


Abstract
The PET radioligand 11C-CUMI-101 was previously suggested as a putative agonist radioligand for the serotonin 1A (5-hydroxytryptamine 1A [5-HT1A]) receptor in recombinant cells expressing human 5-HT1A receptor. However, a recent study showed that CUMI-101 behaved as a potent 5-HT1A receptor antagonist in rat brain. CUMI-101 also has moderate affinity (Ki = 6.75 nM) for α1 adrenoceptors measured in vitro. The current study examined the functional properties and selectivity of CUMI-101, both in vitro and in vivo. Methods: The functional assay was performed using 35S-GTPγS (GTP is guanosine triphosphate) in primate brains. The cross-reactivity of CUMI-101 with α1 adrenoceptors was performed using in vitro radioligand binding studies in rat, monkey, and human brains as well as in vivo PET imaging in mouse, rat, and monkey brains. Results: CUMI-101 did not stimulate 35S-GTPγS binding in primate brain, in contrast to 8-OH-DPAT, a potent 5-HT1A receptor agonist. Instead, CUMI-101 behaved as a potent 5-HT1A receptor antagonist by dose-dependently inhibiting 8-OH-DPAT–stimulated 35S-GTPγS binding. Both in vitro and in vivo studies showed that CUMI-101 had significant α1 adrenoceptor cross-reactivity. On average, across all 3 species examined, cross-reactivity was highest in the thalamus (>45%) and lowest in the neocortex and cerebellum (<10%). PET imaging further confirmed that only preblocking with WAY-100635 plus prazosin decreased 11C-CUMI-101 brain uptake to that of self-block. Conclusion: CUMI-101 behaves as a 5-HT1A receptor antagonist in primate brain, with significant, regional-dependent α1 adrenoceptor cross-reactivity, limiting its potential use as a PET radioligand in humans.
- CUMI-101
- 5-HT1A receptor
- α1 adrenoceptor
- cross-reactivity
- positron emission tomography (PET)
- 35S-GTPγS
- homogenate binding
The serotonin 1A receptor is a G-protein–coupled receptor (GPCR) bound to heterotrimeric α, β, and γ G-protein subunits (1). 5-HT1A receptors exhibit two affinity states—high (coupled to G protein) and low (uncoupled). Agonists bind to the high-affinity state of the GPCR and activate secondary cascades downstream. In contrast, antagonists bind to both the high- and the low-affinity states of the GPCR but do not activate receptor signaling. Of 5-HT’s 16 distinct receptor subtypes, the 5-HT1A receptor is known to play a crucial role in the pathophysiology and treatment of major depressive disorder and is a potential biomarker candidate. Currently, no suitable biomarker for major depressive disorder exists.
5-HT1A receptor density can be measured in intact, living brains using PET (2). To date, several PET radioligands have been developed to measure the 5-HT1A receptor; the five most commonly used are [carbonyl-11C]WAY-100635, 11C-RWAY, 18F-FCWAY, 18F-mefWAY, and 18F-MPPF (3). However, all of these radioligands are antagonists and, as such, do not discriminate between the active (high-affinity) and inactive (low-affinity) state of the receptor. In addition, none of the current 5-HT1A receptor radioligands is sensitive to endogenous fluctuations in intrasynaptic 5-HT. This is particularly relevant to the search for biomarker candidates because the high-affinity states to which an agonist binds may be primarily affected under disease conditions, and agonists might be more sensitive to fluctuations in endogenous 5-HT levels. As a result, an agonist 5-HT1A receptor PET radioligand has been much sought.
A 2007 study reported the initial synthesis and in vivo evaluation of a potential agonist 5-HT1A receptor radioligand, 11C-CUMI-101 (4). In baboons, 11C-CUMI-101 was shown to have optimal brain uptake, good washout, and a plasma free fraction of 60% (5). CUMI-101 exhibited agonist-like properties by dose-dependently stimulating 35S-GTPγS (GTP is guanosine triphosphate) binding in recombinant Chinese Hamster Ovary (CHO) cells expressing human 5-HT1A receptor (4). In contrast, a recent study showed that, in native rat brain tissues, CUMI-101 behaved as a potent 5-HT1A receptor antagonist (6). Thus, it is presently unknown whether CUMI-101 acts as an agonist or an antagonist in human and nonhuman primate brains.
The issue of α1 adrenoceptor binding further complicates the picture. Initially, the affinity of CUMI-101 for 5-HT1A receptors was determined using bovine hippocampal membranes. Competitive binding studies of CUMI-101 with 3H-8-OH-DPAT—a potent 5-HT1A receptor agonist—showed the affinity to be in the subnanomolar range (inhibition constant [Ki], 0.15 nM) (4). Ki is obtained from heterologous displacement, and KD (dissociation constant) is obtained from homologous displacement assays. However, CUMI-101 also had moderate affinity to α1 adrenoceptors (Ki = 6.75 nM). α1 adrenoceptors consist of three highly homologous subtypes: α1A, α1B, and α1C. Studies show that many adrenergic drugs, especially ones targeting α1 adrenoceptors, have a nanomolar affinity for 5-HT1A receptors (7). Whether CUMI-101’s moderate in vitro affinity would cause in vivo binding to brain α1 adrenoceptors is also unknown.
