


Abstract
Protein tyrosine kinase-7 (PTK7), a member of receptor tyrosine kinase superfamily initially identified as colon carcinoma kinase-4, is highly expressed in various human malignancies. Its expression was found to correlate with aggressive biologic behaviors such as increased cell proliferation, invasiveness, and migration. Despite the importance and unmet need of imaging PTK7 in vivo, there is currently no clinically relevant method to visualize tumoral PTK7 expression noninvasively such as PET or SPECT. This study aimed to develop a specific, selective, and high-affinity PET radioligand based on single-stranded DNA aptamer to address this challenge. Methods: Sgc8, a 41-oligonucleotide that targets to PTK7, was labeled with 18F using a 2-step radiochemical synthesis, which featured a direct 1-step radiofluorination on the distinctive spirocyclic hypervalent iodine(III) precursor to give 18F-fluorobenzyl azide followed by copper-mediated click conjugation with Sgc8-alkyne. 18F-Sgc8 was evaluated in vitro and in vivo in 2 cell lines, HCT116 and U87MG, which express high and low amounts of PTK7, respectively. Results: Sgc8 was labeled efficiently with 18F in an isolated radiochemical yield of 62% ± 2%, non–decay-corrected based on 18F-fluorobenzyl azide. 18F-Tr-Sgc8 was found to possess high-affinity binding to both cell lines, with binding affinity values of 2.7 ± 0.6 nM for HCT116 and 16.9 ± 2.1 nM for U87MG. In vivo PET imaging clearly visualized PTK7 expression in HCT116 xenografted mice, with tumor uptake of 0.76 ± 0.09 percentage injected dose per gram (%ID/g) at 30 min after injection for the subcutaneous tumor model and greater than 1.5 %ID/g for the liver metastasis model. U87MG xenograft tumors had much lower tracer accumulation (0.13 ± 0.06 %ID/g at 30 min after injection), which was consistent with the lower expression of PTK7 in this tumor model. The labeled aptamer was rapidly cleared from the blood through the kidneys and bladder to give high tumor-to-blood and tumor-to-muscle ratios of 7.29 ± 1.51 and 10.25 ± 2.08, respectively. Conclusion: The 18F-radiolabeling methodology shown here is a robust procedure for labeling aptamers and similar chemical moieties and can be applied to many different targets. Quantification of PTK7 using 18F-Tr-Sgc8 may be suitable for clinical translation and might help in the future to select and monitor appropriate therapies.
Protein tyrosine kinase-7 (PTK7), a member of receptor tyrosine kinase superfamily initially identified as colon carcinoma kinase-4, is highly expressed in various human malignancies (1,2). Encoding fragments of PTK7 were initially identified in messenger RNA from human melanocytes, and subsequently PTK7 was fully cloned from a colon cancer tissue (3,4). The protein consists of 7 immunoglobulinlike extracellular domains, a single transmembrane domain, and cytoplasmic domain with tyrosine kinase homology that lacks vital catalytic activity but maintains signal transduction (4–7).
Deletion of the PTK7 gene in mice is lethal, and the embryos developed many defects in the gastrulating cell process. Subsequently, it was found that the protein PTK7 is conserved through evolution and plays a pivotal role in cell movement and localization during embryonic development (5,7–9). Endogenous ligands of PTK7 have not yet been identified.
As indicated above, PTK7 was cloned from colon cancer, although not expressed in normal colon. Further research showed that it is also expressed by other cancers including gastric cancer, acute myeloid leukemia, lung cancer, and glioblastoma overexpressing CD44 (6,10–14). The role of PTK7 in cancer is controversial, perhaps because it might play different roles in the biology of cancers that arise from different tissues. Interestingly, whereas in some cancers PTK7 is upregulated and believed to promote tube formation, migration, angiogenesis, and invasiveness of endothelial cells (8), PTK7 is found to be downregulated in other cancers or during advanced stages of the cancer (15–18). Moreover, in lung squamous cell carcinoma PTK7 was suggested to be a tumor suppressor, and when it was transfected into lung cancer cell lines it reduced cell proliferation, invasion, and migration (18). Hence, PTK7 may be a pharmaceutical target in some cancers; however, its role in each specific targeted cancer requires exploration.
One of the limitations in studying PTK7 is the absence of a method for evaluating and quantifying the expression of PTK7 protein in cancer tissue noninvasively over time. Currently there is no radioactive tracer that will allow such studies. To address this need, we developed a PET tracer that targets PTK7, based on a known aptamer sequence named Sgc8 (19,20).
Aptamers are short single-stranded DNA or RNA oligonucleotides that possess high selectivity and specificity to a target molecule. The specific binding of aptamers to their targets depends on a distinctive and unique 3-dimensional folding, and they display high affinities that are similar to antibodies. In contrast to antibodies, the relatively small molecular weight of aptamers allows rapid penetration into tissues and fast clearance from the blood to give a higher target-to-background ratio at early time points. Therefore, aptamers are attractive candidates for in vivo PET imaging (21,22).
Sgc8 aptamer contains 41 oligonucleotides and was selected by cell-based systematic evolution of ligands by exponential enrichment (SELEX) against human T lymphoblast CCRF-CEM cells (19,20). The target of the Sgc8 aptamer was deduced as PTK7 using biotin-Sgc8 and magnetic streptavidin beads for isolation of biomarker complexes and using liquid chromatography-mass spectrometry (LC-MS) to identify the complexed protein. Here we describe radiolabeling of Sgc8 with the PET isotope 18F via a click chemistry reaction and evaluation of 18F-Tr-Sgc8 ex vivo and in vivo for stability and ability to image and quantify PTK7 expression in different mouse tumor models.
