


Abstract
The field of molecular imaging has experienced significant advances in the area of Alzheimer disease (AD), the most significant being the development of PET radiotracers for imaging β-amyloid burden in the brain of individuals at risk for or in the early stages of AD. More recent advances include the development of PET radiotracers for imaging aggregates of hyperphosphorylated tau protein in neurofibrillary tangles, a process that occurs late in the disease process. This article highlights advances in the neurobiology of AD and describes how PET can be used to study the mechanisms of neurodegeneration in AD.
The accumulation of insoluble protein aggregates is a hallmark of neurodegenerative disorders. The most prominent example of this is Alzheimer disease (AD), which is characterized by the formation of 2 different insoluble protein aggregates, β-amyloid plaques, which consist of aggregated β-amyloid protein (Aβ1–42), and neurofibrillary tangles (NFTs), which consist of aggregates of hyperphosphorylated tau protein. For decades, the diagnosis of AD relied on the cognitive assessment of patients exhibiting moderate to severe memory deficits. Progressive cognitive decline resulting in severe memory impairment was consistent with Alzheimer-like dementia, but the clinical diagnosis of probable AD was not confirmed until postmortem analysis demonstrated the presence of β-amyloid plaques and NFTs in the brain. In 2004, a breakthrough in the clinical evaluation of AD emerged with the development of 11C-Pittsburgh compound B (PiB), an analog of thioflavin-T, the fluorescent dye used to visualize Aβ plaques in postmortem samples of AD brain. PET studies have clearly shown that 11C-PiB–positive β-amyloid plaques occur early in the disease process, and amyloid plaques likely represent a preclinical or antecedent biomarker of AD (1,2). The emergence of 18F-labeled β-amyloid imaging agents has enabled PET studies on AD patients at imaging centers without an on-site cyclotron facility. More recently, 18F- and 11C-labeled tau-specific agents for imaging NFTs have been developed (3–6). These agents should provide valuable information on the temporal separation between the formation of Aβ plaques (antecedent biomarker) and NFTs (currently thought to parallel neuronal loss in AD) (1). This ability to measure the pathologic time course of Aβ plaque and NFT formation in AD patients will be critical in the evaluation of disease-modifying therapeutics aimed at slowing the progression of this disease. Therefore, PET radiotracers for imaging Aβ plaques and NFTs will be useful in both the diagnosis and the clinical management of patients at risk for developing AD (1,2).
Although much has been gained in the understanding of the temporal progression of Aβ plaque formation, there is still a significant gap in knowledge of the molecular mechanisms responsible for neurodegeneration leading to severe cognitive impairment in AD. Research in transgenic mouse models of AD, postmortem samples of AD brain, and genetics studies on patients with familial AD have provided insight into possible disease mechanisms. However, these mechanisms must be confirmed in AD patients before they can be widely accepted as providing the neurochemical basis of AD. The development of PET radiotracers capable of studying these processes in cognitively normal age-matched controls and AD patients, and their alteration over time, are clearly needed to fully understand the molecular basis of AD. This review will focus on aspects of the amyloid hypothesis of AD that require further study, including the mechanism of Aβ peptide accumulation in brain, the disruption of neuronal function by Aβ peptide leading to cell death in the central nervous system, and the role of neuroinflammation as either a neuroprotective mechanism or a mediator of neurotoxicity in AD.
