


Abstract
Access to diverse PET tracers for preclinical and clinical research remains a major obstacle to research in cancer and other disease research. The prohibitive cost and limited availability of tracers could be alleviated by microfluidic radiosynthesis technologies combined with a high-yield microscale radiosynthetic method. In this report, we demonstrate the multistep synthesis of 3′-deoxy-3′-18F-fluorothymidine (18F-FLT) with high yield on an electrowetting-on-dielectric (EWOD) microfluidic radiosynthesizer, previously developed in our group. We have identified and established several parameters that are most critical in the microscale radiosynthesis, such as the reaction time, reagent concentration, and molar ratios, to successfully synthesize 18F-FLT in this compact platform. Methods: 18F-FLT was synthesized from the 3-N-Boc-1-[5-O-(4,4′-dimethoxytrityl)-3-O-nosyl-2-deoxy-β-d-lyxofuranosyl] thymine precursor on the EWOD chip starting from the first solvent exchange and 18F-fluoride ion activation step to the final deprotection step. The fluorination reaction was performed in a mixture of thexyl alcohol and dimethyl sulfoxide. The crude product after deprotection was collected from the chip and purified on a custom-made solid-phase extraction cartridge and subjected to quality control testing. The purified 18F-FLT was suitable for small-animal PET studies in multiple nude mice xenografted with the A431 carcinoma cell line. Results: 18F-FLT was successfully synthesized on the EWOD microdevice coupled with an off-chip solid-phase extraction purification with a decayed-corrected radiochemical yield of 63% ± 5% (n = 5) and passed all of the quality control tests required by the U.S. Pharmacopeia for radiotracers to be injected into humans. We have successfully demonstrated the synthesis of several batches of 18F-FLT on EWOD, starting with approximately 333 MBq of radioactivity and obtained up to 52 MBq (non–decay-corrected) of 18F-FLT on cartridge purification. The specific activity of 2 representative preparations of 18F-FLT synthesized on the EWOD chip were measured to be 1,800 and 2,400 GBq/μmol. Conclusion: The EWOD microchip and optimized synthesis method in combination represent an effective platform for synthesizing 18F-FLT with high yield and of good quality for imaging. This compact platform, with configurable synthesis steps, could potentially form the basis of a stand-alone system that decouples PET probe production from the cyclotron and specialized radiochemistry facilities and increases diversity and flexibility in probe production.
PET is an extremely sensitive molecular imaging technique, capable of measuring in vivo metabolism, that is increasingly being used in clinical practice and research to diagnose and study a wide range of diseases including cancer, Alzheimer disease, and Parkinson disease (1–3). However, because of the difficulties and challenges involved in PET probe production (4), most PET imaging studies are limited to 18F-FDG, a glucose analog used to quantify glucose metabolism. Other PET probes that are currently used in clinical trials and research settings, such as 3′-deoxy-3′-18F-fluorothymidine (18F-FLT), 18F-fluoromisonidazole, 18F-fluoroethylcholine, 6-18F-fluoro-3,4-dihydroxy-l-phenylanaline, and many others, are only available at high cost and with limited availability from specialized research laboratories (5). Thus, there is a critical need to develop a new, affordable radiosynthesizer technology coupled with reliable radiosynthetic methodologies that could empower researchers and clinicians to synthesize probes of interest on-demand (at the imaging site) at low cost to address the diversity of biologic events being studied via PET imaging. The new technology platform should produce probes such as 18F-FLT with a high radiochemical yield and a final product that can be purified without the need for additional equipment, for example, via a simple cartridge purification, similar to the synthesis of 18F-FDG (6).
Recently, our group and others (7–11) have investigated microfluidic technology platforms as a means of achieving on-demand radiosynthesis of diverse PET probes (12). Throughout this article, macroscale synthesis refers to any reaction performed with a conventional radiochemistry apparatus, typically in vials at volumes above 250 μL, whereas microscale synthesis refers to reactions performed in microfluidic chips where at least 1 dimension of the reaction volume is on the order of hundreds of microns or less; in the EWOD microfluidic platform, the reaction volume is typically in the range of 1–10 μL. Microfluidic devices that integrate many laboratory functions on a single chip, also known as lab-on-chip, can automate repetitive laboratory tasks and enable users to perform hazardous reactions on chip in a safer manner (13,14). Of particular importance for PET probe synthesis using short-lived radioisotopes, microfluidic reactors enable radiosyntheses to be completed in a shorter time, minimize dilution of the radioisotopes to speed up reaction kinetics (note that only nmol to μmol amounts are typically produced), simplify purification because of the increased reaction selectivity, use smaller amounts of reagents, and have the potential to eliminate the high cost of infrastructure such as hot cells needed in a typical radiopharmacy facility (4,15). Our group has developed an all-electronic (i.e., no fluidic systems external to the chip) microfluidic radiosynthesizer based on the electrowetting-on-dielectric (EWOD) principle (16) and successfully demonstrated reliable synthesis of 18F-FDG (12). EWOD is an exemplary microfluidic platform for performing batch radiosynthesis, where a finite volume of liquid can be manipulated sequentially by applying electrical potential, without the need of moving parts such as pumps and valves. This work focuses on the development of a high yielding and reliable microscale radiochemistry method for the synthesis of a useful tracer with currently limited availability, namely 18F-FLT, a radiolabeled analog of thymidine, on the EWOD chip. The demonstration of this 2-step synthesis in a reliable fashion on the EWOD chip (Fig. 1), combined with previous results (12,17), suggests the capability of this platform to perform diverse syntheses and perhaps form the basis of a compact, benchtop device for producing diverse probes on demand.