MATERIALS AND METHODS
General
The supplemental materials (available at http://jnm.snmjournals.org) describe the procedures for radiolabeling of 18F-fluorobenzyl azide, assays of PTK7 expression, cell-binding procedures, and MR imaging.
Radiochemistry
Sgc8-alkyne aptamer (150 μg in 15 μL of water) was mixed in an Eppendorf tube with 0.7 mg of CuSO4·5H2O in 20 μL of water and 5.6 mg of sodium ascorbate in 200 μL 0.1 M borate buffer (pH 8.6). Then, 50 μL of 18F-benzylazide (185–222 MBq [5–6 mCi]) in CH3CN were added, followed by a short vortex and incubation at 37°C for 15 min. The crude reaction mixture was loaded onto a NAP5 column and eluted with 0.5-mL fractions of H2O. The radiochemical yield of 18F-Tr-Sgc8 was calculated from the combined radioactivity of fractions 4 and 5 divided by the initial quantity of 18F-fluorobenzyl azide without decay correction. Radiosynthesis of 18F-fluorobenzyl azide and quality control of 18F-Tr-Sgc8 are described in the supplemental materials.
Biology
Tumor Xenograft Models
Mice were inoculated subcutaneously with 5 × 106 cells of either HCT116 or U87MG on the right shoulder (n = 5 per group). The tumors were allowed to develop for 2.5–3 wk before PET imaging and biodistribution studies.
Tumor Metastasis Model
The mice were anesthetized with isoflurane/O2 (1.5%–2% v/v) inhalation. For surgical hepatic injection, a small (0.5–1 cm) incision was made in the skin above the liver. The peritoneal membrane was gently lifted, and a small incision was made, avoiding any injury to the liver. Then 2 μL of 1 × 105 HCT116 cells premixed with Matrigel (BD Biosciences) at a 1:1 ratio were injected into the liver of each mouse (n = 4) using a 10-μL syringe (Hamilton) fitted with a 31-gauge needle. After injection of the cells, the syringe was held for 1–2 min, and then withdrawn from the tissue. The abdominal wall was sutured using a 6.0 Vicryl absorbable suture (Ethicon). The tumors were allowed to develop for 4–5 wk before PET studies.
PET and Biodistribution Studies
Tumor-bearing mice were injected intraperitoneally with 0.5 mL of saline 10 min before the intravenous injection of 3.7 MBq (100 μCi) of 18F-Tr-Sgc8 (8 μg, 0.6 nmol) to facilitate urine clearance. For blocking studies, 3.7 MBq (100 μCi) of 18F-Tr-Sgc8 were coinjected with a 100-fold excess (800–850 μg, 60–65 nmol) of unlabeled Sgc8-alkyne (n = 3). Five- to 10-min PET scans were obtained using an Inveon scanner (Siemens Medical Solutions) at 30 min and 1 h after injection. ASI Pro VM software (Siemens Medical Solutions) was used for image analysis. Regions of interest were drawn for each organ on the coronal images to calculate percentage injected dose per gram of tissue (%ID/g), assuming a density of 1 g/cm3 for all tissues. After the 1-h PET scan, the mice were sacrificed; organs were harvested and wet-weighed. The radioactivity of samples was measured using a γ counter (Wallac Wizard 1480; PerkinElmer), and the results were expressed as %ID/g.
18F-Tr-Sgc8 Stability in Blood and Urine
To study the stability in blood, 3.7–5.5 MBq (100–150 μCi) of 18F-Tr-Sgc8 were injected into each mouse. At 5 min after injection, the mice were sacrificed and blood was collected in tubes containing ethylenediamine tetraacetic acid. The plasma was separated from red blood cells by centrifuging at 3,500 rpm for 5 min. The plasma was incubated for 5 min at 95°C, followed by centrifugation at 14,000 rpm for 5 min.
An aliquot of 20–30 μL of each sample was injected into a high-performance liquid chromatograph, and 1-min fractions were collected for 30 min using a fraction collector. The radioactivity of each fraction was measured using a γ counter. Urine was collected at 30-min and 1-h time points. Procedures describing in vitro stability studies of 18F-Tr-Sgc8 are included in the supplemental materials.
Statistical Analysis
Results are presented as mean ± SD. Group comparisons were made using the Student t test for unpaired data. P values of less than 0.05 were considered statistically significant.
RESULTS
Chemistry and Radiochemistry
Sgc8 aptamer, consisting of 41 oligonucleotides, was modified at the 5′ with a terminal hexynyl group (Fig. 1). Conjugation of unlabeled Sgc8-alkyne to a 1.5 equivalent of fluorobenzyl azide in the presence of an excess amount of CuSO4·5H2O and sodium ascorbate resulted in full conversion into the desired product (Supplemental Fig. 1), which was verified by LC-MS analysis (Supplemental Fig. 2). The automated radiochemical synthesis of 18F-fluorobenzyl azide was achieved using an aromatic fluoride substitution on a novel spirocyclic hypervalent iodine(III) precursor (23) (1, Fig. 1). The labeling yield was 32% ± 3% (n = 5), non–decay-corrected based on 18F activity. 18F-fluorobenzyl azide was obtained with a high specific activity of 129.5–148 Gbq/μmol (3.5–4 Ci/μmol, n = 5). Because of its volatility, 18F-fluorobenzyl azide cannot be concentrated via solvent evaporation and, therefore, was used directly from the CH3CN elution of the final C-18 Sep-Pak (Waters). For aptamer conjugation, 50 μL of 18F-fluorobenzyl azide were used, and attempts to increase the volume (and activity) resulted in lower labeling efficiency (Supplemental Table 1).
