


Abstract
The overexpression and excessive signaling of platelet-derived growth factor receptor β (PDGFRβ) has been detected in cancers, atherosclerosis, and a variety of fibrotic diseases. Radionuclide in vivo visualization of PDGFRβ expression might help to select PDGFRβ targeting treatment for these diseases. The goal of this study was to evaluate the feasibility of in vivo radionuclide imaging of PDGFRβ expression using an Affibody molecule, a small nonimmunoglobulin affinity protein. Methods: The PDGFRβ-binding Z09591 Affibody molecule was site-specifically conjugated with a maleimido derivative of DOTA and labeled with 111In. Targeting of the PDGFRβ-expressing U-87 MG glioblastoma cell line using 111In-DOTA-Z09591 was evaluated in vitro and in vivo. Results: DOTA-Z09591 was stably labeled with 111In with preserved specific binding to PDGFRβ-expressing cells in vitro. The dissociation constant for 111In-DOTA-Z09591 binding to U-87 MG cells was determined to be 92 ± 10 pM. In mice bearing U-87 MG xenografts, the tumor uptake of 111In-DOTA-Z09591 was 7.2 ± 2.4 percentage injected dose per gram and the tumor-to-blood ratio was 28 ± 14 at 2 h after injection. In vivo receptor saturation experiments demonstrated that targeting of U-87 MG xenografts in mice was PDGFRβ-specific. U-87 MG xenografts were clearly visualized using small-animal SPECT/CT at 3 h after injection. Conclusion: This study demonstrates the feasibility of in vivo visualization of PDGFRβ-expressing xenografts using an Affibody molecule. Further development of radiolabeled Affibody molecules might provide a useful clinical imaging tool for PDGFRβ expression during various pathologic conditions.
The platelet-derived growth factor (PDGF) signaling axis includes 5 ligands composed of disulfide-bonded dimers of A, B, C, and D polypeptide chains as well as an AB heterodimer and 2 platelet-derived growth factor receptors, PDGFRα and PDGFRβ, which are receptor tyrosine kinases (1). Activation of PDGF receptors by ligand binding triggers intracellular signaling cascade, resulting in cell migration, survival, and proliferation (2). PDGF-A and PDGF-B, and their receptors, PDGFRα and PDGFRβ, are expressed in a largely nonoverlapping pattern, suggesting a paracrine mode of action. PDGF-B and PDGFRβ are mainly expressed in developing vasculature, where PDGF-B is produced by endothelial cells and PDGFRβ is expressed by mural cells (i.e., pericytes and vascular smooth muscle cells) (3). In the adult, PDGF signaling stimulates wound healing and regulates the interstitial fluid pressure of tissues (4).
Excessive signaling of PDGF has been found in diseases involving excessive cell proliferation, such as cancer, atherosclerosis, and various fibrotic conditions (4). PDGFs and their receptors play an important role in many cancers including lung cancer, prostate cancer, renal cell carcinoma, chronic monomyelocytic leukemia, and glioblastoma (5). PDGFR activation can stimulate tumor growth directly by autocrine stimulation. Paracrine stimulation of PDGFRβ on tumor blood vessels pericytes and stromal fibroblasts contributes to tumor growth and homeostasis (4). Not surprisingly, expression of PDGFRβ is a prognostic biomarker for more aggressive disease. In breast and prostate cancer, stromal PDGFRβ expression correlates with shorter survival (6,7). Increased expression of PDGFRβ is also associated with poor overall survival in angiosarcoma (8), hepatocellular carcinoma (9), and a subset of non–small cell lung cancers (10).
Blocking PDGFRβ signaling reduces interstitial fluid pressure in tumors and improves uptake of antitumor drugs (4). PDGFRβ is one of the targets for tyrosine kinase inhibitors (TKI), such as imatinib, sunitinib, and sorafenib. The use of these TKIs improves survival of patients with hepatocellular carcinoma, gastrointestinal stromal tumors, and renal cell carcinoma. Although TKIs generally target several tyrosine kinases, research suggests that PFDGRβ is a key target for imatinib in several malignancies (11–13). There is a well-recognized unmet clinical need for biomarkers for selection of patients who might benefit from such therapy (14). Furthermore, there is a need for biomarkers for the detection of a drug effect on the target (i.e., pharmacodynamic biomarkers) and for the detection of onset of resistance in initial responders.