MECHANISMS OF ACCUMULATION OF Aβ PEPTIDE IN THE CENTRAL NERVOUS SYSTEM
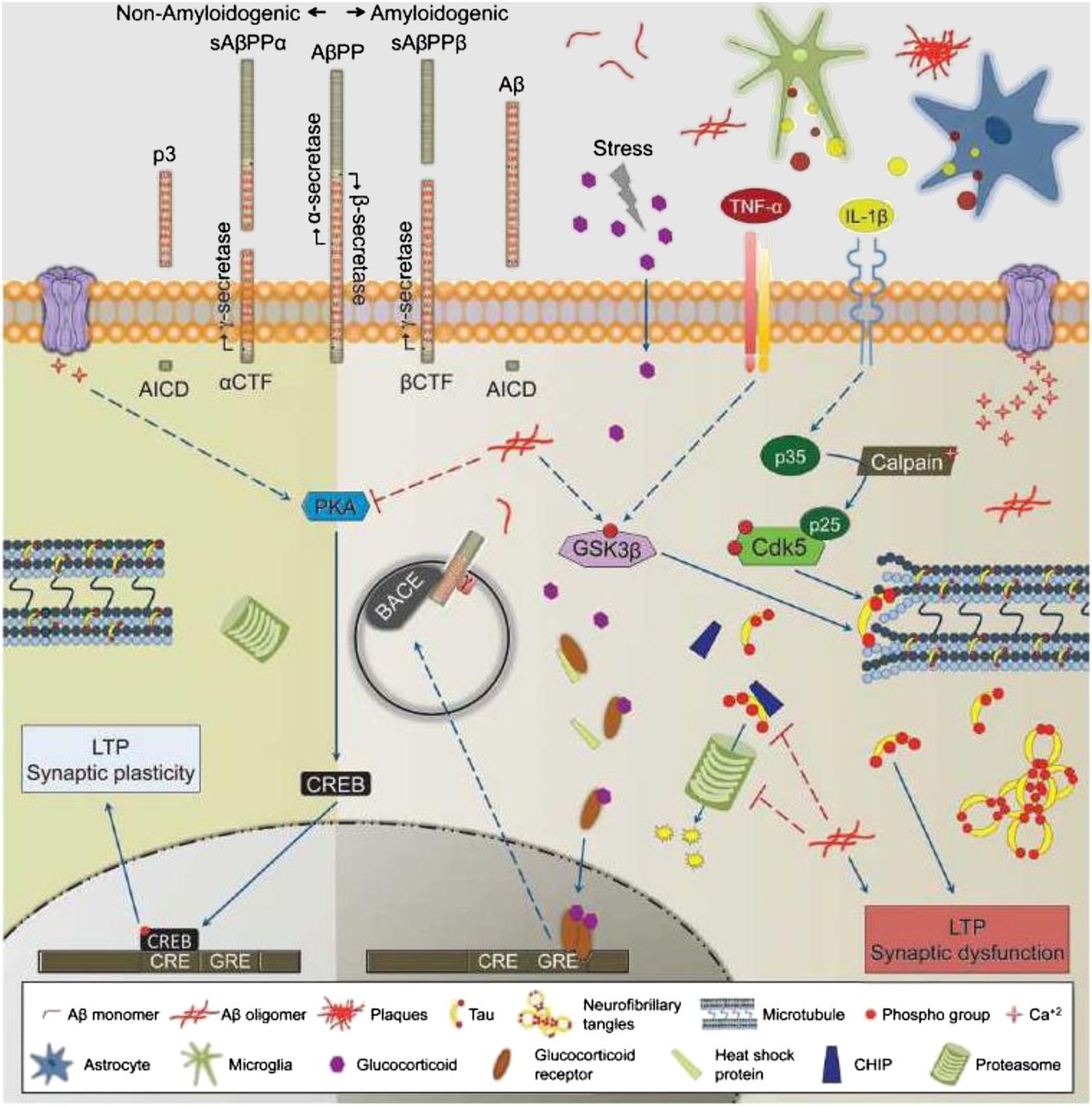
Aβ peptide is formed through the cleavage of a larger, transmembrane-spanning protein named amyloid precursor protein (APP). Although the role of APP in the central nervous system is poorly understood, it has been well established that there are 2 pathways for cleaving this peptide into smaller fragments: amyloidogenic and nonamyloidogenic pathways (Fig. 1) (7). The nonamyloidogenic pathway involves cleavage by the enzyme α secretase; this enzyme cleaves APP within the amyloidogenic Aβ1–42 sequence, resulting in the formation of a soluble species. The amyloidogenic pathway involves the action of 2 enzymes: β secretase, which cleaves APP into a soluble fragment and a larger, membrane-bound fragment called C99, and γ secretase, which cleaves C99 into Aβ peptide and a smaller, membrane-bound fragment called C59. γ-secretase consists of a complex of 4 transmembrane proteins: presenilin, nicastrin, Pen2, and Aph1 (8). The release of Aβ peptides, both amyloidogenic and nonamyloidogenic, is thought to depend on neuronal activity (2). Once released into the extracellular space, Aβ peptides are cleared by one or more transport mechanisms at the level of the blood–brain barrier (BBB), the most prominent being P-glycoprotein (9).