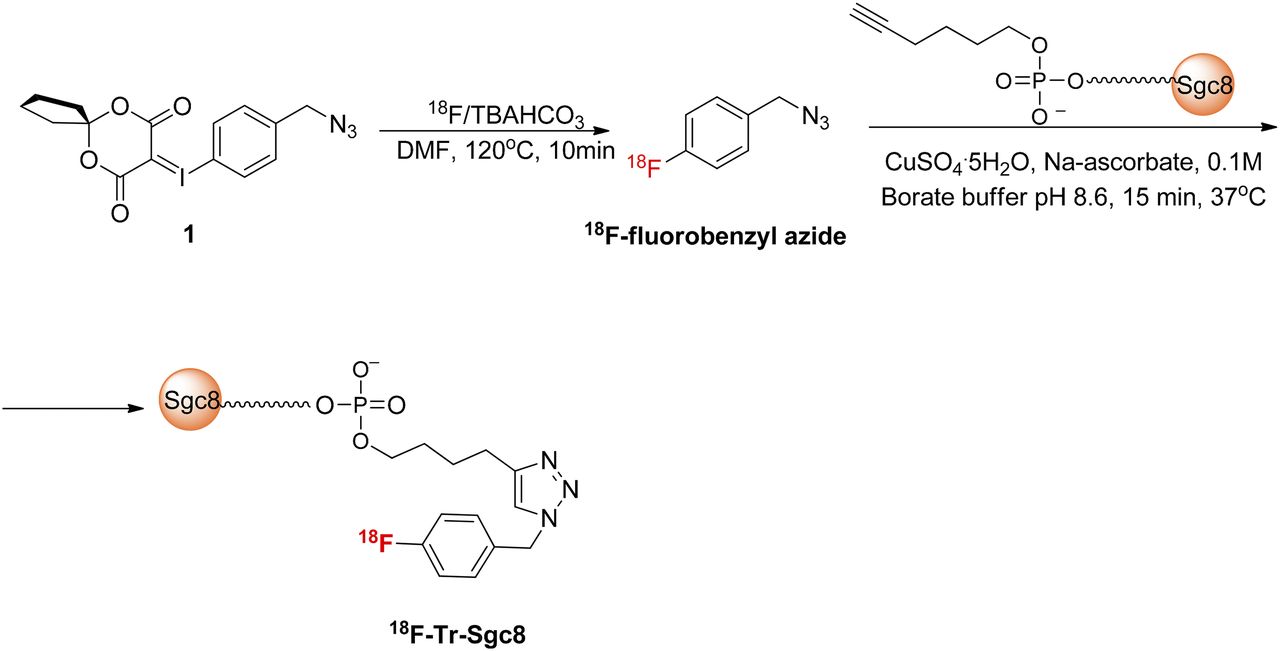
Radiosynthesis of 18F-Tr-Sgc8.
Reaction of Sgc8-alkyne with 18F-fluorobenzyl azide was conducted as described for the unlabeled fluorobenzyl azide and similarly resulted in formation of one major and desired product. The high-specific-activity 18F-fluorobenzyl azide was all consumed (Supplemental Fig. 3). Typically, 150 μg (11 nmol) of Sgc8-alkyne were used for radiolabeling. Attempts to minimize the amount of the aptamer to 50 μg (3.8 nmol) per reaction resulted in the same conversion (Supplemental Table 1). However, using 13 μg (1 nmol) of aptamer resulted in only 48% conversion into the desired labeled aptamer (Supplemental Table 1). The crude reaction was purified on a NAP5 column to give 18F-Tr-Sgc8 with a radiochemical purity of greater than 95% and radiochemical yield of 62% ± 2% non–decay-corrected (n = 5) based on initial 18F-fluorobenzyl azide radioactivity. The final specific activity of the labeled aptamer, calculated by dividing the obtained radioactivity by the total mass of Sgc8-alkyne used for the reaction, was 6.7–7.3 GBq/μmol (182–198 mCi/μmol).
Biology
PTK7 Expression by HCT116 and U87MG Cells
Flow cytometry and Western blot analysis showed higher expression by HCT116 than by U87MG cells (Figs. 2A and 2B). 18F-Tr-Sgc8 binding affinity to HCT116 was slightly better (2.7 ± 0.6 nM, n = 2) than U87MG (16.9 ± 2.1 nM, n = 2, Fig. 2C). The amount of 18F-Tr-Sgc8 captured on each cell line also correlated with expression levels of PTK7 (Fig. 2C). Western blot analysis of proteins from tumor lysates showed that HCT116 retained PKT7 expression in vivo, whereas the protein level was almost undetected in U87MG tumor lysate (Fig. 2D).