The present study sought to answer two key questions regarding the functional property and selectivity of CUMI-101. First, does CUMI-101 behave as an agonist or antagonist? Second, does CUMI-101 demonstrate cross-reactivity with α1 adrenoceptors? In this paper, cross-reactivity was defined as the nonselective property of CUMI-101 to specifically bind to α1 adrenoceptors in addition to its intended target—5-HT1A receptors. To address the first question, functional assays were performed using 35S-GTPγS in brain homogenates of rat, monkey, and human. To assess whether CUMI-101 binds to α1 adrenoceptors, both in vitro radioligand binding studies and in vivo PET imaging studies were done in rodent and primate brains.
MATERIALS AND METHODS
Animal and human postmortem tissues were obtained from either the National Institute of Mental Health or Columbia University. All animals received standard laboratory care, including daily feedings and access to water ad libitum. The study was approved by the Animal Care and Use Committee of the National Institute of Mental Health.
Membrane Homogenization
Brain tissue homogenates were prepared from rat, monkey, and human brains using a Polytron Homogenizer in 50 mM Tris-HCl (pH 7.4) and centrifuged at 25,000g for 30 min at 4°C. The supernatant was discarded, and the pellet was resuspended in the same buffer so that the final concentration was approximately 100 mg of wet tissue per milliliter. Tissues were stored at −80°C until the day of the experiment. We chose brain regions where CUMI’s cross-reactivity could be examined using both in vivo and in vitro techniques. With regard to 5-HT1A receptors, we chose 2 regions with high density—hippocampus and neocortex. Similarly, with regard to α1 adrenoceptors, we chose 2 regions with high density—neocortex and thalamus. We also included the cerebellum because it is commonly used as a reference tissue for 5-HT1A receptors.
Drugs and Radioligands
35S-GTPγS, 3H-CUMI-101, 3H-(±)-8-OH-DPAT, and 3H-prazosin were purchased from PerkinElmer. (+)-8-OH-DPAT, 5-HT, WAY-100635, GTPγS, and prazosin were purchased from Sigma. CUMI-101 was purchased from Alpha Biopharmaceuticals. All other reagents were purchased from Quality Biological.
Agonist-Stimulated 35S-GTPγS Binding
35S-GTPγS binding was performed in brain homogenates, as previously described with minor modifications (8). Briefly, brain tissues were thawed on ice and resuspended in binding buffer (50 mM Tris-HCl, 1 mM MgCl2, 100 mM NaCl, 1 mM EGTA, 1 mM DTT, 300 μM GDP, and adenosine deaminase [10 mU/mL], pH 7.4). Membrane aliquot (50 μg of protein) and drugs of interest were added to borosilicate vials. The reaction was initiated by adding 100 pM 35S-GTPγS, followed by a 30-min incubation in a light-shielded shaker at 30°C. Finally, reactions were terminated by rapid filtration under vacuum in ice-cold buffer (50 mM Tris-HCl, pH 7.4) through Whatman GF/B glass fiber filters. All assays were performed in triplicate.
3H Ligand Binding
Radioligand binding assays were performed as previously described (9). Briefly, brain tissues were thawed on ice and resuspended in binding buffer (5-HT1A receptors: 50 mM Tris-HCl, 10 mM MgCl2, and 1 mM EDTA; α1 adrenoceptors: 20 mM Tris-HCl, 145 mM NaCl; pH 7.4) to a final concentration of 1 mg of wet tissue per milliliter. The radioactivity concentrations for 3H-CUMI-101, 3H-(±)-8-OH-DPAT, and 3H-prazosin were in the range of 0.05–0.2 nM, so that final concentrations were below their KD values (4,10,11). The following were added sequentially to each borosilicate vial: 100 μL of radioligand, 100 μL of buffer/displacer (50 nM 8-OH-DPAT or prazosin; this concentration was ∼15–150 times the Ki values for the respective receptor subtype), and 800 μL of tissues all mixed in binding buffer solution. This was followed by a 30-min incubation in a light-shielded shaker at either 37°C or 23°C. The affinity of receptors can increase or decrease with temperature. We chose 23°C because it was close to the temperature used for the initial characterization of CUMI-101 (25°C) (4). We chose 37°C because it reflects in vivo body temperature. Finally, reactions were terminated by rapid filtration under vacuum in ice-cold binding buffer through Whatman GF/B glass fiber filter (presoaked for 30 min in 0.5% polyethyleneimine).
Radioactivity from 35S and 3H was measured (5 min per vial) in a liquid scintillation Ultima-Gold cocktail (4 mL per vial) in a β counter (PerkinElmer). Protein concentration was determined via the Micro BCA Protein Assay Kit protocol (Thermo Scientific).