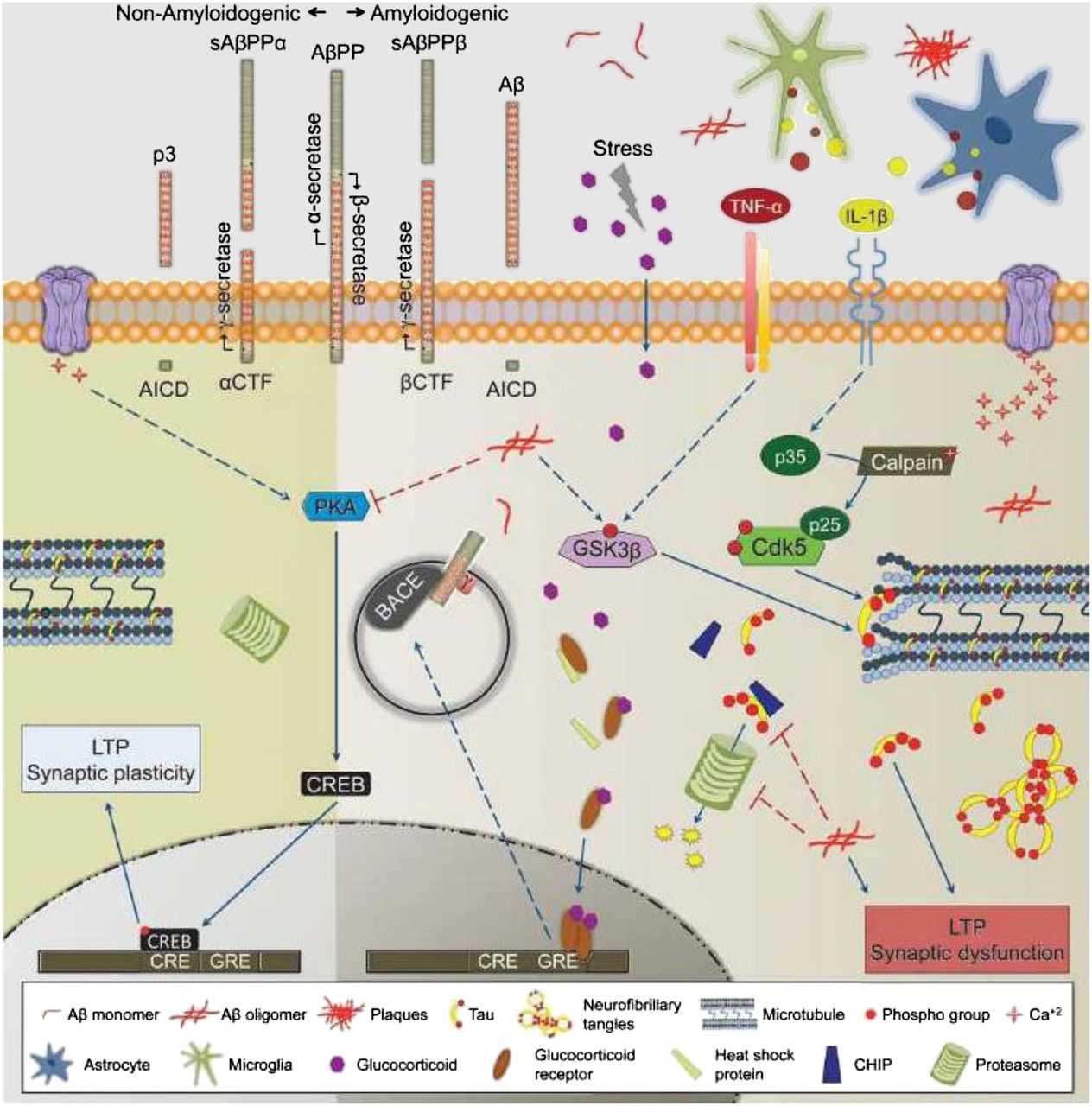
Molecular mechanism leading to formation of Aβ1–42 and subsequent formation of hyperphosphorylated tau. AβPP = Aβ precursor protein; AICD = amyloid precursor protein intracellular domain; BACE = β-site APP cleaving enzyme; Cdk5 = cyclin-dependent kinase 5; CHIP = C-terminus of heat-shock protein 70–interacting protein; CRE = cyclic adenosine monophosphate (cAMP) response element; CREB = cAMP response element-binding protein; CTF = C-terminal fragment; GRE = glucocorticoid response element; GSK3β = glycogen synthase kinase 3β; IL = interleukin; LTP = long-term potentiation; p3 = 3-kDa β-amyloid peptide; sAβPPα = soluble Aβ precursor protein alpha; sAβPPβ = soluble Aβ precursor protein beta; TNF-α = tumor necrosis factor α. (Reprinted with permission of (7).)