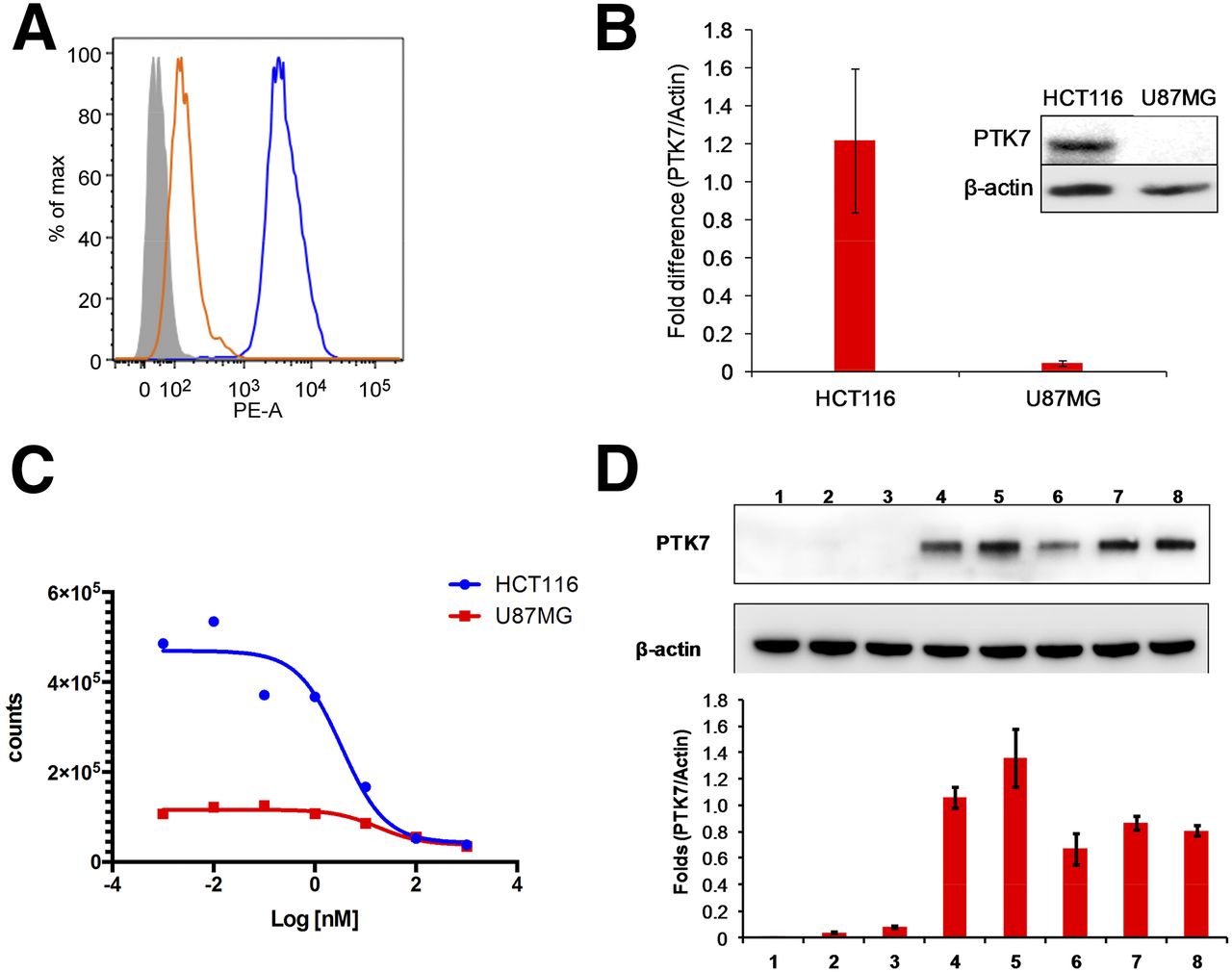
(A) Flow cytometry of PTK7 expression results for HCT116 and U87MG cell lines. (B) Western blot analysis of PTK7 and β-actin expression by cells. (C) Binding affinities of 18F-Tr-Sgc8 in HCT116 and U87MG cells. (D) Representative Western blot analysis of PTK7 and β-actin expression by liver (1), intestine (2), U87MG tumor (3), HCT116 peritoneal metastasis (4), subcutaneous HCT116 tumor (5), HCT116 kidney metastasis (6), HCT116 liver tumor (7), and HCT116 peritoneal wall metastasis (8) (2 upper panels) and quantitative results of PTK7/β-actin ratio from 3 Western blots (lower panel). PE-A = phycoerythrin absorbance.
PTK7 Imaging in Tumor-Bearing Mice
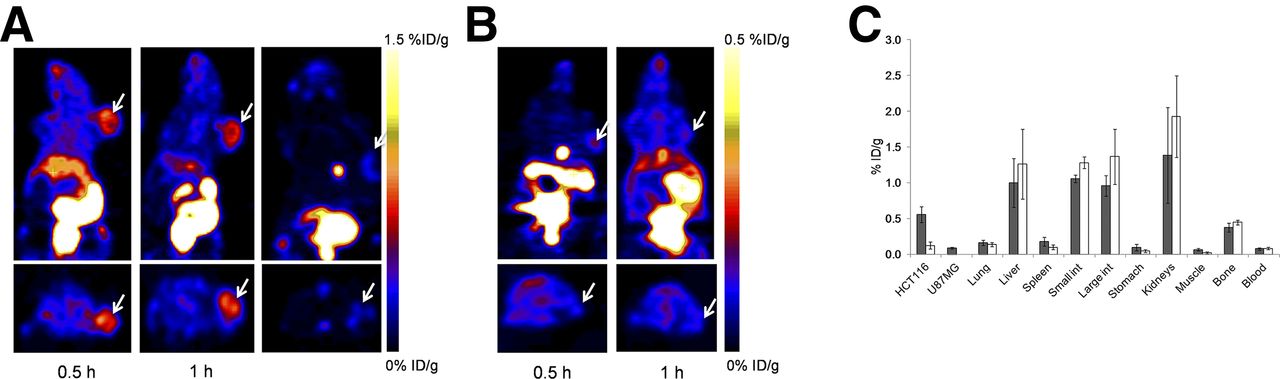
18F-Tr-Sgc8 uptake at all the time points examined was significantly higher in HCT116 than U87MG xenografts (0.76 ± 0.09 vs. 0.13 ± 0.06 %ID/g at 30 min after injection and 0.54 ± 0.12 vs. 0.19 ± 0.06 %ID/g at 1 h after injection, Figs. 3A and 3B). 18F-Tr-Sgc8 uptake in the HCT116 tumors slightly decreased between 30 min and 1 h after injection. The receptor specificity of 18F-Tr-Sgc8 to PTK7 was tested by blocking studies in the HCT116 tumor model. By coinjection of 18F-Tr-Sgc8 with an excess amount of Sgc8-alkyne, the uptake was decreased by approximately 80% (Fig. 3A). Moreover, 18F-Tr-Sgc8 had a faster clearance through the urine when injected with an excess of cold mass. Although 18F-Tr-Sgc8 had an undesired nonspecific uptake in metabolic organs, including gallbladder, intestine, and kidneys (Fig. 3C), 18F-Tr-Sgc8 showed fast clearance from the blood and the optimal imaging time was 1 h after injection with higher tumor contrast and tumor-to-blood and tumor-to-muscle ratios of 7.29 ± 1.51 and 10.25 ± 2.08, respectively. 18F-Tr-Sgc8 had some uptake in the bone (0.37 ± 0.06 %ID/g at 1 h after injection), which was not reduced in the coinjection experiments, suggesting the possibility of slight defluorination in vivo.