The histopathologic evaluation of biopsy samples is considered as a gold standard in the determination of relevant molecular targets and in monitoring of response to therapy. However, morbidity associated with invasive biopsies limits their use. In addition, spatial and temporal heterogeneity in the expression of molecular targets reduces the diagnostic value of biopsies. Radionuclide molecular imaging is an alternative to biopsies, permitting the visualization of molecular events by a noninvasive procedure.
Currently, the response to many TKIs is monitored indirectly, for example, by measuring changes in 18F-FDG uptake in gastrointestinal stromal tumors treated by imatinib (15). This method provides information mainly about metabolism in the lesion and its changes in response to therapy. Imaging of receptor tyrosine kinase expression might provide important additional diagnostic information that cannot be obtained from 18F-FDG imaging. The development of radionuclide-labeled tracers for the visualization of receptor tyrosine kinases is therefore one focus in radiopharmaceutical research (16–19). Radiolabeled TKIs have the potential advantage of enabling direct imaging of the therapeutic target. The synthesis of the 11C-labeled PDGFRβ-targeting TKIs sorafenib (20), sunitinib (21), and imatinib (22) has been reported. However, experiments in baboons demonstrated that 11C-imatinib yields high accumulation of radioactivity in liver and gallbladder (22), reflecting a general problem of radiolabeled TKI. Imaging of intracellular targets requires that a tracer can not only permeate cellular membranes before binding to its target and but also is efficiently washed away from nonspecific compartments lacking the target. The penetration of cellular membranes requires elevated hydrophobicity. However, hydrophobicity is usually associated with an elevated level of hepatic uptake and hepatobiliary excretion (23). Indeed, a high hepatic uptake (exceeding tumor uptake) has been found in all in vivo studies with radiolabeled TKIs. Thus, the use of radiolabeled TKIs might be restricted to imaging of extrahepatic metastases. This is a serious drawback because the liver is a major metastatic site for many cancers.
Imaging of the extracellular domain of PDGFRβ might be a superior alternative to radiolabeled TKIs. A new class of non–immunoglobulin-based targeting proteins, Affibody molecules (Affibody AB, Sweden), have been used for the visualization of several extracellular molecular targets (24). For example, radiolabeled anti–human epidermal growth factor receptor (HER2) Affibody molecules have provided high-contrast imaging of HER2-expressing tumors a few hours after injection both in preclinical (25) and clinical studies (26,27). Recently, we have developed the Affibody molecule Z02465, showing high-affinity binding to both human and murine PDGFRβ (KD = 0.5 and 7.1 nM, respectively) (28). This Affibody molecule competes with the natural ligand, PDGF-BB, for binding to PDGFRβ and inhibits ligand-induced receptor phosphorylation (28).
We hypothesized that radiolabeled derivatives of the Affibody molecule Z02465 would be suitable for radionuclide imaging of PDGFRβ expression in vivo. To test this hypothesis, the Z02465 was redesigned. A unique thiol group was created by introducing a cysteine at the C terminus, enabling site-specific labeling using thiol-directed chemistry. In addition, the first 2 amino acids at the N terminus were changed from VD- to AE-, because such substitution improves biodistribution of Affibody molecules (29). The resulting variant of denoted Z09591 was labeled with 111In and evaluated for targeting of PDGFRβ-expressing U-87 MG cells in vitro and in vivo.
MATERIALS AND METHODS
Statistics
Data on cellular uptake and biodistribution were analyzed by unpaired, 2-tailed t test using GraphPad Prism (version 4.00 for Windows; GraphPad Software) to determine significant differences (P < 0.05).
Affibody Molecule
The Affibody molecule Z09591-Cys was expressed in Escherichia coli and purified essentially as described earlier (30). The maleimido derivative of the DOTA chelator (Macrocyclics) was conjugated to the cysteine thiol of Z09591 as described earlier (30). The identity of the conjugate (designated as DOTA-Z09591) was confirmed and the purity evaluated by high-performance liquid chromatography and in-line mass spectrometry (HPLC-MS) using an Agilent 1100 HPLC/MSD equipped with electrospray ionization and a single quadrupole as described earlier (30). A stock solution of DOTA-Z09591 (1.32 mg/mL in 0.2 M sodium acetate, pH 5.4) was stored frozen.
