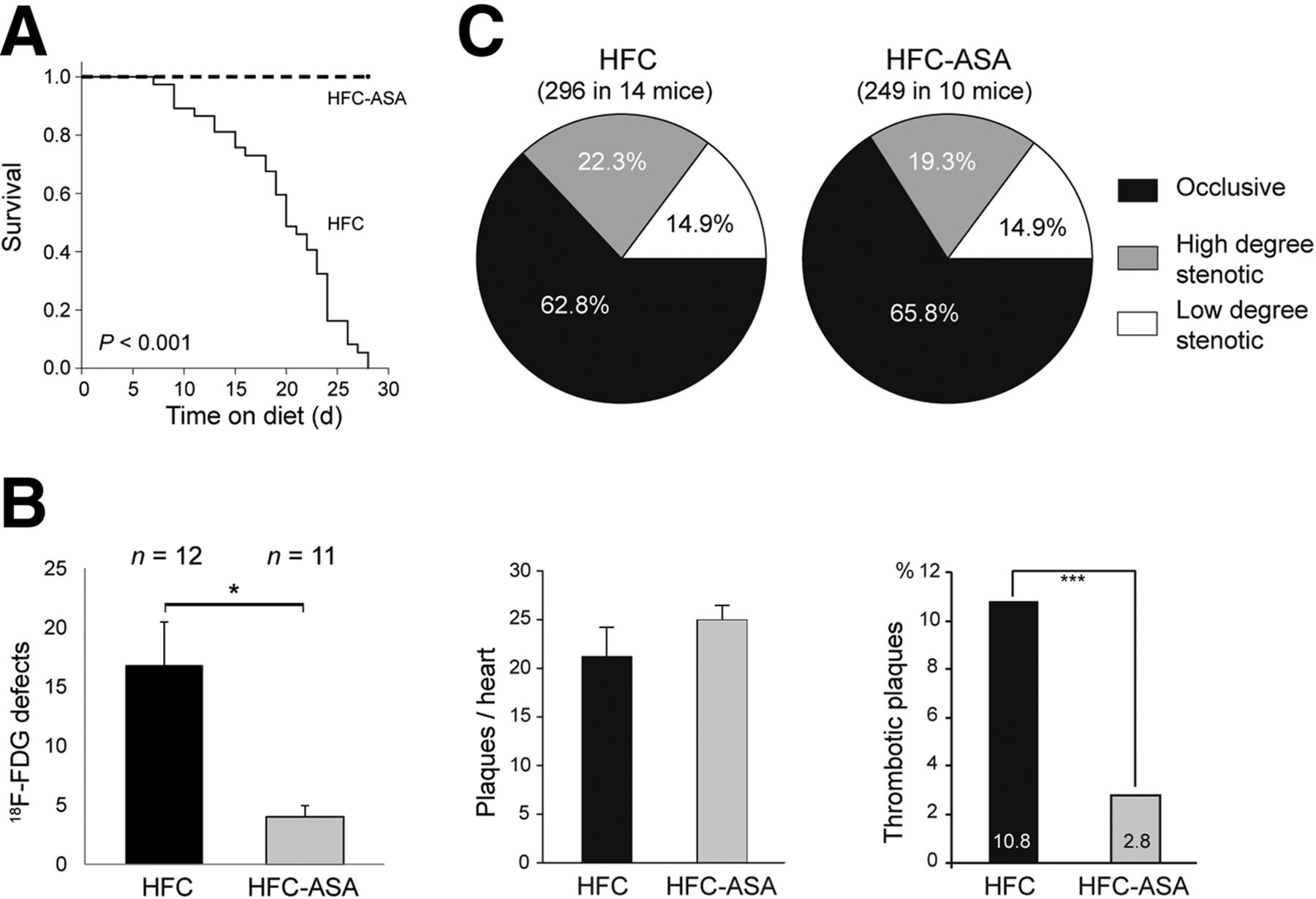
Serial imaging of 18F-FDG defects and plaque analysis in HypoE/SRBI−/− under ASA treatment. HypoE/SRBI−/− mice (n = 11) were put on HFC-ASA diet and imaged by 18F-FDG PET at days 14, 21, and 28 after onset of diet. (A) All mice with ASA treatment survived observation period in contrast to mice under HFC-only diet (n = 37; Mantel–Cox test, P < 0.001). (B) In final scan 18F-FDG defect sizes were significantly larger in HFC-fed HypoE/SRBI−/− (median days on diet, 21 d) than in age-matched ASA-treated HypoE/SRBI−/−. Histologic evaluation of coronary plaques in HFC-ASA–fed mice revealed comparable number of coronary plaques per heart but considerably lower incidence of thrombotic plaques as compared with HFC group. Data are mean + SEM. *P < 0.05, ***P < 0.001 by Mann–Whitney rank-sum test. (C) ASA treatment does not change coronary plaque burden in HFC-fed mice.