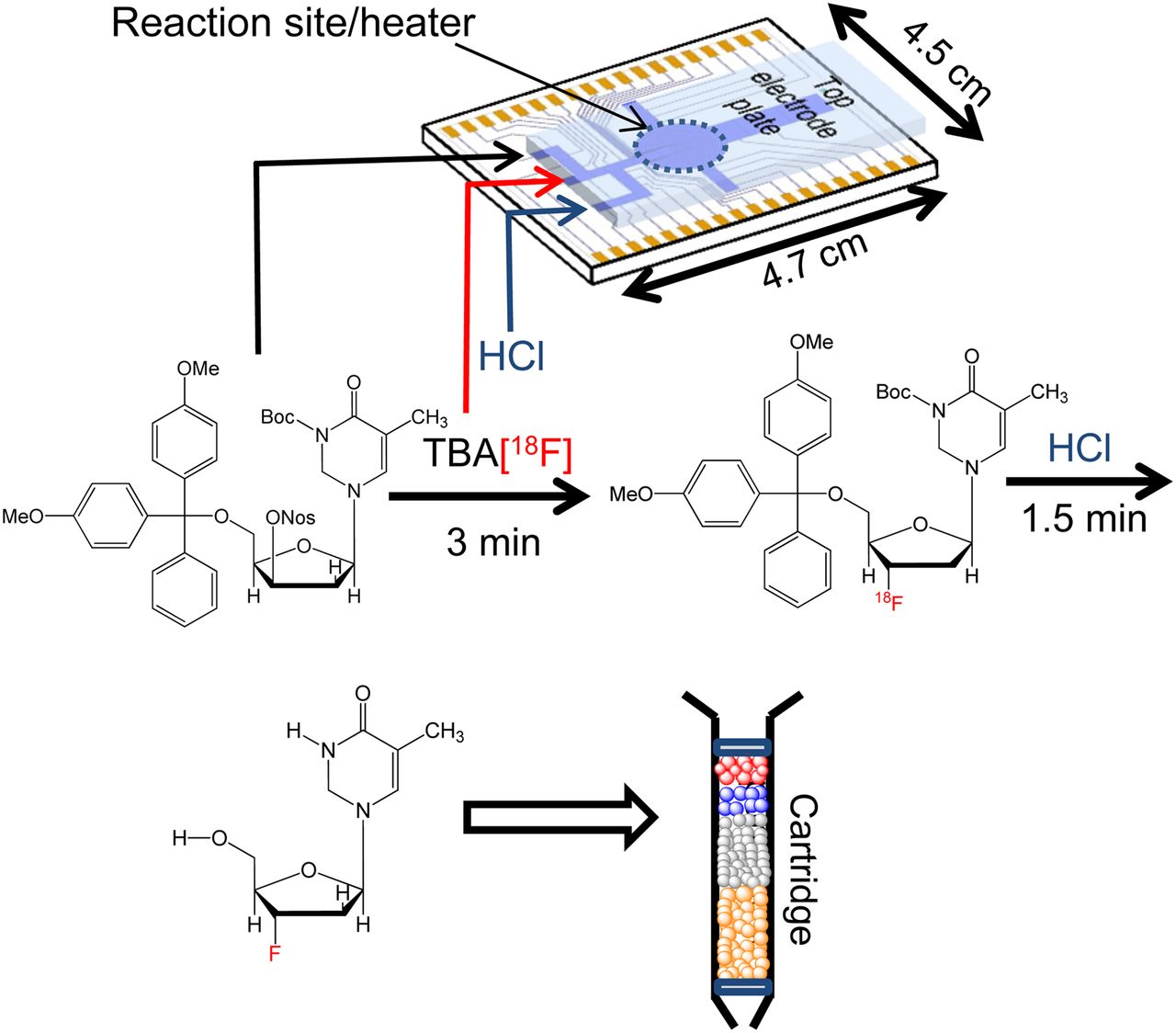
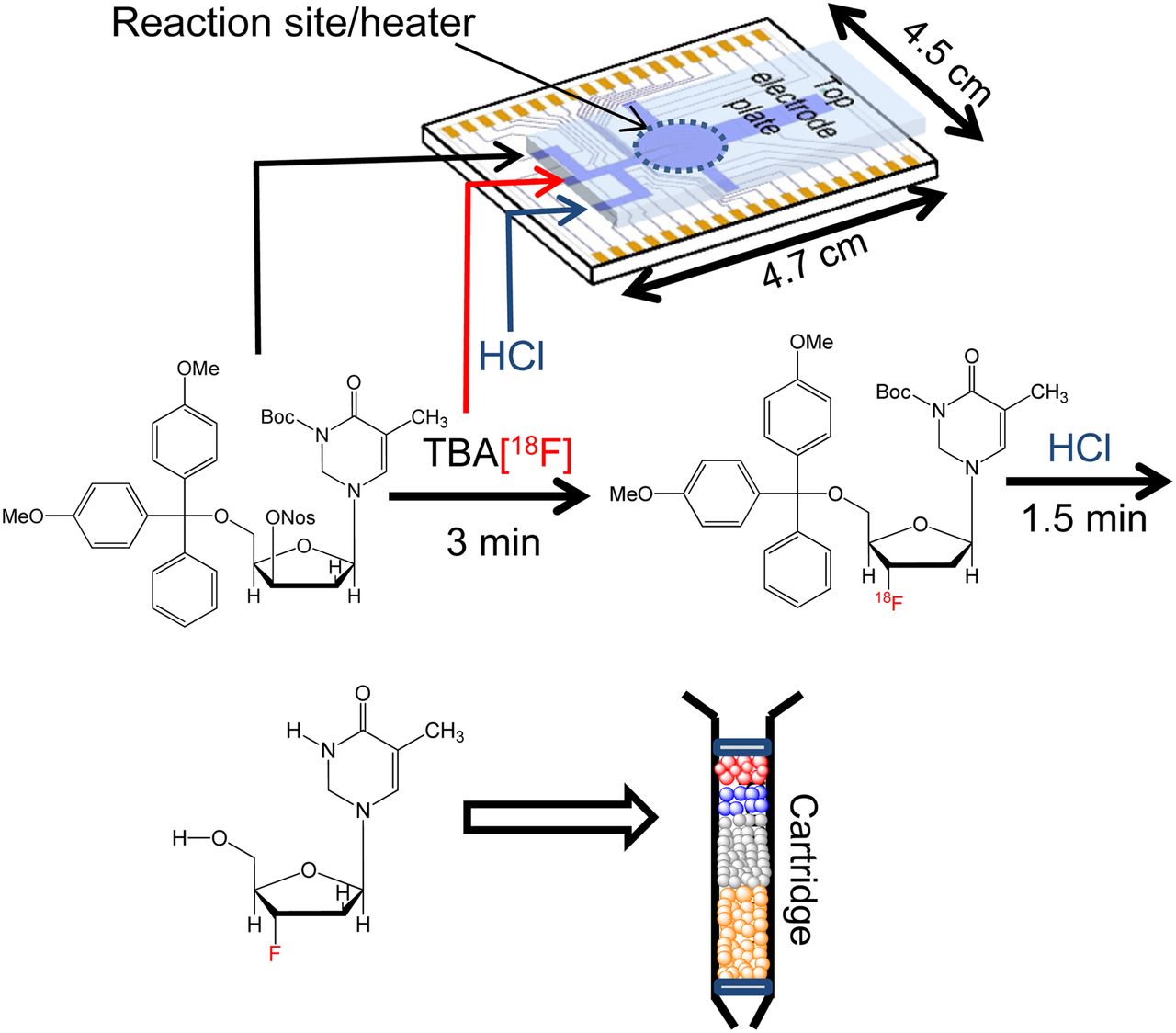
Overall workflow of radiosynthesis of 18F-FLT on EWOD chip, followed by cartridge purification to produce injectable dose of 18F-FLT for mice imaging; synthetic scheme of radiosynthesis of 18F-FLT using mixture of thexyl alcohol and DMSO in fluorination reaction. Crude 18F-FLT product was extracted and purified via simple cartridge purification to yield approximately 52 MBq (non–decay-corrected) of 18F-FLT.