(A) Representative coronal (upper) and transaxial (lower) PET images of mice bearing HCT116 xenografts injected with 18F-Tr-Sgc8 at 30 min (right), 1 h (middle), and 1-h coinjection with excess amount of unlabeled aptamer (left). (B) Representative coronal (upper) and transaxial (lower) PET images of mice bearing U87MG tumors at 30 min and 1 h after injection of 18F-Tr-Sgc8 (3.7 MBq [100 μCi]). (C) Biodistribution of 18F-Tr-Sgc8 at 1 h after injection without addition of unlabeled aptamer (gray columns) and with addition of unlabeled aptamer (white columns). Arrows indicate tumor location.
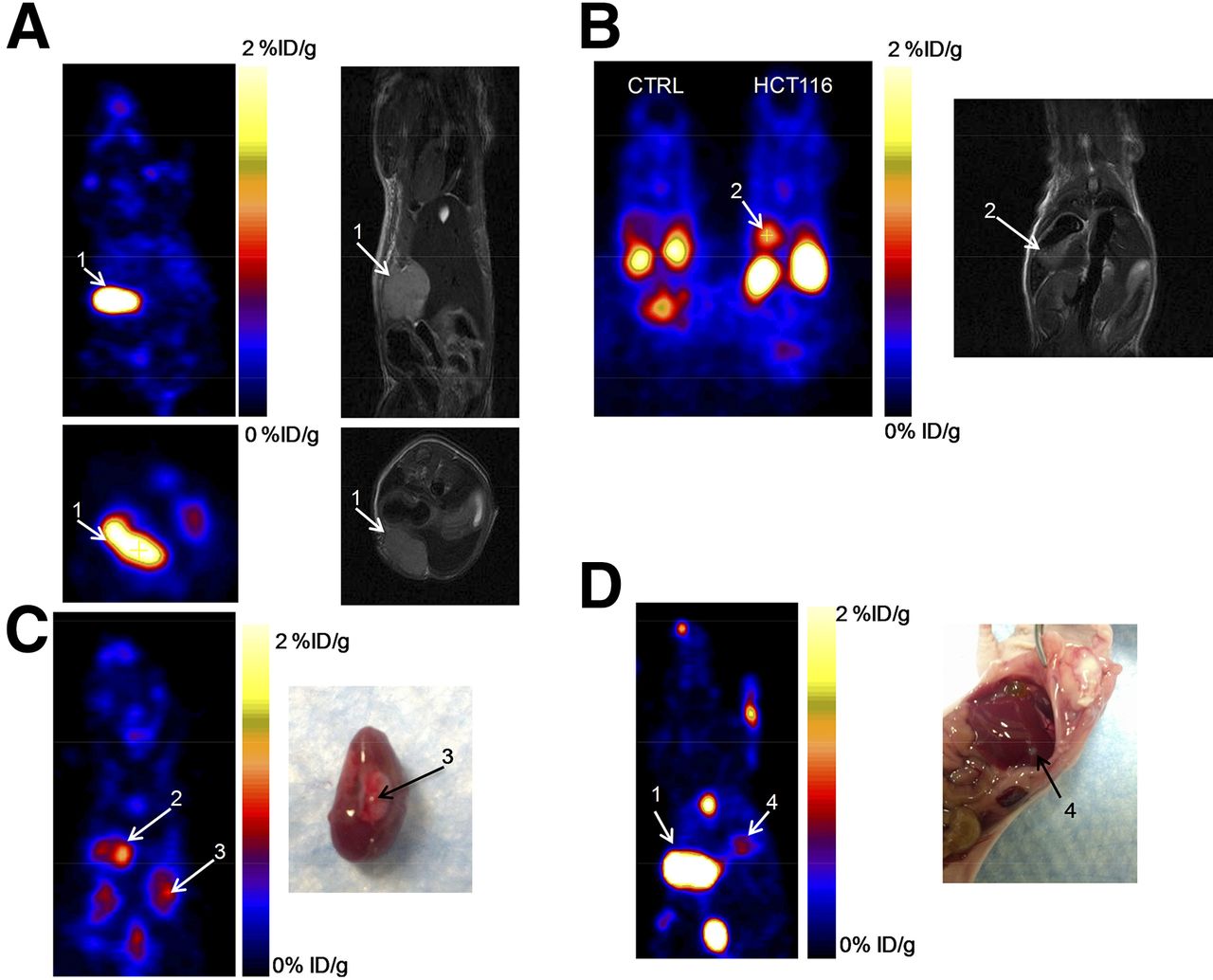
The ability of 18F-Tr-Sgc8 to image tumors in the liver, kidneys, and peritoneal sites was also evaluated (Fig. 4) using a metastatic tumor model. Inoculation of HCT116 cells into the liver resulted in tumors not only in the liver, but also in the kidneys and peritoneum, as confirmed by MR imaging and necropsy. Peritoneal tumors might have formed from some spill of cells from the injection site. The uptake of 18F-Tr-Sgc8 in metastatic lesions in the peritoneum and the kidneys (∼1.5–2 %ID/g at 1 h after injection; Fig. 4; Supplemental Fig. 4) was higher than that of subcutaneous tumors. Liver lesions had an uptake of 0.5–0.7 %ID/g and were small (16–20 mg), possibly because of differential blood flow and vascularity in subcutaneous tumors versus abdominal/liver tumors.