Histologic analysis of 10 HFC-ASA mice sacrificed after 29 d on diet showed 249 coronary atherosclerotic plaques, which corresponds well with HFC-fed animals (Fig. 5B). In addition, the localization (Supplemental Fig. 6) and degree of stenosis of plaques (Fig. 5C) in both groups were similar. However, the incidence of plaque-associated intraluminal thrombosis was significantly reduced from 32 in 296 plaques (10.8%) in HFC-fed mice to only 7 in 249 plaques (2.8%) in HFC-ASA mice (Fig. 5B; Supplemental Figs. 6 and 7).
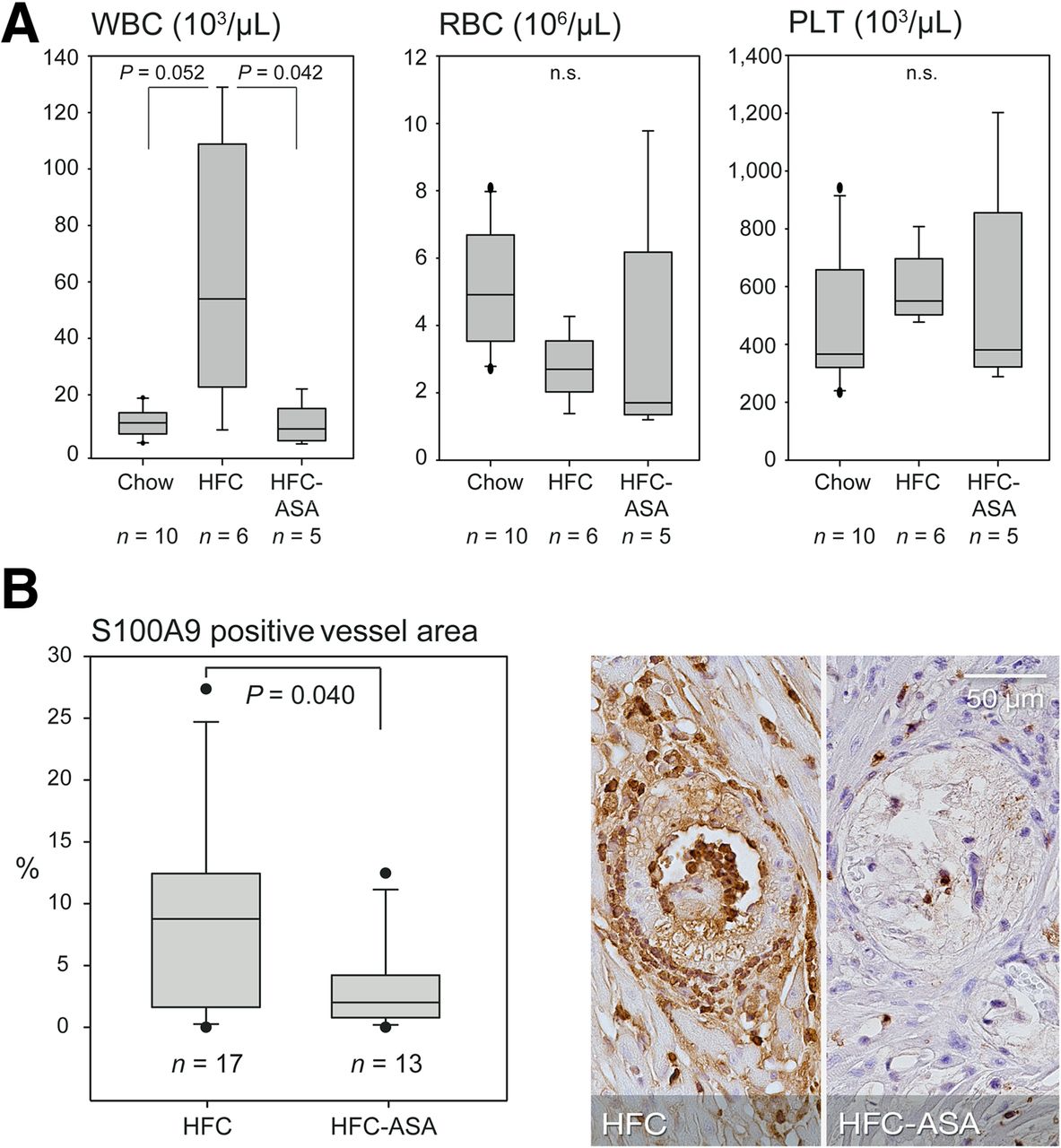
Thrombocyte aggregation assays did not reveal a clear effect of the ASA treatment, although this might be partly due to a low platelet count (median, 550.4 × 103/μL and 381.0 × 103/μL in HFC- and HFC-ASA–fed animals, respectively; Fig. 6A) potentially impairing the assay. In contrast, ASA treatment significantly attenuated the HFC-mediated rise in white blood cells (Fig. 6A), suggesting inhibition of diet-induced systemic inflammatory activity. In addition, S100A9 was significantly reduced locally at sites of atherosclerotic lesions in HFC-ASA mice as compared with HFC mice (Fig. 6B) but not in the postischemic myocardium as assessed by S100A9 and Mac-3 stainings (Supplemental Fig. 8).
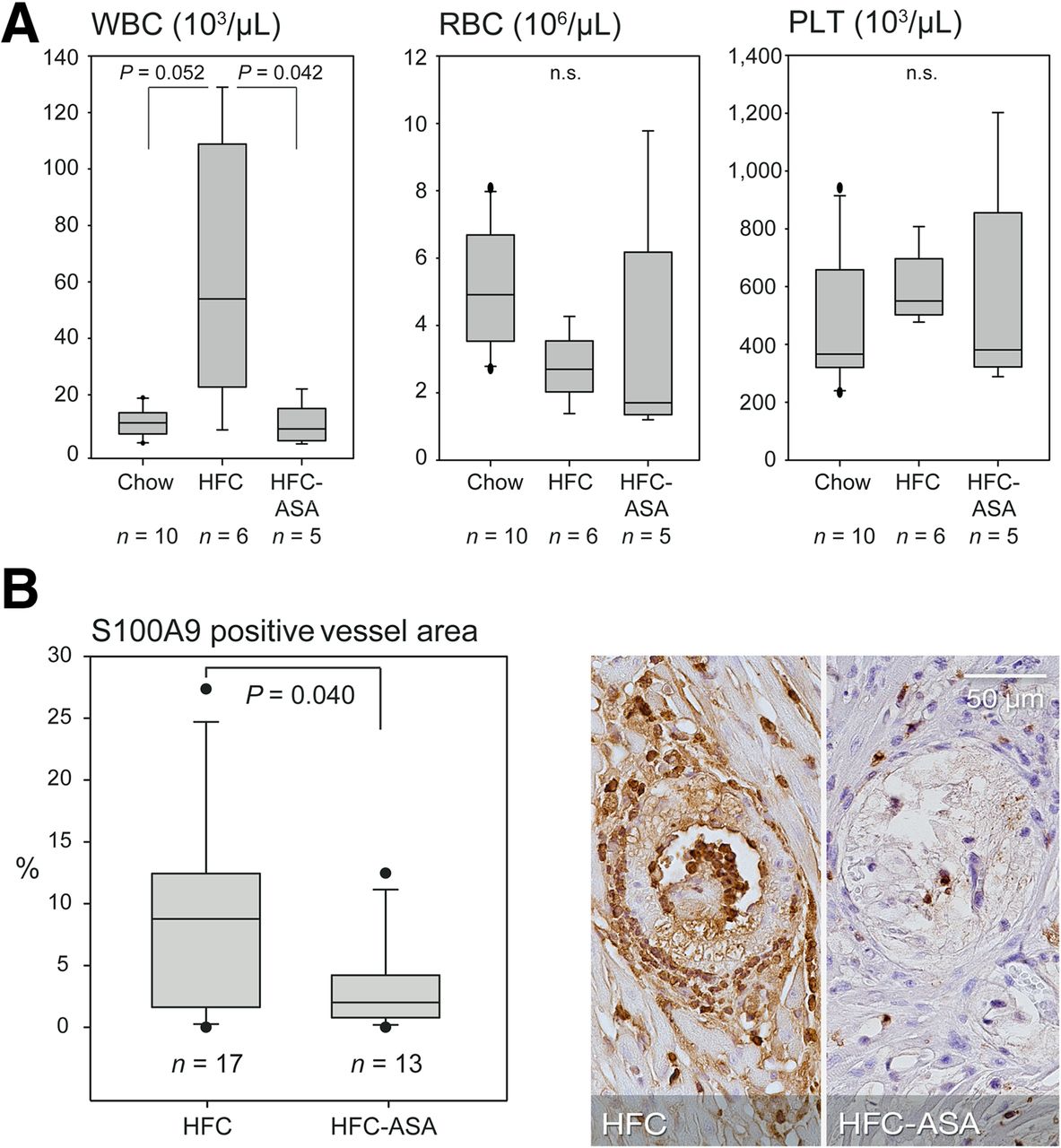