Labeling and Stability Assessment
For labeling of DOTA-Z09591, 40 μL of stock solution (52.8 μg of protein) were mixed with 40 μL of 0.2 M ammonium acetate, pH 5.5, and 80 μL of 111In stock solution (30–60 MBq). The reaction mixtures were incubated at 80°C for 30 min and thereafter analyzed by radio–instant thin-layer chromatography (ITLC). To cross-validate the ITLC results, a sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis and size-exclusion chromatography were performed (supplemental data; supplemental materials are available at http://jnm.snmjournals.org). To evaluate the stability of labeling, the 111In-labeled conjugates were incubated with a 500-fold excess of ethylenediaminetetraacetic acid (EDTA) for 4 h at room temperature and then analyzed using ITLC. Stability in human blood plasma was analyzed using size-exclusion chromatography, as described in the supplemental data.
In Vitro Cell Binding and Processing of 111In-Labeled Affibody Molecules
The PDGFRβ-expressing U-87 MG glioma cell line (American Type Culture Collection) was used for binding specificity and cellular processing studies. Details concerning culturing and seeding are provided in the supplemental data.
An in vitro specificity test was performed, and the rate of internalization of 111In-DOTA-Z09591 by U-87 MG cells was evaluated according to the method described earlier (31). Experimental details are provided in the supplemental data.
The affinity of 111In-DOTA-Z09591 to U-87 MG cells and the number of binding sites were determined using LigandTracer (Ridgeview Instruments AB, Sweden) (32). Experimental details are provided in the supplemental data. The measurements were repeated 6 times at room temperature. Two additional measurements were performed at 4°C.
In Vivo Evaluation of 111In-DOTA-Z09591
All animal experiments were planned and performed in accordance with national legislation on laboratory animals’ protection and were approved by the Ethics Committee for Animal Research in Uppsala. Female BALB/C nu/nu mice were from Taconic M&B, 6–8 wk old at arrival. For xenografting, 2 × 106 U-87 MG cells in phosphate-buffered saline were subcutaneously implanted in the right hind leg. At the time of biodistribution experiments (3 wk after tumor implantation), the average mouse weight was 19.9 ± 0.9 g.
To study the influence of injected DOTA-Z09591 protein dose on targeting, the mice were divided into 4 groups, 4 animals each, and injected with 30 kBq of 111In-DOTA-Z09591 in 100 μL of phosphate-buffered saline. The average tumor weight was 0.34 ± 0.1 g. The injected protein dose was adjusted to 0.1, 0.5, 1, or 70 μg by the addition of nonlabeled DOTA-Z09591. At 2 h after injection, mice were sacrificed by exsanguination under anesthesia. Tumors as well as samples of tissues were collected and weighed, and their radioactivity was measured. The whole gastrointestinal tract (GI tract), with the exception of a small segment of colon, was taken together with its content to assess hepatobiliary excretion. Tissue uptake was calculated as percentage injected dose per gram (%ID/g). Radioactivity in the gastrointestinal tract was calculated as percentage injected dose (%ID) per whole sample.
An additional experiment was performed to assess the potential effects of high doses of the PDGFRβ-specific DOTA-Z09591 on the biodistribution of any type of Affibody molecules. The effect of coinjection of 70 μg of DOTA-Z09591 on the biodistribution of non-PDGFRβ–specific (HER2-binding) 111In-DOTA-Z02395 Affibody molecule was studied in female non–tumor-bearing BALB/C nu/nu mice. In the test group, 111In-DOTA-Z02395 (30 kBq, 1 μg) was coinjected with 70 μg of DOTA-Z09591. In the control groups, only the 111In-DOTA-Z02395 Affibody molecule (30 kBq) was injected at protein doses of 1 and 70 μg per mouse. The biodistribution was measured at 2 h after injection as described above.
The biodistribution of 111In-DOTA-Z09591 at 1, 2, and 4 h in mice bearing U-87 MG xenografts was investigated at an injected dose of 1 μg. The mice were randomized into 3 groups, 4 animals each, and injected with 30 kBq of 111In-DOTA-Z09591 in 100 μL of phosphate-buffered saline and sacrificed at predetermined time points. The biodistribution was measured at predetermined time points as described above.