MATERIALS AND METHODS
Reagents
Tetrabutylammonium bicarbonate (TBAHCO3), 2,3-dimethyl-2-butanol, HCl, anhydrous acetonitrile (99.8%), anhydrous dimethyl sulfoxide (DMSO, 99.9%), hexanes, ethyl acetate, ethanol, and methanol were purchased from Sigma-Aldrich Chemical Co. 3-N-Boc-5′-O-dimethoxytrityl-3′-O-nosyl-thymidine (DMTr-Boc-nosyl precursor), 5′-O-dimethoxytrityl-3′-O-nosyl-thymidine (DMTr-nosyl precursor), and the 3′-deoxy-3′-fluorothymidine (FLT) standard compound were purchased from Advanced Biochemical Compounds (ABX). Ion retardation resin (AG11 A8) and cation exchange (AG-50W-X4) were purchased from BioRad Laboratories. 2′,3′-didehydro-3′-deoxy-thymidine reference standard was purchased from Tokyo Chemical Industry and used as received. Neutral alumina (particle size, 50–300 μm), C-18 (particle size, 55–105 μm), and the Oasis Hydrophilic Lipophilic Balanced (HLB) resin were purchased from Waters Corp.
No-carrier-added 18F-fluoride ion was obtained from the UCLA Crump Institute for Molecular Imaging Cyclotron Facility by irradiation of 97% 18O-enriched water with an 11-MeV proton beam using an RDS-111 cyclotron (Siemens Medical Solution). Radioactivity was measured using a calibrated ion chamber (CRC-15R; Capintec Inc.). A radioactive thin-layer chromatography (TLC) scanner (MiniGITA Star; Raytest USA, Inc.) was used to analyze fluorination efficiency. Samples of the crude fluorination product were spotted on silica TLC plates and developed in a mixture of ethanol and ethyl acetate (50:50 v/v). Analytic high-performance liquid chromatography (HPLC) was performed using a Phenomenex Luna reversed-phase C-18 column (250 × 4.6 mm). Elution was performed at a constant flow rate of 1 mL/min with water:ethanol (90:10 v/v), and the ultraviolet absorbance was measured at 265 nm. Pyrogenicity testing was performed using the Charles River Endosafe-PTS Portable Testing System.
EWOD Chip Fabrication and Operation
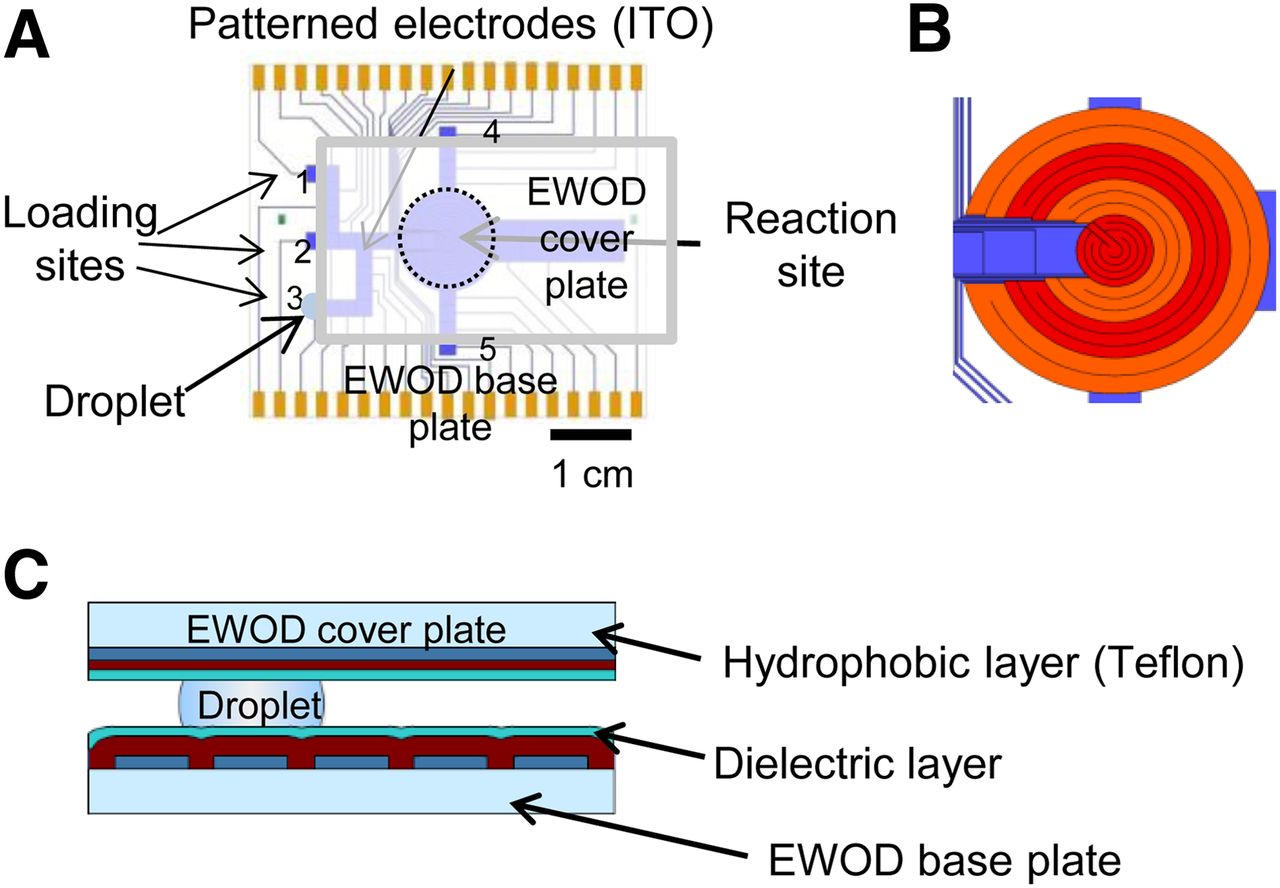
Similar to our previous report (12), the EWOD chip was constructed from 2 parallel plates: the base plate and the cover plate. In comparison to our previous chip design, 2 additional reagent loading sites and inlet pathways were added to increase the flexibility for multistep radiochemistry (Fig. 2). Additional details of EWOD chip fabrication and operation can be found in the supplemental information.