Plaque-associated and systemic inflammatory activity in HFC-fed HypoE/SRBI−/− with and without ASA treatment. (A) HFC induced rise in circulating white blood cells (WBCs) was attenuated by ASA, suggesting systemic antiinflammatory effect whereas concentration of erythrocytes (RBCs) and platelets (PLTs) remained unaffected. (B) S100A9 immunolocalization at site of maximal plaque extend revealed significantly decreased S100A9 expression in septal arteries of ASA-treated animals. P values calculated by Mann–Whitney rank-sum test.
DISCUSSION
Emerging experimental models of atherosclerosis such as the HypoE/SRBI−/− mouse present with diet-induced hyperlipidemia leading to advanced coronary plaque phenotypes, spontaneous myocardial infarction, and sudden death, thus potentially closely mimicking human coronary artery disease (8). We studied here whether ruptures of vulnerable coronary plaques trigger occlusive thrombus formation and cause the observed myocardial infarctions in HFC-fed HypoE/SRBI−/− mice.
In a first step, we used noninvasive imaging to study the temporal and spatial dynamics of spontaneous myocardial infarctions previously described in HypoE/SRBI−/−. Serial 18F-FDG PET demonstrated for the first time in vivo that consecutive myocardial infarctions occur in individual HFC-fed HypoE/SRBI−/− mice with large variations in time point, extent, and fatality. In correlative histologic studies, the typical patterns of recent ischemia with necrosis and immune cell infiltration could be observed side by side with myocardial fibrosis, supporting the notion of myocardial infarctions occurring sequentially in HypoE/SRBI−/− mice. In connection with the functional impairment assessed by echocardiography and the temporal dynamics of mortality, multiple myocardial infarctions are the most likely cause of the spontaneous premature deaths in HFC-fed HypoE/SRBI−/− mice.
However, we used the serial 18F-FDG PET imaging approach not only to assess myocardial infarctions but also to trigger an immediate histologic evaluation of the coronary arteries on the first occurrence of myocardial infarcts. This analysis revealed a connection of myocardial infarctions and atherothrombosis in the supplying coronary arteries. In particular, we found most thrombi in medium and large coronary arteries in the basal LV myocardium. This may also explain the predominantly septal location of the thrombotic coronary arteries because the septal arteries are rather direct proximal branches of the right or left coronary artery or the aortic sinus itself (16). To further study the connection between coronary atherothrombosis, myocardial infarctions, and spontaneous deaths in HFC-fed HypoE/SRBI−/− mice, we treated a subset of animals with ASA, which significantly reduced coronary atherothrombosis (78% compared with nontreated) and premature deaths in the 4-wk treatment interval.