Radiochemistry
11C-CUMI-101 was synthesized by methylation of the desmethyl precursor with 11C-MeI using the autoloop method as previously described (12). Desmethyl precursor (0.7 mg, 1.9 μmol) and tetrabutylammonium hydroxide (1.0 M in MeOH, 3 μL) were dissolved in DMF (anhydrous, 80 μL). The solution was loaded into an autoloop (Bioscan) and reacted with 11C-MeI for 5 min at room temperature. All contents inside the loop were loaded onto a semipreparative scale high-performance liquid chromatography column (Luna C18, 10 μ, 250 × 10 mm; Phenomenex) and eluted with an isocratic mixture of MeCN (40%) and aqueous ammonium hydroxide (1 mM, 60%) at a flow rate of 6 mL/min monitored with UV (254 nm) and radioactivity. Radioactive product (tR = 12.5 min) was collected, and solvents were removed under vacuum. Residue was redissolved in a formulation vehicle (10 mL) of saline containing 5% (v/v) ethanol and passed through an MP sterile filter (0.22 μ, Millex) to obtain 11C-CUMI-101 for injection. Radiochemical purity was greater than 99%, and chemical purity was greater than 98%.
PET Imaging
A 90- or 120-min dynamic PET scan was acquired in rat or monkey on a Focus 220 scanner (Siemens Medical Solutions) as previously described (13). For rat imaging, 15 male Sprague–Dawley rats (336 ± 66 g) were anesthetized with 1.5% isoflurane. 11C-CUMI-101 was injected (intravenously) through a penile vein catheter (29 ± 6 MBq; specific activity at time of injection, 67 ± 53 GBq/μmol). Dynamic PET scans were acquired after a bolus injection of 11C-CUMI-101 over 30 s. The dynamic frame sequences for PET imaging were 6 × 20, 5 × 60, 4 × 120, 3 × 300, 3 × 600, and 2 × 1,200 s in rats and 6 × 30, 3 × 60, 2 × 120, and 16 × 300 s in monkeys. To determine whether 11C-CUMI-101 binds to 5-HT1A or α1 adrenoceptors, we obtained PET scans under the following 5 conditions: baseline; preblocking with the 5-HT1A receptor antagonist, WAY-100635 (2.0 mg/kg); preblocking with the α1 adrenoceptor antagonist, prazosin (2.0 mg/kg); preblocking with WAY-100635 and prazosin; and self-blocking with CUMI-101 (2.0 mg/kg).
For monkey imaging, 5 rhesus monkeys (8.5 ± 2.4 kg) underwent a total of 6 PET scans with 11C-CUMI-101 (193 ± 30 MBq; specific activity at time of injection, 120 ± 46 GBq/μmol). Each session consisted of a baseline scan followed by a blocked scan with a 3-h interval to allow for radioactivity decay. Before scanning, each monkey was immobilized with ketamine (10 mg/kg, intramuscularly) to allow for endotracheal intubation. All animals were placed under isoflurane anesthesia (1.5%) throughout the scans. Before radioligand injection, a 10-min transmission scan was acquired using a 57Co point source for attenuation correction. The blocked scans were acquired under the following conditions: preblocking with WAY-100635 (0.5 mg/kg; n = 3), preblocking with prazosin (1.0 mg/kg; n = 2), and preblocking with WAY-100635 and prazosin (n = 1). Previously published studies noted that approximately complete receptor occupancy was achieved at these doses (4,14). For rats, we used 2.0 mg/kg, which was slightly higher than values reported in the literature, to ensure maximum receptor blockade. The estimated baseline occupancy of 5-HT1A receptors in the hippocampus was approximately 1.2% in rats and approximately 1% in monkeys (15,16). Arterial blood samples were obtained in all but preblocking with WAY-100635 and prazosin.
Plasma radiometabolites were separated using high-performance liquid chromatography (17). Parent plasma concentration was obtained as an input function for compartmental modeling. All preblocking agents were administered intravenously 30 min before radioligand injection. Data were reconstructed using 3-dimensional filtered backprojection with an image resolution of 1.7 mm in full width half maximum.
Image Analysis
With regard to the location of the 5-HT1A receptors, we selected the neocortex, thalamus, and hippocampus as our regions of interest. The cerebellum was used as the nonspecific reference region. In monkeys, our reference region contained 2 regions of interest (1 for each hemisphere) centered in the cerebellar white matter excluding the vermis. However, the partial-volume effect in PET may have resulted in some spillover from adjacent gray matter and vermis. The neocortex reflects a weighted combination of 5 different cortical regions: frontal, cingulate, temporal, parietal, and occipital cortices. For rats, the brain regions were drawn directly on coronal sections of the summed PET images. For monkeys, dynamic PET images were coregistered directly to an averaged template created from 6 individual monkey MR imaging scans in standardized space. Time–activity curves (TACs) were generated using predefined regions of interest for both the neocortex and the hippocampus (18). The concentration of radioactivity was expressed as standardized uptake value (SUV), a unitless value that is normalized for weight and injected activity. SUV = concentration (kBq/mL)/injected activity (kBq) × body weight (g). TACs were obtained and expressed as SUV. For monkeys, distribution volume (VT), which is the ratio of brain uptake to parent plasma, was calculated using a one-tissue-compartment model (19). Although the two-tissue-compartment model had a slightly better fit, a small k4 affected the stability of VT in many regions. Binding potential (BPND), which is the ratio at equilibrium of specific binding to nondisplaceable uptake, was also calculated using a simplified reference tissue model with cerebellar white matter as the reference region. All image analyses and compartmental modeling were performed with the FSL library (FMRIB Software Library) and PMOD 3.1 (PMOD Technologies Ltd.).