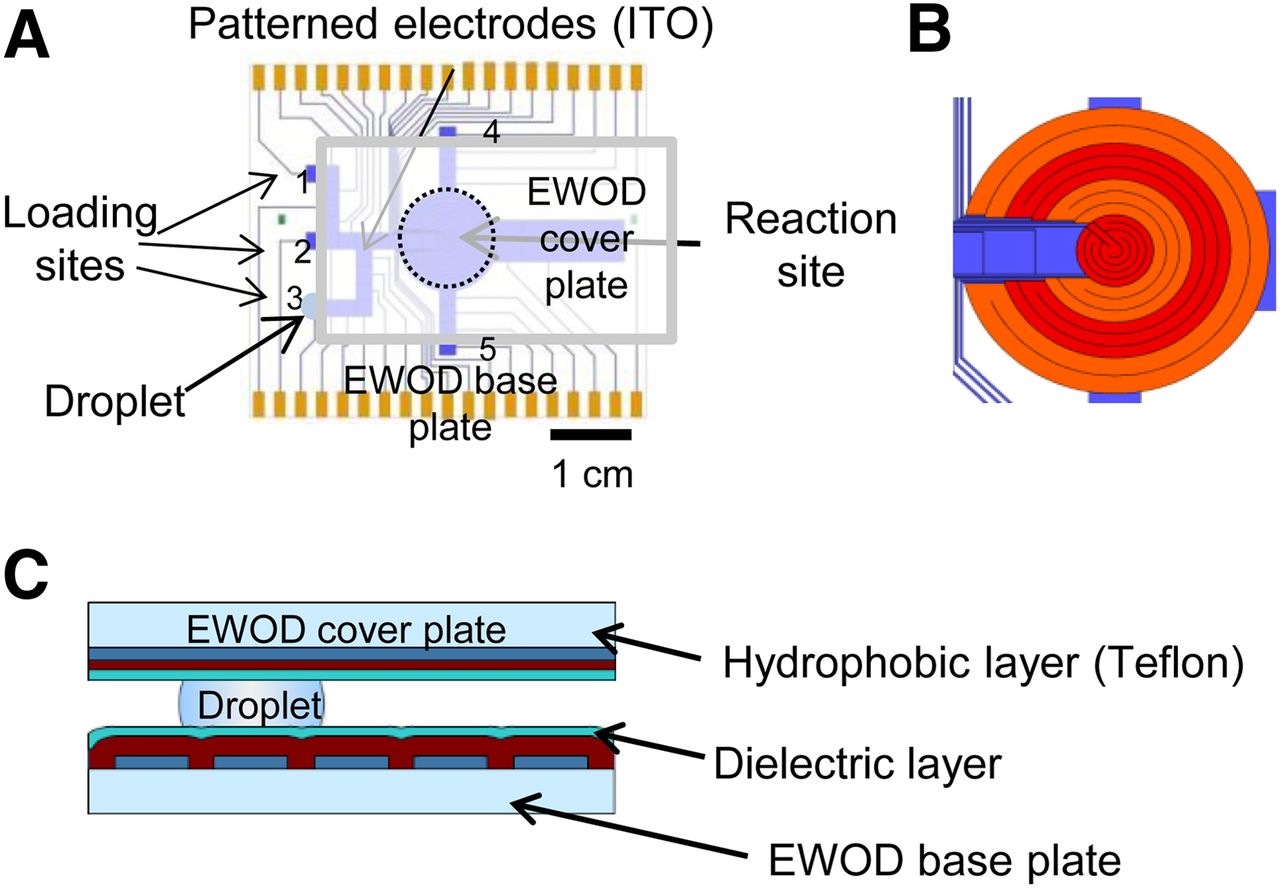
(A) EWOD chip base plate design with reaction site and multiple loading sites. Reagent droplet (blue circle), sandwiched between EWOD device plate and cover plate, is shown at loading site 3. (B) Detail of multifunctional electrowetting, heating, and temperature sensing electrodes. (C) Cross-sectional view of EWOD chip with sandwiched droplet.
Radiosynthesis of 18F-FLT on EWOD Chip
Stock solution of TBAHCO3 (5 μL, 75 mM) was mixed with the no-carrier-added 18F-fluoride (20 μL; ∼740 MBq) solution to form the 18F-tetrabutylammonium fluoride (TBAF) complex. Three droplets (2 μL each) of the 18F-TBAF complex were pipetted to loading site 1 and then transported to the reaction site by EWOD actuation, followed by the addition of a 3-μL droplet of MeCN (acetonitrile). The 18F-TBAF complex was heated to 105°C and held at 105°C for 1 min to evaporate the solvent. Subsequently, 1 cycle of azeotropic distillation was performed by adding a 9-μL MeCN droplet via loading site 1 to the dried residue and heating to 105°C for 1 min. On completion of this drying step and thus activation of the 18F-TBAF complex, DMTr-Boc-nosyl precursor (4.5 mg) was dissolved in a mixture of DMSO (20 μL) and 2,3-dimethyl-2-butanol (40 μL) to achieve a final concentration of 90.4 mM. One droplet of the DMTr-Boc-nosyl precursor solution (2 μL) was pipetted to loading site 2 and transferred to the dried 18F-TBAF complex on the reaction site at room temperature, followed by the addition of a 3-μL droplet of MeCN. The reaction mixture was gradually heated to 120°C and held at 120°C for 3 min to perform the fluorination reaction. After the fluorination reaction, two 3.5-μL droplets of a 4:1 mixture of 1N HCl/MeCN were pipetted to loading site 3 and transported to and mixed with the crude intermediate product at the reaction site. The reaction mixture was slowly heated to 95°C and held for 1.5 min to complete the hydrolysis reaction. The cover plate was removed, and the crude product was extracted using 200 μL of H2O, 50 μL of MeCN and 18 μL of DMSO, sequentially. A small amount of the crude product was used for radio–thin-layer chromatography (radio-TLC) and radio-HPLC analyses, whereas the remainder of the crude product was purified using a miniaturized cartridge as described in the following sections. The total synthesis time and cartridge purification was about 63 min.