Intraplaque and intraluminal thrombi have been described in larger arteries of ApoE−/− mice on high-cholesterol diets before (3,6). However, in none of these models were thrombotic plaques in coronary arteries reported. In addition, no clinical events such as stroke or myocardial infarction arising from the thrombotic plaque events were observed.
On the basis of these striking differences between the HypoE/SRBI−/− mouse studied here and classic mouse models of atherosclerosis, we thoroughly investigated the plaque phenotype of culprit lesions of myocardial infarctions occurring under an HFC diet. In particular, we looked for vulnerable plaque phenotypes, which are considered a primary cause of plaque rupture events. Hallmarks of vulnerable plaques have been derived mainly from human autoptic studies of sudden death cases. There, typically only 1 culprit plaque has been identified as the primary cause of the fatal atherothrombosis despite multiple other coronary artery plaques (17,18). In our study, HFC-fed HypoE/SRBI−/− mice exhibited several key features of human vulnerable plaques, as recently defined (19): they are found in vessels of larger size and proximal segments of the coronary tree, are lipid- and cholesterol-rich, present with perivascular inflammation, exhibit thrombi communicating with the necrotic core, and most importantly are associated with clinical events—myocardial infarctions or spontaneous deaths.
ASA treatment did not significantly interfere with plaque development in HFC-fed HypoE/SRBI−/−: no difference in the number and extent of coronary plaques between treated and nontreated mice was observed, suggesting no substantial antiatherosclerotic effects of ASA in this mouse model. In agreement with this finding, it was shown that ASA had no effect on the development of atherosclerotic lesions in ApoE−/− mice (6,20). In these studies, in which plaque rupture of carotid artery plaques was either mechanically induced or spontaneously occurring, it was additionally demonstrated that ASA directly reduced atherothrombosis by its antiplatelet action. This finding could not be confirmed in our study, most probably due to technical issues.
Other effects exerted by ASA might contribute to the reduction of thrombotic events in HypoE/SRBI−/−. Cyrus et al. reported that low-dose ASA treatment in low-density lipoprotein receptor–deficient under a high-fat diet suppressed vascular inflammation (15). This observation is in line with our finding of reduced systemic and local inflammatory activity under ASA treatment. Considering that inflammation is among the main drivers for plaque vulnerability, suppression of inflammatory activity in HypoE/SRBI−/− mice by ASA might induce plaque stabilization and therefore might result in fewer plaque ruptures and thrombotic events.
Given the humanlike nature of spontaneous plaque rupture and myocardial infarctions, the HypoE/SRBI−/− mouse should also provide an excellent basis for studying pharmaceutical interventions. Recently, immunosuppression by the sphingosine-1 phosphate analog FTY720 has been shown to improve LV function in HypoE/SRBI−/− put on an HFC diet for only 3 wk (21). In a recent publication, the same group used HypoE/SRBI−/− with an inducible Cre-mediated gene repair of the HypoE allele together with switching mice to a normal chow diet. A subgroup of mice surviving the HFC diet was subsequently given orally FTY720 in drinking water and compared with nontreated mice with respect to LV function up to 15 wk. As expected, untreated mice showed a progressive impairment of LV function over time. In FTY720-treated mice, initially LV function also dropped but was almost completely restored by 15 wk. As in our ASA-treatment intervention, FTY720 did not reduce atherosclerosis but resulted in systemic immunosuppression and reduced cardiac inflammation (22). Further studies should investigate the immunosuppressive effect of ASA accordingly. Other clinically approved pharmaceutical interventions such as treatment with statins, which is known to reduce cardiovascular events in humans (23), are warranted to further evaluate the humanlike nature of the HypoE/SRBI−/− model. If these are successful, the HypoE/SRBI−/− model could be used for testing emerging pharmaceutical interventions in a preclinical setting.