(A and B) Representative PET and MR images of mice with metastatic HCT116 tumors at 1 h after injection of 18F-Tr-Sgc8. (C and D) PET image and necropsy of indicated tumors. Arrows and numbers indicate different tumors shown in PET image and matching MR imaging/necropsy. CTRL = control.
Ex Vivo and In Vivo Stability of 18F-Tr-Sgc8
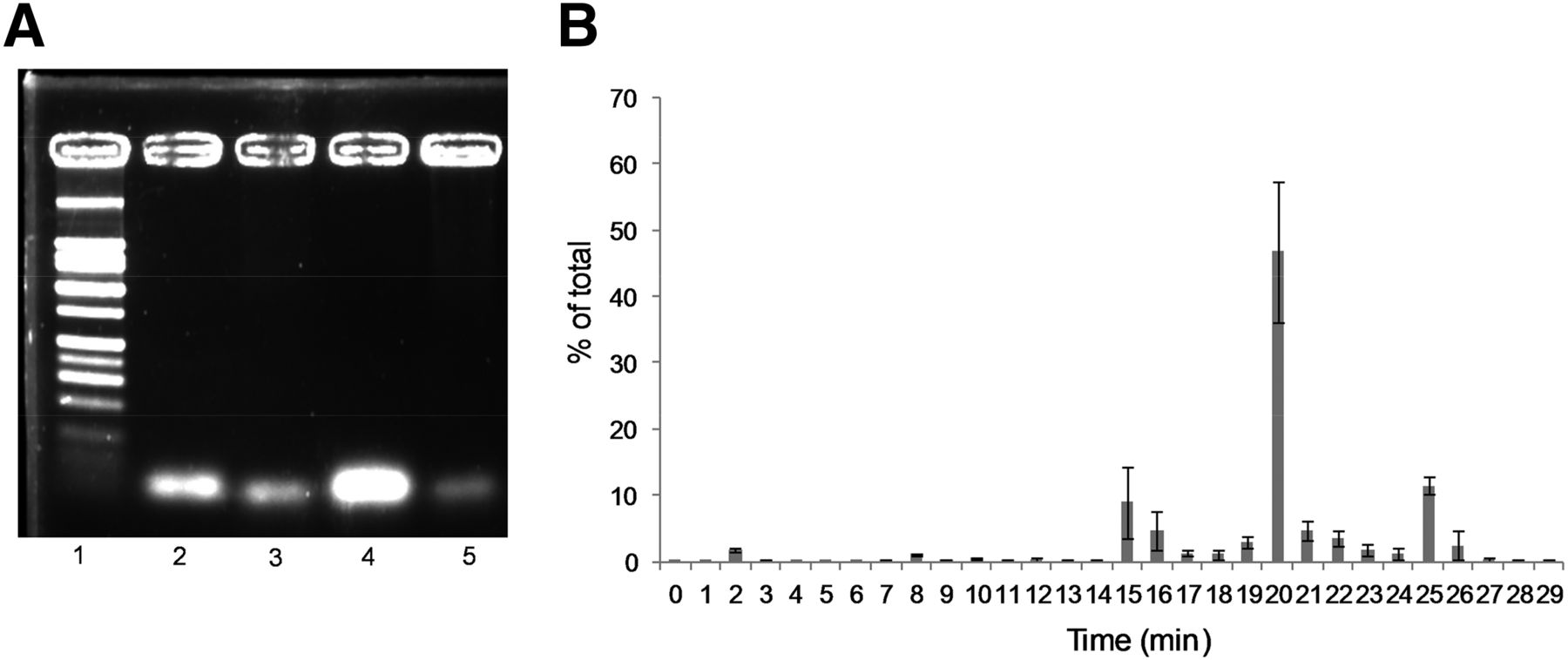
18F-Tr-Sgc8 stability was initially evaluated ex vivo in mouse serum. An aliquot of the serum was taken at 30 min and 1 h after incubation, and the radioactivity was diluted using saline. The aptamer readily bound to serum components, and thus most of the radioactivity remained in the pellet. Denaturation of the proteins at 95°C increased aptamer recovery but remained low with 80% of 18F-Tr-Sgc8 in the pellet. To evaluate the formation of small molecules from aptamer degradation, the mixture of 18F-Tr-Sgc8 in serum was loaded onto a NAP5 column and the activity of different fractions was measured. Most of the activity (>80%) came off the column in fraction 4, suggesting that the aptamer was intact but did not exclude its binding to proteins. Gel electrophoretic analysis showed that no significant amount of metabolites was formed in the serum up to 1 h after incubation (Fig. 5A). Activity extracted from the 30-min time point was injected into the high-performance liquid chromatograph (Supplemental Fig. 5A); no degradation of the labeled aptamer was observed. 18F-Tr-Sgc8 stability was further evaluated in vivo in the blood and urine. Because 18F-Tr-Sgc8 rapidly clears from the blood, the blood was withdrawn at 5 min after injection. An aliquot of the extracted plasma was injected into the high-performance liquid chromatograph, and the major component in this aliquot eluted at 19.7 min, 2 min later than the retention time of 18F-Tr-Sgc8 (Fig. 5B; Supplemental Fig. 5B). Moreover, urine samples that were taken 30 min and 1 h after injection showed no traces of 18F-Tr-Sgc8 but the appearance of 2 new peaks—one was more hydrophilic, with a retention time of 15.2 min, and the other was more hydrophobic, with a retention time of 24.9 min (Supplemental Fig. 5C). It is important to emphasize that traces of unlabeled aptamer could be detected in the urine samples, according to the ultraviolet chromatogram (Supplemental Fig. 5C).