RESULTS
CUMI-101 Did Not Stimulate 35S-GTPγS Binding in Primate Brain
Unlike the 5-HT1A receptor agonist 8-OH-DPAT, CUMI-101 did not behave as an agonist in either monkey or human hippocampal tissue (Fig. 1). That is, CUMI-101 did not stimulate 35S-GTPγS binding in either monkey or human brain tissue. On the contrary, CUMI-101 dose-dependently blocked 8-OH-DPAT–stimulated 35S-GTPγS binding in human hippocampal tissue (Fig. 2). Similar to WAY-100635, a potent 5-HT1A receptor antagonist, CUMI-101 also blocked 8-OH-DPAT–induced 35S-GTPγS stimulation in both monkey and human hippocampal tissue.

Both 8-OH-DPAT (1 μM) and 5-HT (1 μM), but not CUMI-101 (10 μM), stimulated 35S-GTPγS binding in rat, monkey, and human hippocampal tissue. Similar to WAY-100635 (10 μM), a potent 5-HT1A receptor antagonist, CUMI-101 blocked 8-OH-DPAT–stimulated 35S-GTPγS binding in all 3 species. Bars represent mean ± SEM.
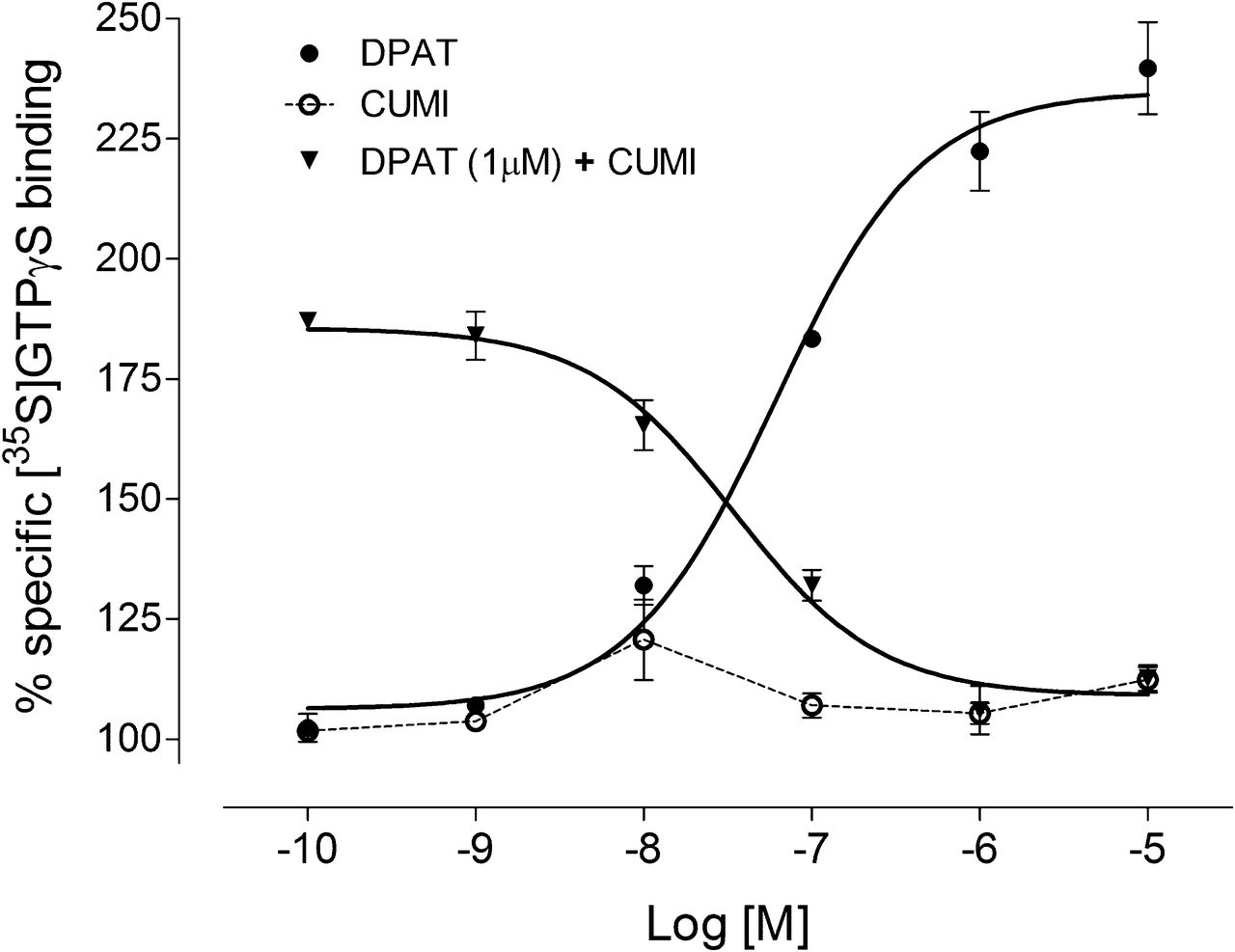
CUMI-101 dose-dependently blocked 8-OH-DPAT–stimulated 35S-GTPγS binding in human hippocampal tissues. Unlike 8-OH-DPAT, a potent 5-HT1A receptor agonist, CUMI-101 did not stimulate 35S-GTPγS binding. Bars represent mean ± SEM.