There is strong evidence suggesting that AD is caused by a shift from nonamyloidogenic to amyloidogenic cleavage of APP. The main support for this hypothesis is familial or early-onset AD. Familial AD is characterized by mutations in APP and presenilin (the catalytic subunit of γ secretase), which result in an increase in the formation of Aβ1–42 peptide (1,7). Other mutations causing a reduction in α-secretase activity lead to elevated Aβ1−42 formation by increasing cleavage of APP via the amyloidogenic pathway. Since it represents the first step in the amyloidogenic pathway, β-secretase has been a primary target for drug development, and many small-molecule β-secretase inhibitors are undergoing evaluation in clinical trials (8). These inhibitors could serve as lead compounds for PET radiotracer development for studying the role of β-secretase activity in AD.
Another source of increased amyloid burden in AD patients may be reduced clearance of Aβ peptide from the brain. For example, a strong risk factor for sporadic AD is the e4 allele encoding apolipoprotein E4. A recent study on transgenic mice demonstrated that animals homozygous for this allele have a slower clearance of Aβ peptide than mice carrying other apolipoprotein E genotypes (10). Three BBB-related transport proteins for Aβ peptide have been identified: lipoprotein receptor protein, receptor for advanced glycation end products (RAGE), and P-glycoprotein (9). Lipoprotein receptor protein is thought to remove Aβ peptide from the brain, whereas RAGE transports Aβ peptide from the circulation to the brain. P-glycoprotein is a 170-kDa protein that belongs to the adenosine triphosphate–cassette transporters. In the BBB, it is expressed at the luminal side of the endothelial cells and removes substances from the brain to the vascular compartment. Recent studies have shown that there is a decrease in P-glycoprotein in postmortem samples of AD brain relative to age-matched controls (11). The loss of P-glycoprotein was not uniform in the brain of AD patients and matched Aβ plaque density; that is, brain regions having the greatest loss of P-glycoprotein also had the highest plaque density. Imaging studies with R-11C-verapamil, a substrate for P-glycoprotein, showed an increase in binding potential in AD patients versus controls, which is consistent with a reduction in P-glycoprotein in AD patients (12). The regional uptake of R-11C-verapamil matched that of 11C-PiB, but there was no correlation between the binding potentials of the 2 radiotracers. This was attributed to a ceiling effect in the 11C-PiB studies, since 11C-PiB binding does not dramatically increase over the course of AD. This study indicates that loss of P-glycoprotein activity may contribute to increased amyloid burden in AD patients. It is of interest to see if other Aβ peptide transport mechanisms are similarly affected in AD patients and if there is a difference in Aβ transporter function in individuals carrying the e4 allele of apolipoprotein E.