Cartridge Preparation
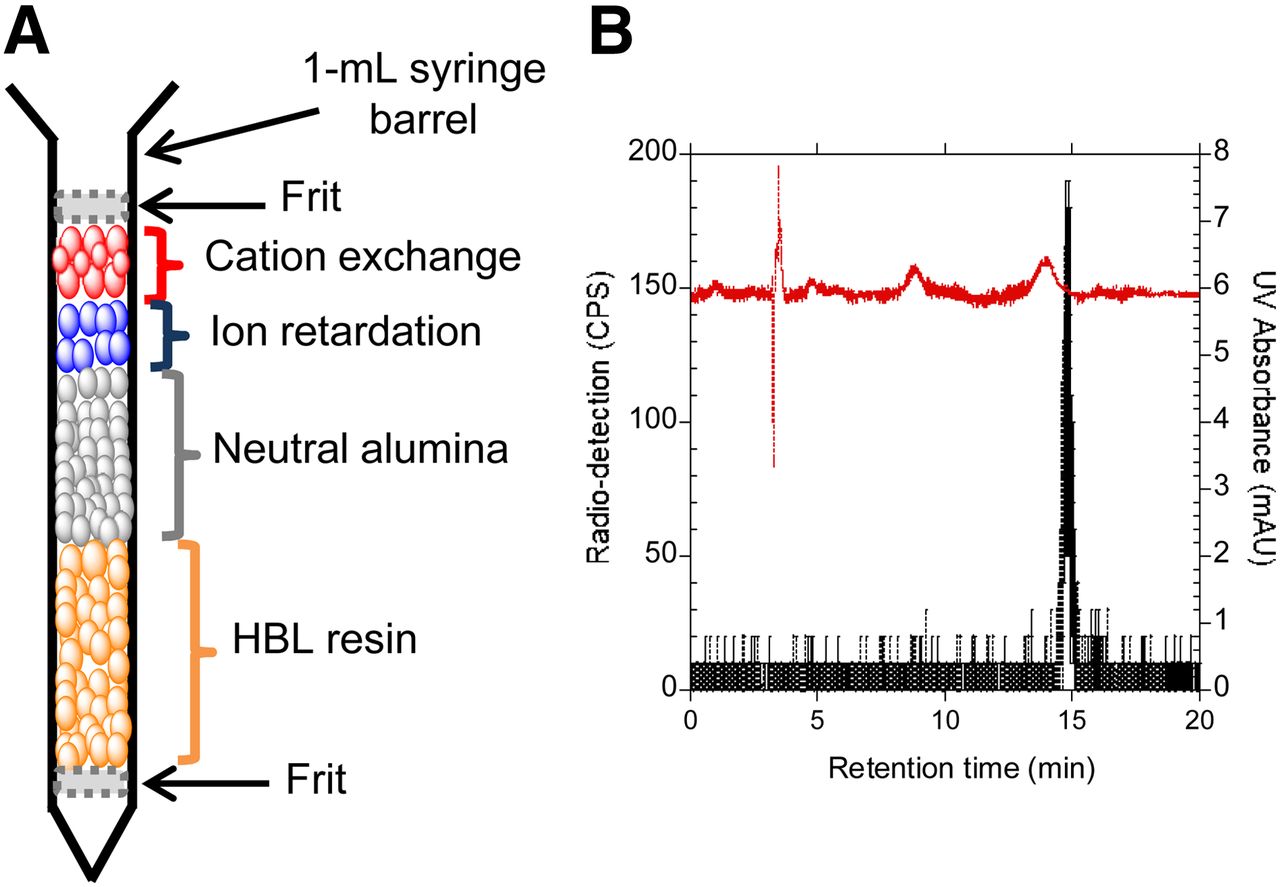
A miniaturized purification cartridge was designed on the basis of our previous report (12) to achieve a high purification efficiency (Fig. 3A). The custom-made cartridges consisted of 5 mg of ion retardation resin, 5 mg of cation exchange, 30 mg of neutral alumina (mesh size, 50–100), and 150 mg of Oasis HLB resins packed within a 1-mL syringe barrel (Becton, Dickinson and Co.). To prevent the formation of air bubbles, 2 polyethylene frits (pore size, 20 μm) were used to sandwich the resins. The cartridge was conditioned with methanol (1 mL) and water (2 mL) before use.

(A) Custom-made purification cartridge used to purify 18F-FLT that was synthesized on EWOD chip. (B) High-performance liquid chromatogram of cartridge-purified 18F-FLT. Red chromatogram represents absorbance in ultraviolet range, and black chromatogram represents radio detection.
Cartridge Purification of 18F-FLT
The crude reaction mixture collected from the chip was passed through the conditioned miniature cartridge to trap the desired product using a 1-mL syringe, followed by sequential elution with 1% ethanol in water (9 mL) and 5% ethanol in water (6 mL) to release the side products to waste. A final washing step was performed using 100% ethanol (0.2 mL, 0.1 mL at a time). After removal of all the side products (analyzed by HPLC), the final 18F-FLT product was eluted from the cartridge using 0.5 mL of ethanol and collected into a sterile empty vial. The ethanol was evaporated by heating at 75°C and blowing nitrogen for 5 min into the vial, and the resulting dried 18F-FLT residue was redissolved in 0.2 mL of saline.
The residual solvents (ethanol, acetonitrile, DMSO, thexyl alcohol) in the formulated 18F-FLT were analyzed via gas chromatography following the method reported in our previous work (details are provided in the supplemental data; available at http://jnm.snmjournals.org) (12). The chemical and radiochemical purity of the formulated 18F-FLT was analyzed using an analytic HPLC with ultraviolet detection at 265 nm and a radiodetector. The product and impurities in the final sample were identified by comparing their retention times to known standards. A calibration curve for 18F-FLT and several chemical impurities, such as 2′,3′-didehydro-3′-deoxy-thymidine (stavudine) and thymine, was performed to quantitate the amount of impurities present in the final product.