In Vitro CUMI-101 Bound to α1 Adrenoceptors in Brain Homogenates
CUMI-101 showed significant cross-reactivity with α1 adrenoceptors in vitro in rat, monkey, and human (Table 1). In rats, α1 adrenoceptor cross-reactivity was 45% in the thalamus and 42% in the neocortex. In monkeys, α1 adrenoceptor cross-reactivity was 50% in the thalamus and 12% in the neocortex. In humans, α1 adrenoceptor cross-reactivity was 43% in the thalamus and 10% in the neocortex. α1 adrenoceptor cross-reactivity was greater at 23°C than 37°C.
Percentage Displacement of 3H-CUMI-101 by (+)-8-OH-DPAT (Selective for 5-HT1A Receptors) and Prazosin (Selective for α1 Adrenoceptors)
In Vivo C-CUMI-101 Was Displaced by Prazosin
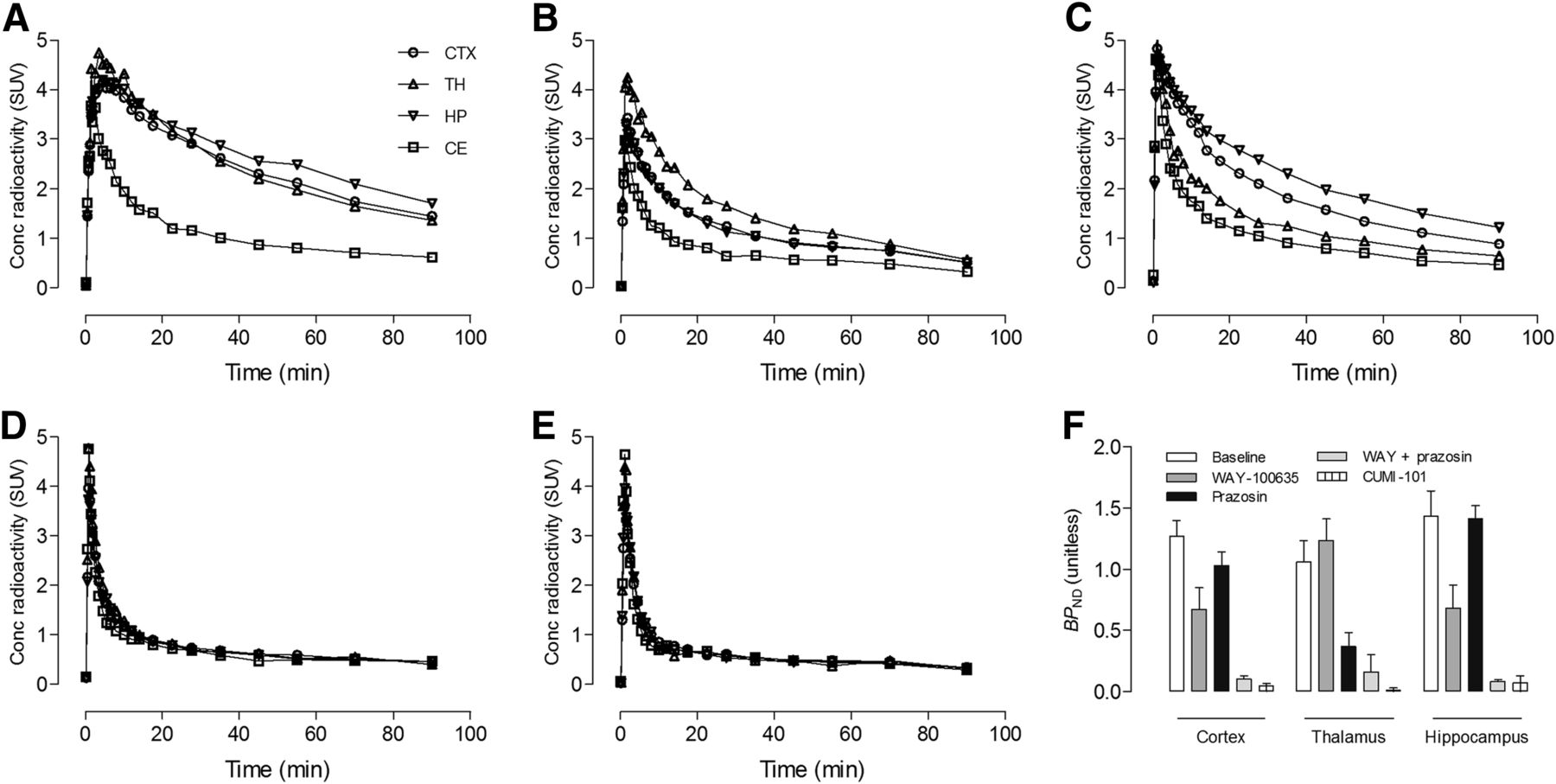
In rats, 11C-CUMI-101 had good uptake in the neocortex, thalamus, and hippocampus (Fig. 3A). Preblocking with WAY-100635 decreased brain uptake in all regions but not to levels obtained with self-block (i.e., CUMI-101) (Fig. 3B). Furthermore, WAY-100635 decreased hippocampal uptake by a greater extent than thalamic uptake. In contrast, preblocking with prazosin decreased thalamic uptake by a greater extent than hippocampal uptake (Fig. 3C). Only preblocking with WAY-100635 plus prazosin decreased brain uptake in all regions to that of self-block (Figs. 3D–3F). In mice, α1 adrenoceptor cross-reactivity was similar to that of rats, and only preblocking with WAY-100635 plus prazosin decreased brain uptake to that of self-block (Supplemental Fig. 1; supplemental materials are available at http://jnm.snmjournals.org).