NEUROTOXICITY OF Aβ PEPTIDE
11C-PiB studies on aged people have revealed that up to approximately 33% of cognitively normal older adults are PiB-positive (2). Furthermore, 11C-PiB studies on noncarrier relatives of individuals with familial AD have shown that β-amyloid plaques appear up to 20 y before the onset of cognitive impairment (1,2). These studies strongly suggest that β-amyloid plaques are not neurotoxic and that another species, possibly Aβ oligomers, represents the toxic form of Aβ peptide. One study supporting this theory compared both β-amyloid plaque and Aβ oligomer levels in postmortem brain samples of control aged (clinical dementia rating 0) and early AD (clinical dementia rating 1) patients (13). The authors found no difference in Aβ plaque density between the 2 groups; however, a higher Aβ oligomer–to–plaque ratio was found in the early AD brain samples than in the clinical dementia rating 0 samples. These data suggest that an oligomeric form of Aβ peptide plays a role in initial cognitive impairment in AD and may very well trigger neurotoxicity by a currently unknown mechanism.
Aβ plaques are extracellular, whereas NFTs are formed inside neurons. An unresolved question in AD is the molecular mechanisms leading to NFT formation and neuronal loss and then severe cognitive impairment. Several studies have identified putative receptors that are capable of binding Aβ peptide/oligomers, including α7 nicotinic receptors, N-methyl-d-aspartate receptors, tumor necrosis factor receptor 1, cellular prion protein, and, more recently, epidermal growth factor receptor, ephrin type B receptor 2, and leukocyte immunoglobulinlike receptor B2 (14–16). Finally, lipoprotein receptor protein, the transport protein on endothelial cells of the BBB described above, is also expressed on neurons and microglia and is thought to lower extracellular Aβ levels via receptor-mediated endocytosis (17). Binding of Aβ oligomers to membrane-bound receptors either disrupts receptor-mediated signaling pathways (e.g., N-methyl-d-aspartate receptors and Ca2+ signaling) or results in Aβ internalization via receptor-mediated endocytosis. Alternatively, intracellular Aβ peptide may be produced via endosomal processing of APP by β secretase (7) (β-site APP cleaving enzyme intracellular pathway in Fig. 