Specific Activity Analysis
For specific activity analysis, 2 batches of 18F-FLT were prepared on the EWOD chip, with all volumes doubled to obtain sufficient mass for ultraviolet detection on the HPLC. For these experiments, the crude 18F-FLT was first purified via analytic HPLC, and the fraction containing the 18F-FLT product was collected. The collected product was diluted in water and passed through the custom-made purification cartridge. The cartridge-purification procedure was identical to the single batch experiment. On partial evaporation of the ethanol, the concentrated 18F-FLT (∼0.15 mL) was injected into the analytic HPLC for quantification of the cold mass of 18F-FLT (ultraviolet detection at 254 nm) using the previously obtained calibration curve for 18F-FLT. The decay-corrected radioactivity of the purified 18F-FLT was used for specific activity analysis.
Pyrogenicity Test
An aliquot (500 μL) of the final 18F-FLT product in saline was tested for the presence of bacterial endotoxin using a Limulus Amebocyte Lysate (LAL) Test. The sample was further diluted with saline by 20 times before the LAL analysis. The test was performed using a Charles River Endosafe-PTS Portable Testing System.
Small-Animal PET and CT Imaging
Details of the small-animal imaging can be found in the supplemental data.
RESULTS
The multistep on-chip reaction begins with the 18F-fluoride ion activation step, followed by the radiofluorination of DMTr-Boc-nosylate FLT precursor, and finally the deprotection of the tert-butyloxycarbonyl (Boc) and the 4,4′-dimethoxytriphenylmethyl (DMTr) groups via acid hydrolysis. A systematic optimization of various reaction parameters including reagent concentration, precursor, precursor-to-base ratio, reaction temperature, reaction time, and phase transfer catalyst was performed on Teflon (DuPont)-coated glass substrates, which mimic the microdroplet reaction on the EWOD chip. In the first phase of the method development, we found that the optimal fluorination condition used the DMTr-Boc-nosylate FLT precursor and the TBAHCO3 as the phase transfer catalyst in 2:1 molar ratio to achieve 80% ± 7% (n = 10) fluorination efficiency. Efficiencies and yields are reported with SD based on the number of experiments, n.
On further investigation, we found that a significant percentage of radioactivity was lost during the fluorination and hydrolysis step. In our attempt to minimize the loss by reducing the reaction time to 3 min, the fluorination efficiency decreased to 51% ± 7% (n = 3). To counteract this effect, the overall reagent concentration was doubled, and the reaction temperature was increased from 110°C to 120°C, resulting in an increase in fluorination efficiency to 79% ± 6% (n = 2). The fluorination efficiency was further increased to 94% ± 3% (n = 9) when the reaction droplet volume was reduced from 4 to 2 μL, while maintaining the 2-fold increase in the reagent concentrations. Following the optimized fluorination protocol, the protected 18F-FLT intermediate was hydrolyzed in a mixture of HCl and MeCN at 95°C for 1.5 min to obtain 18F-FLT in 82% ± 10% (n = 10) crude radiochemical yield. The crude radiochemical yield is defined as (radioactivity remaining on chip after synthesis × conversion to 18F-FLT as determined by radio-TLC)/estimated radioactivity loaded onto chip. In comparison to the initial method, the crude radiochemical yield obtained from the optimized method improved from 49% ± 11% (n = 10) to 82% ± 10% (n = 10).
After the radiosynthesis of 18F-FLT on the EWOD chip, the crude mixture was first collected using a mixture of DMSO, MeCN, and water with 84% ± 2% (n = 5) collection efficiency. The collected crude reaction mixture (∼270 μL) was then purified on a custom-made miniaturized cartridge to obtain 18F-FLT in greater than 99% radiochemical purity and an overall decay-corrected radiochemical yield of 63% ± 5% (n = 5). The cartridge-purification efficiency was 89% ± 2% (n = 5). This efficiency is defined by the radioactivity of purified 18F-FLT recovered after cartridge purification divided by the total 18F-FLT collected from the EWOD chip (i.e., radioactivity of collected crude product × conversion to 18F-FLT as determined by radio-TLC). Starting with approximately 333 MBq of radioactivity (18F-fluoride ion) on the EWOD chip, we have successfully prepared 52 MBq (non–decay-corrected) of 18F-FLT on cartridge purification and formulation.
The purified 18F-FLT sample was subjected to a set of quality control procedures required for testing purity and safety before administering to humans. The final product solution was observed to be clear and free of particulates. The pH was measured to be between 6.5 and 7 using pH paper. The gas chromatography analysis showed that the final product contained less than 20 ppm of MeCN, DMSO, ethanol, and thexyl alcohol. The allowable limits for the residual organic solvents are as follows: 400 ppm, MeCN; 5,000 ppm, DMSO; 5,000 ppm, ethanol; and 5,000 ppm, thexyl alcohol. The chemical purity of the formulated product was analyzed using analytic HPLC. On the basis of the standard ultraviolet calibration curves for FLT and the other known impurities, we found that the final sample contained 0.3 ppm of stavudine, 0.05 ppm of thymine, and 0.07 ppm of FLT. Other ultraviolet active peaks that eluted around 3.5 min were unidentified and do not match the retention times of any of the known impurities reported in the literature. The LAL test detected less than 1 EU/mL in concentration of the final sample, which is lower than the established U.S. Pharmacopeia endotoxin limit of 175 EU/mL per dose for radiopharmaceuticals. The specific activities of 18F-FLT synthesized on the EWOD chips were measured to be 1,800 and 2,400 GBq/μmol.
DISCUSSION
Since the first radiosynthesis of 18F-FLT reported by Grierson et al. (18), there have been a multitude of methods developed to achieve higher and more reliable yield (19). Notably, Eisenhut et al. developed a new FLT precursor, DMTr-Boc-nosyl, to facilitate synthesis automation, and it has become the precursor of choice to achieve high radiosynthesis yield (20). Since then, multiple research groups have reported the radiosynthesis of 18F-FLT using this DMTr-Boc-nosyl FLT precursor based on the conventional no-carrier-added radiofluorination method, with radiochemical yields ranging from 23% to 50% (19,21–24). Particularly, the use of a bulky alcohol as cosolvent in assisting the no-carrier-added radiofluorination is attractive and has been reported to attain 18F-FLT in a high radiochemical yield (60%–65%) (25–28). We adapted this high-yielding, high-selectivity protic solvent chemistry to the EWOD microdevice and developed a miniature-cartridge-purification method to eliminate the expensive and complicated HPLC purification.