In the imaging experiment, a U-87 MG xenograft–bearing mouse was injected with 1 MBq (amount of peptide, 1 μg) of 111In-DOTA-Z09591. Immediately before imaging, 3 h after injection, the mouse was sacrificed. The SPECT/CT imaging experiment was performed using the Triumph Trimodality system (Gamma Medica), an integrated small-animal SPECT/PET/CT platform. A CT image (field of view, 53.82 mm, 512 projections; scanning time, 2.13 min) was acquired to position the body of the animal in the scanner and for assisting in image analysis. SPECT acquisition was performed using 2 solid-state cadmium zinc telluride camera heads equipped with 5-pinhole 75A10 collimators. The following acquisition parameters were used for SPECT: nongated; field of view, 53.82 mm; tomographic; 32 projections; 1,000 s. SPECT raw data were reconstructed with FLEX SPECT software (Gamma Medica), which uses an ordered-subset expectation maximization iterative reconstruction algorithm. CT raw files were reconstructed by filtered backprojection. SPECT and CT data were fused and analyzed in PMOD (PMOD Technologies).
RESULTS
Labeling and Stability Assessment
According to mass-spectrometry analysis, 95% of the Affibody molecules were conjugated with a single DOTA chelator. Five percent remained unconjugated and exist in dimeric form. The radiochemical yield of 111In-DOTA-Z09591 was 99.3% ± 0.6% (specific radioactivity ≤ 1.1 MBq/μg) after labeling at 80οC for 30 min. Both radio–SDS-PAGE and size-exclusion chromatography analysis showed a radioactivity peak corresponding to Affibody molecules (Supplemental Figs. 1 and 2). A challenge with a 500-fold molar excess of EDTA during 4 h demonstrated high stability of the label. The radiochemical purity of the EDTA-treated samples was 96.6% ± 0.4% whereas the purity of the untreated control was 97.8% ± 0.4%—that is, the difference was within the accuracy of the analytic method. After 1- and 4-h incubation in human blood plasma, the Affibody-associated fraction of radioactivity was 99.2% ± 0.6% and 97.3% ± 1.1%, respectively (Supplemental Fig. 2).
Because of the high purity, 111In-DOTA-Z09591 was used in further experiments without purification after labeling.
In Vitro Cell Binding and Processing of 111In-Labeled Affibody Molecules
The preincubation of PDGFRβ-expressing U-87 MG glioma cells with nonlabeled conjugates significantly (P < 0.05) decreased the binding of the radiolabeled Affibody molecules, suggesting that the binding of 111In-DOTA-Z09591 to the receptor was saturable (Fig. 1A).
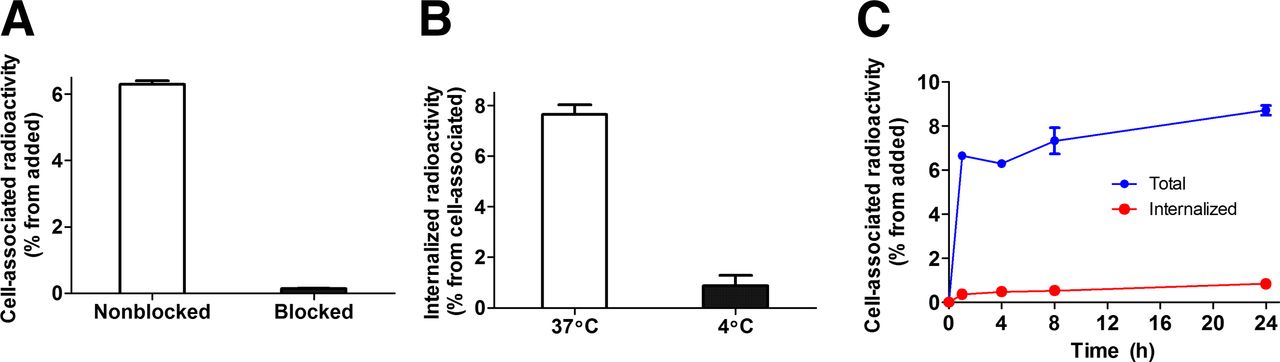
(A) In vitro binding specificity of 111In-DOTA-Z09591 to glioma U-87 MG cell line. In blocking group, receptors were presaturated by 200-fold excess of nonlabeled DOTA-Z09591. (B) Internalization of 111In-DOTA-Z09591 by glioma U-87 MG cells after 4-h incubation at 37°C and 4°C. (C) Internalization of 111In-DOTA-Z09591 by glioma U-87 MG cells during continuous incubation. Cells were incubated with conjugate (0.5 nM) at 37°C. Data presented as mean values from 3 cell dishes ± SD. Error bars might be not seen because they are smaller than symbols.