1). Internalized Aβ peptide is thought to interact with a host of targets, including disruption of mitochondrial function (leading to oxidative stress), interference with proteosomal function (leading to disrupted protein turnover), or activation of enzymes such as cyclin-dependent kinase 5 and glycogen synthase kinase 3β (resulting in the phosphorylation of tau and formation of NFTs) (Fig. 1) (7). As to which of these effects leads to altered neuronal function and neurodegeneration, that subject is open to debate.
NEUROINFLAMMATION: NEUROTOXIC OR NEUROPROTECTIVE?
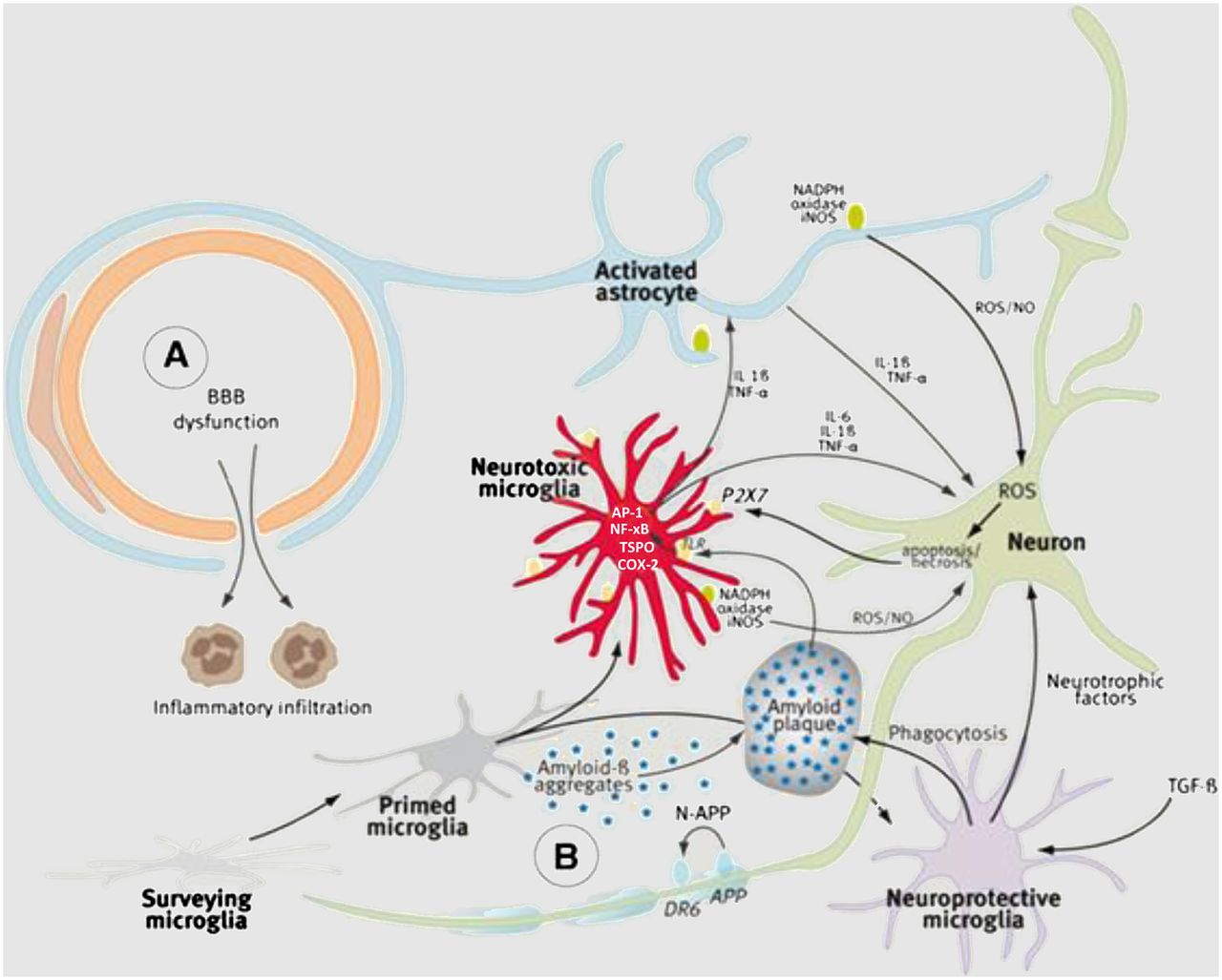
Microglia are the major resident immune cells in the central nervous system. Their primary function is to survey the microenvironment and release factors that influence surrounding neurons and astrocytes, a second type of glial cell in the central nervous system having support functions. Microglia are normally in a resting, or ramified, state. Under pathologic conditions, they lose their ramified morphology and convert to an amoeboidlike, or activated, state (18). During activation, microglia are polarized into 2 forms, M1 and M2 (Fig. 2) (19). M1 microglia are proinflammatory and release reactive oxygen and reactive nitrogen species (ROS/RNS). M2 microglia are neuroprotective and release growth factors. Both M1 and M2 microglia are thought to remove Aβ peptide by phagocytosis. It has also been suggested that there is an age-related shift in polarization of M1 and M2 microglia, with M1 > M2 being prominent in aged brain (19). Prolonged activation of microglia results in a second wave of inflammation via infiltration of monocyte-derived macrophages from blood (Fig. 2) (19).

Microglial activation leading to neuroprotective (M2) and proinflammatory, neurotoxic (M1) microglia. Second wave of inflammation comes from infiltration of peripheral monocyte-derived macrophages from blood. AP-1 = activator protein 1; COX-2 = cyclooxygenase 2; DR6 = death receptor 6; IL = interleukin; iNOS = inducible macrophage-type nitric oxide synthase; NADPH = reduced nicotinamide adenine dinucleotide phosphate; NF-xB = nuclear factor xB; NO = nitric oxide; P2X7 = purinogenic 2X7 receptor; TGF-β = tumor growth factor-β; TLR = toll-like receptor; TNF-α = tumor necrosis factor α; (Reprinted with permission of (22).)