During this developmental procedure, most of the processes were performed manually, such as reagent loading, product collection, and cartridge purification, which limits the amount of radioactivity that was used in this work. Although this report focused on the development of a reliable microscale radiochemistry on the EWOD chip, we are also currently developing an automated reagent delivery, product collection, and purification system (29–32). We anticipate that high-activity production within microliter volume can easily be achieved based on the recent demonstration of the production of clinical dose amounts of 18F-FDG and 18F-fallypride in batch microfluidic devices (9,33). For example, a miniaturized anion exchange cartridge was able to concentrate approximately 32 GBq of 18F-fluoride/18O-H2O into a 5-μL volume of eluent. Such volume is commensurate with the design of EWOD chip reaction site, which can accommodate up to an approximately 17-μL droplet size.
Starting from the original report of the radiosynthesis of 18F-FLT in protic solvent (26), we first performed systematic screening of various reaction parameters including reagent concentrations, precursors, precursor-to-base ratios, and phase transfer catalysts on the microscale using Teflon-coated glass substrates. In contrast to the macroscale method, we chose to use a mixture of DMSO and thexyl alcohol to improve the solubilization of the precursor throughout the entire fluorination reaction, which is critical for synthesis automation and to increase the reaction reliability. Although macroscale syntheses generally avoid the use of DMSO (boiling point, 182°C) and thexyl alcohol (boiling point, 120°C) because of the difficulty in removing these solvents after the synthesis, the much smaller volume used in the microscale synthesis (2 vs. 500–1,000 μL) facilitates the rapid removal of these solvents. At the end of the synthesis, the droplet volume has already shrunk to approximately 0.2 μL because of solvent evaporation during the fluorination reaction (Supplemental Fig. 1). Without further evaporation, we have confirmed that the level of residual solvents of the final purified product is below the recommended limits for injection. A summary of results from the optimization studies performed on Teflon-coated glass is presented in Table 1. We found that the precursor-to-base concentration ratio is one of the critical parameters affecting the fluorination efficiency, which is consistent with the work by Suehiro et al. (34). In addition to the precursor-to-base ratio, we also investigated the use of the cryptand complex as the phase transfer catalyst. As shown in Table 1, the fluorination yield using the cryptand complex is lower (58%; Table 1, condition 2) in comparison to the TBAHCO3 (80%; Table 1, condition 4) when performed under similar conditions (i.e., precursor-to-base ratio, ∼2), which is also consistent with the report by Kim et al. (35). To confirm the catalytic effect of the alcohol in the radiosynthesis, we also performed a control experiment by replacing the DMSO/thexyl alcohol with an aprotic solvent mixture of DMSO and MeCN. The DMSO/MeCN solvent ratio was determined empirically such that the droplet size at the end of fluorination reaction was a similar size for the 2 cases. We observed significantly lower fluorination efficiency of 38% ± 4% (n = 2) when DMSO/MeCN was used versus 80% ± 7% (n = 10) when thexyl alcohol was used in the fluorination reaction (Table 1, conditions 4 and 5), confirming the advantage of thexyl alcohol in assisting the nucleophilic substitution reaction. In our attempt of using the DMTr-nosyl FLT precursor, we found that the fluorination yield was lower and less reliable in comparison to the DMTr-Boc-nosylate FLT precursor under similar conditions (Table 1, condition 1 vs. 3). The final hydrolysis step was performed at 95°C for 5 min. The completion of the hydrolysis reaction was tentatively confirmed by the emergence of a single radio peak at around 15 min, which corresponds to 18F-FLT in the radio-HPLC (Supplemental Fig. 3). This first phase of optimized method is referred to as method 1 in Table 2.
Optimization of Radiofluorination with Varying Parameters and Precursors on Teflon–Glass Substrates
Summary of Reaction Conditions for Radiosynthesis of 18F-FLT on EWOD Chip Based on Method 1
Though method 1 exhibited high fluorination and hydrolysis efficiencies, we found that the overall crude radiochemical yield of 18F-FLT at the end of the synthesis was relatively low (47% ± 19%; n = 7). To understand the discrepancy between overall efficiency and yields of individual steps, we measured the radioactivity losses after each step of the synthesis on a Teflon-coated glass substrate. The sandwiched Teflon–glass substrate was removed from the Peltier heater, and the radioactivity on the substrate was measured using the dose calibrator after each step of the method 1 synthesis (Table 2). On the basis of this study, we found that the major loss occurred after both the fluorination and the hydrolysis steps. Although the exact mechanism of the radioactivity loss is yet to be determined, our initial approach was to reduce the reaction times for both the fluorination and the hydrolysis reaction to decrease the overall radioactivity losses, while ideally not diminishing the yields.
To do so, we first investigated the reaction kinetics on the Teflon–glass substrate and found that a reduction of the fluorination reaction time from 5 to 3 min resulted in a significant decrease in the fluorination efficiency. Through a series of experiments, we found that the fluorination reaction kinetics can be improved by increasing the reagent concentration and the reaction temperature. Interestingly, we also found that the fluorination yield was further increased (from 79% to 94%) by simply reducing the droplet size from 4 to 2 μL, while maintaining the 2-fold increase in the concentration of the reagent. The enhanced fluorination yield could be speculated to be attributable to the increase in the 18F-fluoride ion concentration as the reaction volume was decreased. Although consistent with speculations that this is an advantage of microfluidic synthesis platforms (15), further investigation of this effect is necessary.
On the basis of new optimized synthesis (method 2, Table 3), we indeed observed a reduction in radioactivity loss from 33% to only about 15% (Table 2). The new microscale 18F-FLT conditions also yielded higher fluorination efficiency (91% ± 4%; n = 10). Overall, the crude radiochemical yield of 18F-FLT increased from 49% ± 11% (Table 2) to 82% ± 10% (Table 3).
Summary of the reaction conditions for the radiosynthesis of [18F]FLT on EWOD chip based on Method 2.