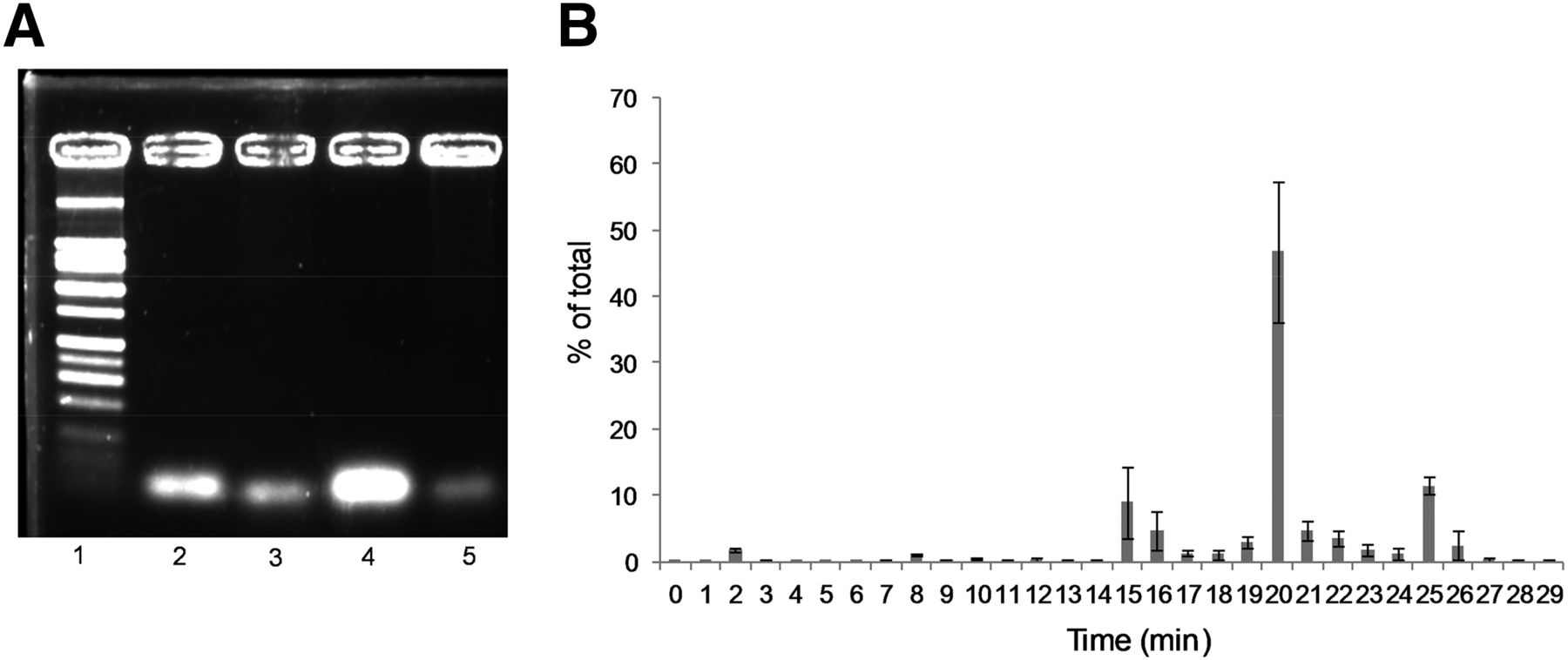
(A) Representative agarose gel electrophoresis of 18F-Tr-Sgc8 after mouse serum incubation and extraction as described in “Materials and Methods” section. Lane 1 = ladder; lane 2 = aptamer standard; lane 3 = 30-min incubation; lane 4 = 1-h incubation after NAP5; lane 5 = 1-h incubation. (B) In vivo blood stability analysis of 18F-Tr-Sgc8 by γ counter.
DISCUSSION
PTK7 expression was shown to have controversial roles in different tumors and seems to be dependent on several conditions, which are in part tissue-specific. Hence, the study of clinical significance and function of PTK7 in cancers is an ongoing research effort. However, it is at least partially hampered by the lack of a reliable method to evaluate PTK7 expression noninvasively in whole tumors (12,24). An example of the above-mentioned controversy is a study done in adenocarcinoma patients that suggests that PTK7 expression predicts a favorable prognosis (13). On the other hand, evaluation of acute myeloid leukemia patients showed that PTK7-positive patients had higher resistance to anthracycline-based cancer chemotherapy and a significantly reduced rate of relapse-free survival (25). The methods for determination of PTK7 expression by tumors and tumor cells are limited to immunohistochemistry and polymerase chain reaction, which are conducted on biopsy samples. A method to evaluate the receptor expression in the whole tumor noninvasively could be a great asset to study the significance and functions for PTK7 in a wide variety of cancers. In this study, we address this unmet need through the development of a PET tracer for PTK7 based on a DNA aptamer.