There are 2 current theories on the role of activated microglia in AD: the neuroinflammatory hypothesis and the microglial dysfunction hypothesis (20). The neuroinflammatory hypothesis states that over-activated microglia caused by elevated Aβ levels result in uncontrolled inflammation and elevated ROS/RNS leading to neuronal cell death. In this case, neuroinflammation is believed to have a causal role in AD. The microglial dysfunction hypothesis states that activated microglia have a neuroprotective effect by reducing Aβ levels and releasing growth factors. Prolonged activation of microglia results in the formation of dystrophic microglia that have lost their neuroprotective function. The presence of dystrophic microglia in postmortem samples of AD seems to support the microglial dysfunction hypothesis (18). Increased formation of Aβ peptide/oligomers disrupts neuronal function through the mechanisms described above, elevates neuronal ROS/RNS (Fig. 2), and triggers cell death via apoptosis or necrosis (21).
Many labs have focused on developing PET radiotracers for imaging neuroinflammation in AD. Most of this effort has been directed toward imaging the 18-kDa translocator protein (TSPO) in activated microglia (22,23). One such radiotracer, 11C-PK11195 (1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-1-isoquinoline carboxamide), exhibits a low signal-to-noise ratio, which limits the sensitivity for imaging moderate levels of microglial activation and led to the preparation of several second-generation analogs possessing a higher affinity for TSPO and lower nonspecific binding. Although many promising ligands have been identified (23), the main limitation of these second-generation analogs is the low-binder effect, which is caused by a single-nucleotide polymorphism that reduces the affinity of these radioligands for TSPO (24). Oddly enough, 11C-PK11195 does not display the low-binder effect (24), suggesting that it should be possible to prepare a radioligand that does not display reduced affinity to TSPO in subjects having the low-binder single-nucleotide polymorphism.
The low-binder effect with the TSPO ligands has resulted in the investigation of other molecular targets for the imaging of activated microglia (22). As with the TSPO, these newer targets appear to be expressed on proinflammatory (M1) microglia and not on neuroprotective (M2) microglia. Clinical studies with radiotracers for these newer targets will reveal if these represent an advantage over the TSPO for imaging neuroinflammation. However, it will be critical to develop a method for imaging M2 microglia with PET to fully understand the role of neuroinflammation in AD.
NEUROMECHANISMS AND TEMPORAL DYNAMICS OF AD
From the above discussion, it is possible to divide the progression of AD into 3 critical phases (Fig. 3).
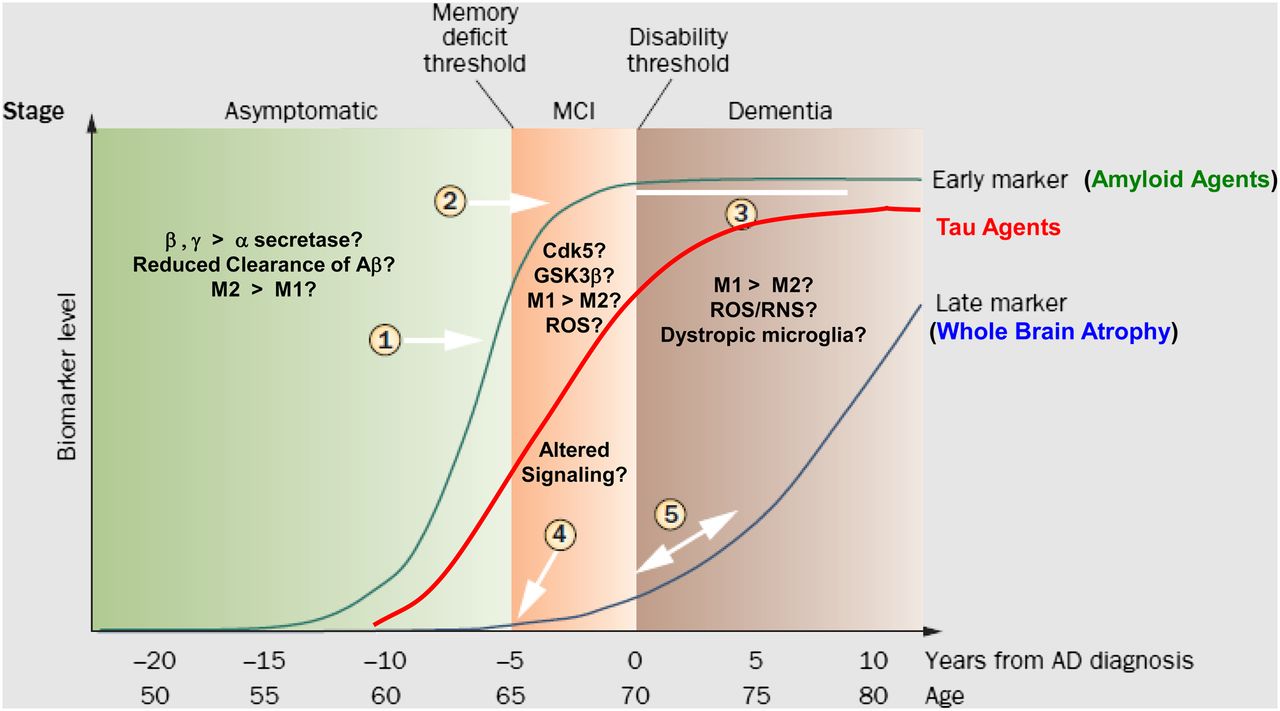