With the exception of 18F-FDG and 18F-NaF, other PET probes generally require final purification via HPLC to remove excess precursor or other radiolabeled or toxic side products that cannot be easily removed via solid-phase extraction. However, this technique is not easily miniaturized as would be needed for a benchtop radiosynthesis platform. These shortcomings of HPLC have led to the emergence of several HPLC-free radiochemistry methods (36–38).
From the point of view of the cartridge purification, the most desirable solvent to collect the product from the chip is water. However, in the microscale, where the surface-to-volume ratio is large, we found that the collection efficiency (ratio of radioactivity recovered from the chip vs. the total radioactivity that was measured on-chip after the synthesis) of the final crude 18F-FLT from the EWOD chip was below 15% when only water was used as the extraction solvent. With 50:50 v/v MeCN/H2O as the extraction solvent, the collection efficiency improved to 47% ± 10% (n = 5). Finally, we attempted a mixture of DMSO, MeCN, and water (18, 50, and 200 μL, respectively) and obtained 84% ± 2% (n = 5) collection efficiency. However, in this solvent mixture, we found that the 18F-FLT was not able to be separated from the other side products when the conventional reversed-phase C18 resins were used in the cartridge purification. This observation can be explained by the increasing composition of nonpolar solvent during the cartridge purification, which is sufficient to disrupt the van der Waals forces between the 18F-FLT analyte and the reversed-phase resin (39). To address this issue, we investigated a new type of sorbent known as the Oasis HLB. On the basis of systematic cartridge-purification studies, we determined the optimal HLB resin to be 150 mg to efficiently retain 18F-FLT from a solvent mixture of MeCN, water, and DMSO, while enabling elution of the other side products from the cartridge. This resin was combined in a miniaturized cartridge with additional resins for removal of other impurities. On the cartridge purification of 18F-FLT, the final purified 18F-FLT product was collected in 500 μL of 100% ethanol. On evaporation and reformulation in saline, 52 MBq of 18F-FLT was obtained for small-animal PET imaging studies of several A431 tumor–bearing mice. As expected, the small-animal PET images showed a high accumulation of 18F-FLT in the tumor due to the high level of expression of the thymidine kinase-1 enzyme in rapidly proliferating cells (Supplemental Fig. 7). No adverse effect on the physiology of the mice was observed after injection.
On the basis of several 18F-FLT samples that were subjected to a standard quality control procedure, the final 18F-FLT solutions were found to have a negligible amount of impurities on formulation in approximately 0.2 mL of saline. The only radioactive component present in the final compound was 18F-FLT on successful removal of the unreacted 18F-fluoride ion using the custom-made cartridge (Fig. 3B; radio-TLC in Supplemental Fig. 6). Because of the minute amount of reagent used on the EWOD microfluidic device for radiosynthesis, the absolute amounts of impurities and residual solvent reported here and in our previous report (12) were extremely small. Additionally, the level of impurities reported here would be approximately 50 times lower on diluting the single dose of 18F-FLT from the EWOD chip in 10 mL of saline for clinical PET imaging. For clinical production, we anticipate to increase only the amount of radioactivity loaded onto the EWOD chip while keeping all other aspects of the synthesis process the same. Therefore, there will be a significant reduction in the level of impurities, bacterial endotoxins, and residual solvents that are present in a single dose of PET radiopharmaceuticals for clinical studies.
The specific activity of 18F-FLT synthesized on the EWOD chip was measured to be more than 10 times higher than literature reports using conventional macroscale radiosynthesizers. The high specific activity of 18F-FLT synthesized on the EWOD chip suggests that the Teflon layer on the chip does not lead to a significant amount of carrier fluoride contamination through radiolysis or other mechanisms. This result is consistent with the recent report by Rensch et al. (40) on the reduction of radiolysis on microfluidic devices due to the geometric confinement. The simulation and experimental studies conducted by Rensch suggested that most of the energy of positrons is deposited into the walls of microfluidic chips rather than in the reaction mixture when the dimension of the microfluidic channel or gap is smaller than the positron range (∼400 μm). The high specific activity suggests an advantage of microfluidic platforms for the production of high-specific-activity radiopharmaceuticals for imaging low-abundance receptors. We are currently investigating key contributing factors that lead to this high specific activity.
CONCLUSION
In this report, we have developed an optimal 2-step, 1-pot procedure to synthesize 18F-FLT on the EWOD radiosynthesizer. The method uses a bulky alcohol (thexyl alcohol) as a cosolvent and exhibits a high and reliable decay-corrected radiochemical yield of 63% ± 5% (n = 5). The synthesis time (including cartridge purification) was 63 min. The optimized microscale radiofluorination strategy yielded up to 52 MBq (non–decay-corrected) of 18F-FLT. The final product passed all quality control tests, including pH, chemical purity, residual solvent analysis, and pyrogenecity tests, that are required before administering to humans.
The small size of the microfluidic platform suggests that the overall need for radiation shielding could be dramatically reduced in comparison to the size of hot cells and mini cells. The reduction in the overall size and shielding enable the synthesizer to be self-shielded and placed on a standard laboratory benchtop, thus eliminating some of the barriers to PET probe production. With an increasing degree of automation of the EWOD microfluidic platform, such as radioactivity concentration, reagent delivery, cartridge purification, and an integrated quality control module, a compact, robust radiosynthesizer that produced probes on demand on a benchtop may be possible. Currently, the EWOD chip is designed to be 1-time-used, similar to the disposable reagent cassettes used in many recent macroscale radiosynthesizers. This report demonstrates that a similar microfluidic chip design as reported in our previous publication can be used to synthesize different PET tracers.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This work was supported in part by funds from the UCLA Department of Molecular and Medical Pharmacology and the UCLA Foundation from a donation made by Ralph and Marjorie Crump for the Crump Institute for Molecular imaging. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Dr. David Stout at the Crump Radiochemistry Cyclotron facility and Dr. Saman Sadeghi at the UCLA Biomedical Cyclotron facility for providing 18F-fluoride ion for these studies and performing the pyrogenecity test, Dr. Caius Radu for performing the 18F-FLT in vivo studies, and Jeffrey Collins for performing residual solvent analysis.
Footnotes
Published online Jan. 16, 2014.
- © 2014 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication February 1, 2013.
- Accepted for publication July 24, 2013.





{kind=link}
{kind=link}
{kind=link}