Ruptures of inflammatory plaques might not be solely responsible for the formation of intravascular thrombi in HypoE/SRBI−/−. Liu et al. reported that the incidence of atherothrombosis in their ApoE−/−-based model of vulnerable plaque formation was highly dependent on the concomitant increase in blood thrombogenicity introduced by adenovirus-mediated prothrombin overexpression (6). SRBI deficiency has been previously shown to alter platelet function, resulting in a higher susceptibility for thrombosis and therefore further contributing to atherothrombotic events in HFC-fed HypoE/SRBI−/− (24). The cholate fraction of the HFC diet, which was reported to be hepatotoxic and alter lipid metabolism (25), is rather unlikely to substantially promote atherothrombosis in HypoE/SRBI−/−. Nakagawa-Toyama et al. recently reported that myocardial infarctions, cardiomegaly, and premature death can be induced in HypoE/SRBI−/− mice using the HFC diet even without cholate (9).
Although the concept of plaque ruptures of vulnerable plaques underlying clinical events such as myocardial infarctions in humans is well accepted, the hypothesis of a straightforward single and first plaque rupture in a patient leading to the clinical event has been recently debated. More likely and in accordance with the notion, that atherosclerosis is a systemic vascular disease, multiple plaque ruptures could occur at various vessel sites in individuals, followed by healing of the vessel wall. These multiple plaque ruptures might be associated with subclinical, maybe unspecific symptoms while only under certain circumstances, which still have to be finally studied, a plaque rupture would result in a clinical event (2). Interestingly, the HypoE/SRBI−/− mouse would support the multiple-plaque-rupture hypothesis: using serial 18F-FDG PET imaging, we detected multiple infarctions in individual HFC-fed HypoE/SRBI−/− mice, which were well tolerated in most cases, therefore possibly resembling subclinical events in humans. Only a few myocardial infarctions led to the clinical event of sudden death.
Studies in humans suggest that atherosclerotic plaque development and rupture may be sex-related (26). The investigation of sex effects in this study was severely hampered by the tedious and inefficient breeding of HypoE/SRBI−/− mice that finally resulted in only small groups of animals available for the various experiments. However, mortality in the largest group of HFC-fed HypoE/SRBI−/− in our study, which had comparable numbers of male and female mice, did not show a difference in the clinical endpoint mortality, suggesting no differences in plaque vulnerability. Future studies in larger cohorts of HypoE/SRBI−/− are warranted to finally assess whether sex effects on plaque phenotypes and vulnerability exist.
CONCLUSION
The HypoE/SRBI−/− mouse model mimics a broad range of characteristic features of human coronary artery disease encompassing the development of advanced/vulnerable plaques, plaque ruptures, coronary atherothrombosis, and myocardial infarction. In contrast to other models and previous studies, we discovered here that there is a causal connection between plaque ruptures and myocardial infarctions, and most interestingly, multiple subclinical events may occur before it comes to sudden death. Thus, the HypoE/SRBI−/− mouse might have a great potential as a preclinical animal model for developing new diagnostic and therapeutic strategies toward the prevention of plaque rupture and myocardial infarctions.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This work was partly supported by the Deutsche Forschungsgemeinschaft, Collaborative Research Center SFB 656 ‘Cardiovascular Molecular Imaging’, projects A6, C3, C6, and Z2; the Interdisciplinary Center for Clinical Research (IZKF, core unit PIX) Münster, Germany; a research grant from Siemens Medical Solution, Erlangen, Germany; and a fellowship of the NRW research school “Cell Dynamics and Disease – CEDAD.” No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We gratefully acknowledge Monty Krieger, Massachusetts Institute of Technology, Cambridge, Massachusetts, for providing mice for breeding of HypoE/SRBI−/−, providing excellent scientific discussions, and sharing unpublished results on the HypoE/SRBI−/− mice. HypoE/SRBI−/− mice were also provided by J. David Gladstone Institutes, San Francisco, California. We acknowledge the scientific support of Augusto Martins Lima, Silke Niemann, and Jerzy-Roch Nofer and the technical assistance of Irmgard Hoppe, Christa Möllmann, Christine Bätza, and Sandra Höppner.
Footnotes
↵* Contributed equally to this work.
Published online Apr. 28, 2016.
- © 2016 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication December 16, 2015.
- Accepted for publication March 21, 2016.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.