Data concerning the binding and internalization of 111In-DOTA-Z09591 by PDGFRβ-expressing U-87 MG glioma cell lines are presented in Figures 1B and 1C. The radioactivity in the alkaline fraction was 0.8% ± 0.4% of the cell-associated radioactivity after incubation at 4°C (Fig. 1B), reflecting the accuracy of the method. The radioactivity in the alkaline fraction after incubation at 37°C was significantly higher, 7.6% ± 0.4% of the cell-associated radioactivity. The kinetics of binding was rapid up to around 1 h, then slower. The internalization of 111In-DOTA-Z09591 was relatively inefficient, with a slow increase of the internalized fraction to around 10% after 24 h.
According the LigandTracer measurements, the association rate was (2.0 ± 0.3) × 105 M−1s−1 and dissociation rate (1.8 ± 0.4) × 10−5 s−1. The dissociation constant for 111In-DOTA-Z09591 binding to living U-87 MG cells was 92 ± 10 pM at ambient temperature (22°C) and 94 ± 17 pM at 4°C. The cell line displayed 36,000 ± 4,000 binding sites per cell.
In Vivo Evaluation of 111In-DOTA-Z09591
Data concerning the biodistribution of 111In-DOTA-Z09591 in BALB/C nu/nu mice bearing U-87 MG xenografts at 2 h after injection of 0.1, 0.5, 1, or 70 μg are presented in Table 1. The biodistribution of 111In-DOTA-Z09591 was characterized by a rapid disappearance from blood and normal tissues and build-up in the kidney, probably resulting from reabsorption in proximal tubuli after glomerular filtration. The accumulation of radioactivity in the gastrointestinal tract with its content (a measure for hepatobiliary excretion) was low (<3% of injected radioactivity). At all protein doses, the uptake in tumor was appreciably higher than uptake in any other organ or tissue except for the kidney. Hepatic uptake was significantly higher after injection of 0.1 μg than other doses. There were tendencies to decreasing uptake in the lung, stomach, and muscle with the increase injected protein dose from 0.1 to 1 μg per mouse, but the difference was not significant. Similarly, a tendency of increased tumor uptake with increased dose in this range did not reach significance. The tumor uptake (5.6 ± 1.1 to 7.2 ± 2.4 %ID/g) was in the expected range for Affibody molecules binding to a molecular target presented at this level. Increasing the injected protein dose to 70 μg per mouse had a strong effect on the biodistribution (Table 1; Supplemental Fig. 4). Tumor uptake was reduced to 0.89 ± 0.04 %ID/g, showing that uptake of 111In-DOTA-Z09591 in tumor xenografts is saturable. There was also a significant decrease of uptake of other organs and tissues, except for the kidneys.
Biodistribution of 111In-DOTA-Z09591 at 2 Hours After Injection in BALB/C nu/nu Mice Bearing U-87 MG Xenografts
The coinjection of HER2-targeting 111In-DOTA-Z02395 Affibody molecule with 70 μg of DOTA-Z09591 was performed to further elucidate the nature of this phenomenon, for example, potential effects on vascular permeability or glomerular filtration rate. The results are presented in Table 2. The coinjection of DOTA-Z09591 did not cause significant difference in uptake of the HER2-binding 111In-DOTA-Z02395 in any normal organ or tissue, except for the stomach and colon, in which uptake was increased. The coinjection of the same amount of cold DOTA-Z02395 did not have any effect on 111In-DOTA-Z02395 uptake, in agreement with earlier data (27), suggesting that injection of 70 μg of DOTA-Z09591 does not influence vascular permeability or excretion rate of Affibody molecules.
Influence of Coinjection of DOTA-Z09591 on Biodistribution of Anti-HER2 111In-DOTA-Z02395 at 2 Hours After Injection in BALB/C nu/nu Mice
The data concerning tumor-to-organ ratios at 2 h after injection of 0.1, 0.5, and 1 μg of 111In-MMA-DOTA-Z9591 in BALB/C nu/nu mice bearing U-87 MG xenografts are presented in Table 3. There is a tendency of increased ratios with increased dose in this range. However, a significant difference was observed only for tumor-to-stomach, tumor-to-colon, and tumor-to-muscle ratios between 0.1 and 1 μg of injected protein. Because of a more favorable biodistribution profile, an injected dose of 1 μg was selected for further studies.