α1 adrenoceptor cross-reactivity in rat brain. Shown are TACs for 11C-CUMI-101 under the following five conditions (n = 3 for each condition): baseline (A), preblocking with 5-HT1A receptor antagonist WAY-100635 (2 mg/kg) (B), preblocking with α1 adrenoceptor antagonist prazosin (2 mg/kg) (C), preblocking with WAY-100635 (2 mg/kg) plus prazosin (2 mg/kg) (D), and self-blocking with CUMI-101 (2 mg/kg) (E). (F) BPND values using cerebellum (CE) as reference region were compared for each condition in 3 brain regions: cortex (CTX), thalamus (TH), and hippocampus (HP). A–E show representative TACs from 1 animal for each condition. F shows mean BPND values ± SEM for each condition in A–E. Bars represent mean ± SEM.
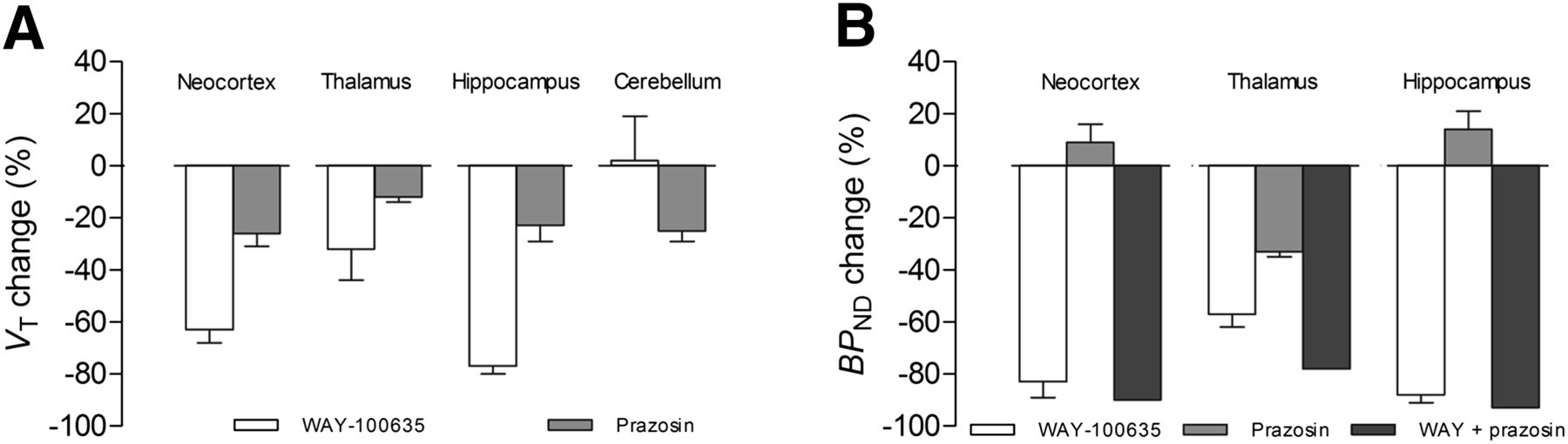
In monkeys, preblocking with WAY-100635 decreased VT in all regions (Fig. 4A). A decrease in specific binding as indicated by BPND was 83% ± 6% in the neocortex, 88% ± 3% in the hippocampus, and 57% ± 5% in the thalamus (Fig. 4B). However, VT also decreased after preblocking with prazosin, although to a lesser extent. The change in BPND with prazosin preblocking was greatest in the thalamus (33% ± 2%) where α1 adrenoceptors are abundant. Preblocking with WAY-100635 plus prazosin (n = 1) further decreased BPND values of 11C-CUMI-101 by 78% in the thalamus, 90% in the neocortex, and 93% in the hippocampus. In addition, prazosin also displaced VT values of 11C-CUMI-101 in the cerebellum by 25% ± 4% (Fig. 4A). To note, this decrease in cerebellar VT by prazosin preblocking may have led to the small increase in BPND for both the neocortex and the hippocampus (Fig. 4B).