The 3 neurochemical phases of AD. Asymptomatic or prodromal phase is characterized by increased formation of Aβ plaques by either overproduction or reduced clearance of Aβ1–42. PET imaging studies with 11C-PiB confirm presence of Aβ plaques in this prodromal phase (1) and leveling off in MCI (2). Ceiling effect is shown (3). Red line shows theoretic time course for imaging with tau imaging agents or 18F-FDG. Late markers such as whole-brain atrophy are represented by blue line (4, 5) and can be followed with MR imaging. Cdk5 = cyclin-dependent kinase 5; GSK3β = glycogen synthase kinase 3β; MCI = mild cognitive impairment. (Adapted with permission of (25).)
Prodromal or Preclinical Phase
This phase is characterized by increased levels of Aβ1–42 in the brain, which can occur either by a shift in cleavage of APP from the α to the β/γ secretase pathway or by the reduced clearance of Aβ peptide across the BBB. Possible protective mechanisms include microglial activation favoring neuroprotection (i.e., M2 > M1) and sequestering of Aβ1–42 in the form of Aβ plaques. During this stage, imaging of Aβ plaques serves as an antecedent biomarker for identifying patients at risk for developing AD. Cognitive function is normal since oligomeric Aβ1–42 is not present in high concentrations.
Cognitive Impairment Phase
During this phase, neuroprotective mechanisms are overwhelmed and Aβ1–42 oligomers are present. The oligomers bind to proteins residing on the neuronal cell membrane, disrupt signaling, and undergo receptor-mediated internalization into neurons. Trafficking to the mitochondria generates ROS, which further disrupts signaling pathways and activates kinases leading to the phosphorylation of tau. Cognitive impairment results from reduced synaptic function and altered kinase activity.
Neurodegeneration Phase
This phase is caused by a shift in neuroinflammation from a neuroprotective (M2 > M1) to a proinflammatory profile (M1 > M2) leading to a large increase in ROS/RNS, neuronal damage, and activation of apoptosis or necrosis. Alternatively, there may be a complete loss of microglial activation resulting in dystrophic, nonfunctional glial cells. Invasion of peripheral monocyte-derived macrophages results in a second wave of inflammation. Progressive formation of NFTs causes cell death via apoptosis or necrosis, which can be imaged with tau radiotracers (NFTs) and 18F-FDG (reduced uptake due to neuronal loss).
FUTURE DIRECTIONS
Many of the steps outlined above were identified using transgenic models of AD or data obtained from postmortem samples of AD brain. PET radiotracers capable of imaging the functional activity of the key pathways (secretase activity, kinase activity) or mechanisms (Aβ clearance, M1 vs. M2 microglial activation) will provide a more comprehensive understanding of the molecular basis of neurodegeneration in AD. Such information will be critical in identifying therapeutic strategies for the treatment of AD.
DISCLOSURE
No potential conflict of interest relevant to this article was reported.
Footnotes
Published online Jul. 17, 2014.
- © 2014 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication May 1, 2014.
- Accepted for publication July 7, 2014.





{kind=link}
{kind=link}
{kind=link}