Tumor-to-Organ Ratios of 111In-DOTA-Z09591 Affibody Molecule (2 Hours After Injection) in BALB/C nu/nu Mice Bearing U-87 MG Xenografts
The data concerning the biodistribution of 111In-DOTA-Z09591 in BALB/C nu/nu mice bearing U-87 MG xenografts at 1, 2, and 4 h after injection of 1 μg are presented in Table 4. The data demonstrate a rapid clearance from all organs and tissues except from the kidney. The radioactivity uptake in tumor was equal at 1 and 2 h after injection but decreased significantly at 4 h in comparison with 1 h after injection. Tumor-to-organ ratios (Table 5) appear to increase over time, although the only significant increase between 2 and 4 h was for the tumor-to-colon ratio. On the basis of these data, an optimal imaging time was determined to be between 2 and 4 after injection.
Biodistribution of 111In-DOTA-Z09591 (Injected Dose 1 μg) at 1, 2, and 4 Hours After Injection in BALB/C nu/nu Mice Bearing U-87 MG Xenografts
Tumor-to-Organ Ratios for 111In-DOTA-Z09591 Affibody Molecule (Injected Dose 1 μg) at 1, 2, and 4 Hours After Injection in BALB/C nu/nu Mice Bearing U-87 MG Xenografts
Small-animal SPECT/CT imaging, performed 3 h after injection (Fig. 2), confirmed the results of the biodistribution experiments. A substantial renal retention of radioactivity was visualized. Other organs and tissues were not seen. 111In-DOTA-Z09591 enabled imaging of the PDFGRβ-expressing U-87 MG xenograft. Rapid clearance of the conjugates provided low background radioactivity and ensured a high imaging contrast.

Imaging of PDFGRβ expression in U-87 MG xenograft (arrow) in BALB/C nu/nu mouse using small-animal SPECT/CT (maximum-intensity projection). Image was acquired at 3 h after injection. SPECT scale, 0.02–0.04; CT scale, 400–2000 HU.
DISCUSSION
A first imaging agent for noninvasive visualization of PDGFRβ expression is presented in this report. To our knowledge, no adequate imaging agent for PDGFRβ has previously been presented. PDGFRβ inhibition by imatinib has been assessed indirectly, by monitoring the tumor uptake of an anti–carcinoembryonic antigen antibody (33) or by studying the decrease of 18F-FDG uptake (15). Askoxylakis et al. (34) have reported the selection and evaluation of unrestricted dodecapeptide-targeting PDGFRβ. The reported affinity of this peptide is low, showing an IC50 (half maximal inhibitory concentration) of 1.4 μM. In vivo, the accumulation of radioactivity in tumor was lower than in blood and liver, excluding high-contrast imaging. In contrast, 111In-DOTA-Z09591 yielded a tumor-to-blood ratio of 28 ± 14, a tumor-to-liver ratio of 9 ± 4, and a tumor-to-bone ratio of 13 ± 4 at 2 h after injection, indicating that this tracer might be used for the detection of an increased level of PDGFRβ in all major metastatic sites by molecular imaging.
A unique cysteine at the C terminus of the Affibody molecule Z09591 may be applied for labeling using a variety of alternative radionuclides. In this study, thiol-directed chemistry was used for site-specific conjugation of the versatile DOTA chelator, which was used to chelate 111In. Radiolabeling did not affect the specificity of 111In-DOTA-Z09591 binding to PDGFRβ-expressing U-87 MG cells (Fig. 2A). The internalization of 111In-DOTA-Z09591 by U-87 MG was rather slow (Fig. 2C). This has been seen with Affibody molecules binding other receptors (30,31). With PDGFRβ, slow internalization may be due to the monovalent mode of binding of the Affibody molecule, because dimerization has been shown to drive endocytosis of PDGFRβ (35). With slow internalization, a strong binding to the cell surface target is required for obtaining high accumulation of radioactivity. The dissociation constant of 111In-DOTA-Z09591 for binding to U-87 MG cells was assessed as 92 ± 10 pM. Previous studies suggest that a tracer displaying an affinity in this range meets the requirement for visualization of targets with relatively low expression and slow internalization rate (36). We used a residualizing 111In label in this first study, because it permits the detection of sites of uptake and catabolism in vivo. However, this was associated with a high renal retention of radioactivity. A slow internalization rate suggests that nonresidualizing labels, for example, radiohalogens or some forms of technetium labels, might be used in the future studies. Experience with derivatives of slowly internalizing HER2-binding Affibody molecules demonstrated that this enables a substantial reduction of renal retention of radioactivity without compromising tumor uptake (37,38).