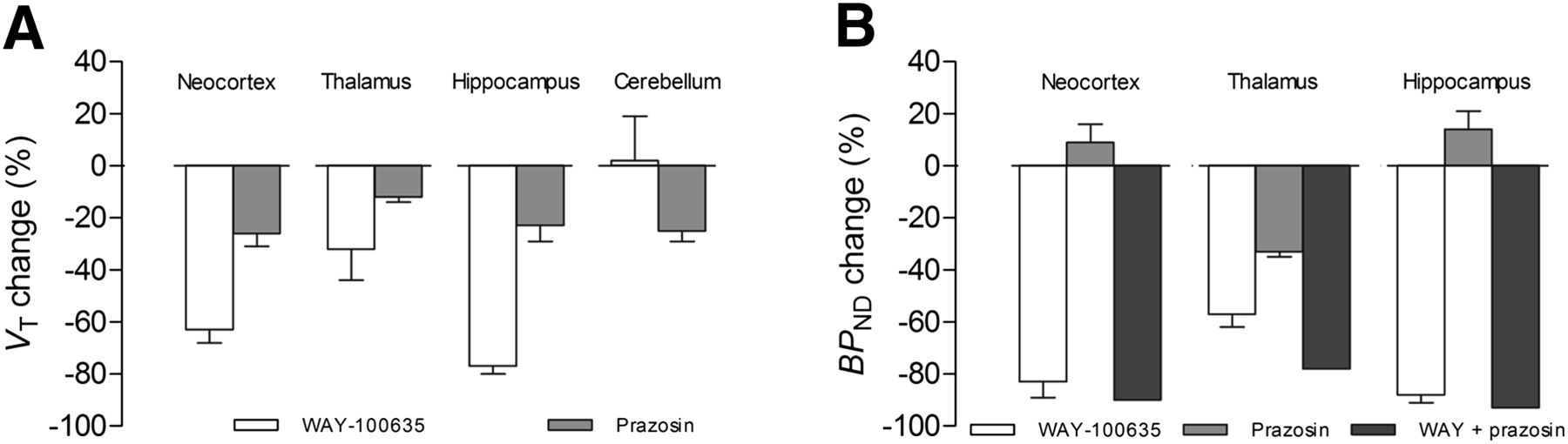
α1 adrenoceptor cross-reactivity in monkey brain. Shown is 11C-CUMI-101 displacement after preblocking with either WAY-100635 (0.5 mg/kg; n = 3), prazosin (1 mg/kg; n = 2), or WAY-100635 plus prazosin (n = 1). (A) VT decreased in all regions after preblocking with prazosin. (B) BPND decreased more after preblocking with WAY-100635 plus prazosin than after only preblocking with prazosin.
DISCUSSION
We found that CUMI-101 behaves as a potent 5-HT1A receptor antagonist in monkey and human brains. Furthermore, we replicated the previous finding that CUMI-101 behaves as a potent 5-HT1A receptor antagonist in rat brain (6). Similar to WAY-100635, a known potent 5-HT1A receptor antagonist, CUMI-101 blocked 8-OH-DPAT–stimulated 35S-GTPγS binding in monkey and human hippocampal tissue. CUMI-101 also had significant α1 adrenoceptor cross-reactivity in rat, monkey, and human brains. In addition, both in vitro radioligand binding and in vivo PET imaging studies demonstrated that CUMI-101 had significant cross-reactivity with α1 adrenoceptors, primarily in the thalamus. In vitro homogenate binding studies found that α1 adrenoceptor cross-reactivity in the thalamus was approximately 45% in rats, 50% in monkeys, and 43% in humans. In vivo PET studies showed that prazosin decreased thalamic uptake by approximately 50% in rats and approximately 33% in monkeys.
Initial studies of CUMI-101’s functional properties used CHO cells expressing human 5-HT1A receptors and found that, similar to 5-HT, CUMI-101 dose-dependently increased 35S-GTPγS binding (4). As a result, CUMI-101 was thought to be a 5-HT1A receptor agonist. However, another study conducted with rat brain tissue showed that CUMI-101 behaved as a potent antagonist and dose-dependently inhibited 5-HT–stimulated 35S-GTPγS binding (6). The present study replicated this finding.
To extend this work in other species, we further evaluated the functionality of CUMI-101 in human and monkey brains and found that it did not stimulate 35S-GTPγS binding even at concentrations of up to 10 μM. In the present study, CUMI-101 behaved as a typical antagonist by blocking 8-OH-DPAT–stimulated 35S-GTPγS binding in rat, monkey, and human hippocampal tissues. The results suggest that whether CUMI-101 behaves as an agonist or an antagonist at 5-HT1A receptors depends on the biochemical background in which it is tested. For example, the composition and density of G proteins varies between cell lines and between organs and will affect how readily a receptor stimulates agonist or antagonist pathways. Our findings underscore the importance of testing radioligands and drugs in native tissue from both the species (human) and the organ (brain) of interest.