We have previously published the labeling of tenascin-C aptamer with 18F via N-succinimidyl 4-18F-fluorobenzoate (18F-SFB) (26), which resulted in a low radiochemical yield. In an effort to improve radiochemical yields for this study, we applied the click reaction between 18F-fluorobenzylazide and Sgc8-alkyne. The radiosynthesis of 18F-fluorobenzylazide prepared via spirocyclic iodonium ylide and the subsequent click reaction with Sgc8 proved to be robust and reliable and resulted in a high radiochemical yield. Although usage of 11 nmol of Sgc8-alkyne for the click reaction provided complete conversion of 18F-fluorobenyzlazide, a small amount of Sgc8-alkyne (as little as 1 nmol) also resulted in nearly 50% conversion into the labeled aptamer (Supplemental Table 1), which is obviously better than 18F-SFB conjugation yields realized for tenascin-C aptamer. Our preliminary in vivo PET studies showed suboptimal images when a small mass of aptamer was injected into the mice, whereas higher mass amounts resulted in better images (Supplemental Fig. 6), possibly because of the rapid clearance of the radioactive aptamer through the gallbladder and kidneys. We believe that an increase in injected mass slowed the clearance or metabolism just enough to allow higher uptake in target tissues. Indeed, our optimized conditions used 11 nmol of Sgc8-alkyne, and purification of radiolabeled product from unreacted precursor was not necessary. This phenomenon of lower uptake in the gallbladder coupled with higher uptake in the target organ/tumor might be beneficial to other tracers. However, our experience is limited to this aptamer, and further studies on this phenomenon should be performed with other tracers. If this phenomenon is seen with other tracers, then tracer development studies should also include experiments to optimize the unlabeled-to-labeled tracer ratio, to circumvent gallbladder accumulation and improve target uptake. In our standard radiolabeling method, 18F-Tr-Sgc8 was injected with 5–8 μg of unreacted Sgc8-alkyne per mouse.
PET studies showed specific accumulation of 18F-Tr-Sgc8 in HCT116 tumor–bearing mice (Figs. 3A and 3C) that express a high level of PTK7 (Fig. 2D). U87MG tumors express a lower level of PTK7 (Fig. 2D) and had relatively lower uptake of 18F-Tr-Sgc8 (Fig. 3B). The specificity of the uptake was demonstrated by an 80% inhibition of uptake on coinjection of 60 nmol of Sgc8-alkyne. Moreover, images of 18F-Tr-Sgc8 uptake were obtained after inoculation of HCT116 cells into the liver (Fig. 4). Although there was relatively high background due to uptake in the gallbladder, intestine, and kidneys, the tumors in the liver, peritoneum, and above the kidneys were clearly visualized with good tumor-to-background ratios (Fig. 4; Supplemental Fig. 4; Supplemental Video 1). The uptake in the intestine, which was higher than that in the tumor, suggests that 18F-Tr-Sgc8 is not suitable for imaging PTK7 expression by colon tumors. Lower uptake seen in the subcutaneous tumors of HCT116 when compared with the tumor lesions elsewhere might be due to different blood flow and not attributed to different receptor expression (Fig. 2D).
To gain some insight into the biologic clearance mechanism, we compared the biodistribution of 18F-Tr-Sgc8 with 18F-SFB–labeled Sgc8. Radiolabeling of Sgc8 with 18F-SFB required acquisition of Sgc8 with a primary amine at the 5′ end. Formation of the benzyl triazole ring during the click reaction makes 18F-Tr-Sgc8 more hydrophobic than aptamer conjugated to 18F-SFB. 18F-Tr-Sgc8 had considerable uptake in the intestine (Fig. 3C) and also showed some bone uptake (0.37 ± 0.06 %ID/g), which suggests metabolic defluorination (Fig. 3C) or, perhaps, formation of 18F-clicked phosphate metabolite, which accumulates in the bone. When Sgc8 was labeled with 18F-SFB, the bone uptake was much lower (0.11 ± 0.02 %ID/g at 1 h after injection) (Supplemental Fig. 7). A more detailed study of the effect of different chemical linkers on defluorination may be required.
18F-Tr-Sgc8 stability was further tested in vitro in mouse serum and in vivo in blood and urine. 18F-Tr-Sgc8 proved to be stable in mouse serum, with no significant metabolite formation (Fig. 5A). About 25%–30% of the total injected radioactivity was collected from the urine at 30 min after injection, which indicated a fast clearance of 18F-Tr-Sgc8 through the kidneys. Radio–high-performance liquid chromatography (HPLC) showed 2 small-molecule-based components but no parent 18F-Tr-Sgc8. These products were probably caused by the metabolism of the phosphodiester bond between the aptamer and the prosthetic group (Supplemental Fig. 5C). Typically, large molecules such as aptamers or proteins are eluted in fraction 4 (0.75–1.0 mL). However, we observed that most of the radioactivity remained on the column and could elute only with extensive washing, which is indicative of small molecules. One would be an alcohol, which is consistent with the more hydrophobic peak seen in HPLC at 24.9 min (Supplemental Fig. 5C), and the other would contain a phosphate group and peaked at 15.4 min (Supplemental Fig. 5C). On analysis by LC-MS, we observed not only unreacted Sgc8 but also 1 large molecule with a deconvoluted mass of 18 kDa, which is 6 kDa higher than the unreacted Sgc8 (Supplemental Fig. 8). This observation supports our hypothesis that the aptamer or its fragments bind to some blood component (lipids/small proteins) in vivo.
Although we observed some degradation of the 18F-Tr-Sgc8 or the unreacted Sgc8, it does not hamper the use of 18F-Tr-Sgc8 as a PET imaging tracer in the visualization and quantification of PTK7 expression of the tumors in vivo, evidenced by the rapid clearance of 18F-Tr-Sgc8 from the blood via the kidneys into urine and excellent target-to-background signal ratios.
CONCLUSION
18F-labeling methodology that uses 4-18F-fluorobenzyl azide is efficient for labeling aptamers and other biomolecules. 18F-Tr-Sgc8 was specific and sensitive to the PTK7 level as shown by the higher uptake in HCT116 tumors with high expression of PTK7 compared with low uptake in U87MG tumors with low expression of the protein. Quantification of PTK7 using 18F-Sgc8 will be valuable for future studies of PTK7 expression and its different roles in various cancers. It is expected that this tracer will be suitable for clinical translation and help select and monitor appropriate therapies that are PTK7-related.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This work was supported by the intramural research program of the National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH). No other potential conflict of interest relevant to this article was reported.
Footnotes
↵* These authors contributed equally.
Published online Aug. 27, 2015.
- © 2015 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication May 18, 2015.
- Accepted for publication August 12, 2015.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}