In humans, and probably also in mice, there is expression of PDGFRβ in normal organs and tissues (in normal vasculature and, according to Human Protein Atlas, http://www.proteinatlas.org/, in a subset of cells in the stomach, colon, and kidney). Therefore, we were concerned that it might be challenging to obtain a sufficient contrast for imaging the PDGFRβ overexpressing tumor in mice using a tracer that binds both human and murine PDGFRβ. Increasing the injected dose of unlabeled protein may provide preferential block of binding to normal tissues with low antigen density (39). This requires that the receptor density is higher in the tumor, sufficiently high to avoid PDGFRβ saturation at a protein concentration providing low uptake in normal tissues. However, the expression level in U-87 MG cells is rather low (36,000 receptors/cell). Earlier, we studied targeting of HER2-expressing LS174T xenografts, which display a target level similar to the level of PDGFRβ in the current tumor cell line U-87. With the monomeric HER2-binding Affibody molecule, an increase of the injected protein dose above 1 μg per mouse was shown to cause an abrupt decrease of the tumor uptake (40). For this reason, the dose range tested in the present study was 0.1–1 μg of protein. Except for the liver, no difference in accumulation of radioactivity in various tissues reached significance in this range (Table 1). However, tumor-to-stomach, tumor-to-colon, and tumor-to-muscle ratios were higher after an injected dose of 1 μg than after a dose of 0.1 μg. In contrast, increasing the protein dose to 70 μg caused a significant decrease of uptake in all organs and tissues except for the kidney (Table 1; Supplemental Fig. 4). Tumor uptake was reduced more than 7-fold in comparison with the 1-μg injected dose, which is taken as evidence of saturable binding of 111In-DOTA-Z09591 to PDGFRβ in the U-87 MG xenografts. The decrease of uptake in all organs and tissues except for the kidney indicated saturable binding to murine PDGFRβ receptors at these sites, likely including the vasculature. However, PDGFRβ is involved in the regulation of vascular permeability and is also expressed in glomeruli. The parental PDGFRβ binder Z02465 possesses some antagonistic properties, for example, it inhibits PDGF-B–induced phosphorylation of PDGFRβ (28). Therefore, interaction of large doses of DOTA-Z09591 with murine PDGFRβ might alter vascular permeability or glomerular filtration rate and influence the biodistribution of 111In-DOTA-Z09591. If the organ uptake reduction is due to such effects, injection of 70 μg of DOTA-Z09591 should have the same influence on the biodistribution of any Affibody molecule. However, the control experiment showed that 70 μg of DOTA-Z09591 did not have any effect on the level of nonspecific uptake of the HER2-binding 111In- DOTA-Z02395 in any tissue (Table 3). Taken together, these data support the hypothesis that the uptake of 111In-DOTA-Z09591 in various nontumor tissues was PDGFRβ-specific.
There was a significant increase of the tumor-to-organ ratios between 1 and 2 h after injection. However, there was little difference in tumor-to-organ ratios between 2 and 4 h after injection (Table 5), suggesting that an optimal imaging time is between the later time points. This assumption is supported by clear visualization of the PDGFRβ-expressing xenograft at 3 h after injection. This high contrast shortly after injection should enable the use of short-lived positron emitters, such as 68Ga or 18F, as labels. The use of PET might provide a further increase of imaging sensitivity and improve quantification accuracy.
CONCLUSION
This study has demonstrated the feasibility of in vivo targeting of PDGFRβ-expressing xenografts using Affibody molecules. Further development of radiolabeled Affibody molecules might provide a useful clinical tool for imaging of PDGFRβ levels during various pathologic conditions.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This research was financially supported by grants from the Swedish Cancer Society (Cancerfonden) and Swedish Research Council (Vetenskapsrådet). Fredrik Y. Frejd and Per Jonasson are employees of Affibody AB. Affibody AB holds intellectual property for Affibody molecules. Academic partners have control of any data that might present a conflict of interest for employees or consultants. No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Jan. 9, 2014.
- © 2014 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication February 19, 2013.
- Accepted for publication October 21, 2013.





{kind=link}
{kind=link}