Two recent PET studies in humans examined the sensitivity of 11C-CUMI-101 to changes in endogenous, intrasynaptic 5-HT levels before and after intravenous injection of citalopram, a highly selective serotonin reuptake inhibitor. In the first study, no difference in 11C-CUMI-101 binding was noted before or after citalopram infusion (20). In contrast, the second study showed a mean 7% increase in postsynaptic binding after citalopram infusion (21); the authors suggested that this increase in postsynaptic binding might reflect activation of the inhibitory 5-HT1A autoreceptor, which decreases both firing and 5-HT release after acute administration of selective serotonin reuptake inhibitors. The significance of these two conflicting results is limited by the small sample size and the relatively small effect (compared, for example, to the test–retest reproducibility of CUMI, which is ∼10%) (22).
Our initial study in rats showed that WAY-100635 incompletely blocked 11C-CUMI-101 brain uptake. Because the next highest affinity of CUMI-101 was to α1 adrenoceptors, this study examined α1 adrenoceptor cross-reactivity in rat, monkey, and human brains. We found that when WAY-100635 and prazosin were coadministered, 11C-CUMI-101 brain uptake was blocked to that of the nonspecific, cerebellar level. This contrasted with the findings of Kumar et al. (4), who had found a 45-fold difference in selectivity between 5-HT1A and α1 adrenoceptors using bovine hippocampal tissue (4). Indeed, the present study found cross-reactivity with α1 adrenoceptors across all 3 species examined, both in vitro and in vivo. In vitro radioligand binding studies using 3H-CUMI-101 showed that, on average across all 3 species, 50 nM 8-OH-DPAT displaced 56% of thalamic binding, whereas 50 nM prazosin displaced 46%. In vivo PET studies showed similar α1 adrenoceptor cross-reactivity of approximately 40% in the thalamus in rats and monkeys. Because of specific binding of 11C-CUMI-101 to α1 adrenoceptors in the cerebellum, some concern exists regarding the accurate quantification of BPND values. Interestingly, a recent study demonstrated that 11C-CUMI-101 had high uptake in the striatum, potentially due to cross-reactivity with α1 adrenoceptors (23); in contrast, neither of the two 5-HT1A receptor PET radioligands—[carbonyl-11C]WAY-100635 or 18F-MPPF—displayed such properties.
Another recent study evaluated the selectivity of CUMI-101 to α1 adrenoceptors using autoradiography in baboon and human brains and found that prazosin (up to 1 μM) did not significantly displace 3H-CUMI-101 binding in either the neocortex, the thalamus, or the cerebellum (24). However, minimal binding was seen in the molecular layer of the dentate gyrus. In contrast, our current study found that prazosin, even at a 20-fold lower concentration (50 nM), displaced 3H-CUMI-101 binding in the neocortex, thalamus, and cerebellum in rat, monkey, and human. We do not know the causes of these discrepant findings, which may be related to the different techniques used (e.g., homogenate binding vs. autoradiography). Unlike homogenate binding, autoradiography uses tissue sections with intact membranes that might maintain conditions that better reflect the in vivo state. However, our in vivo PET studies in rats and monkeys found that 11C-CUMI-101 had significant cross-reactivity with α1 adrenoceptors and correlated with our in vitro binding results.
The use of anesthesia in animal research is a limitation in this PET study. Ketamine is routinely used in veterinary medicine to immobilize nonhuman primates for PET imaging and has many other applications (e.g., anesthesia, immobilization for vaccinations). To minimize the effects of ketamine, radioligands were not injected until at least 120 min after ketamine injection. As regards the possible impact of isoflurane (1.5%) anesthesia on results, we assume that cross-reactivity was not altered. Moreover, blocking both 5-HT1A receptors and α1 adrenoceptors (WAY-100635 + prazosin) provided complete blockade, which was similar to self-block with cold CUMI-101 alone.
CONCLUSION
We found that, similar to WAY-100635, CUMI-101 acts as a potent antagonist at the 5-HT1A receptor in monkey and human brains. We further found that CUMI-101 has significant cross-reactivity with α1 adrenoceptors in rat, monkey, and human brain, primarily in the thalamus (>35%). CUMI-101 has approximately 10% cross-reactivity with α1 adrenoceptors in other regions (e.g., neocortex, cerebellum), making quantification problematic, especially using the cerebellum as the reference region. These findings suggest that the utility of 11C-CUMI-101 as a PET radioligand in humans is limited.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. No potential conflict of interest relevant to this article was reported.
Acknowledgments
We gratefully acknowledge the support of the Intramural Research Program of the National Institute of Mental Health, National Institutes of Health (IRP-NIMH-NIH). PMOD Technologies (Zurich, Switzerland) graciously provided image analysis and modeling software. We thank Dr. Ramin Parsey and Dr. Victoria Arango for providing human brain tissues, Ioline Henter for excellent editorial assistance, Kacey Anderson for assisting with PET imaging, and the joint National Institutes of Health-Karolinska Institutet Doctoral Program in Neuroscience.
- © 2014 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication May 6, 2013.
- Accepted for publication August 14, 2013.





{kind=link}
{kind=link}
{kind=link}
{kind=link}