


Abstract
2-Deoxy-2-18F-fluoro-d-mannose (18F-FDM) is an 18F-labeled mannose derivative and a stereoisomer of 18F-FDG. Our preliminary study demonstrated that 18F-FDM accumulated in tumors to the same extent as 18F-FDG, with less uptake in the brain and faster clearance from the blood. However, detailed studies on the uptake of 18F-FDM in tumors have not been conducted. We undertook this study to establish a practical method of 18F-FDM synthesis based on an 18F-nucleophilic substitution (SN2) reaction and to advance the biologic characterization of 18F-FDM for potential application as a tumor-imaging agent. Methods: We synthesized 4,6-O-benzylidene-3-O-ethoxymethyl-1-O-methyl-2-O-trifluoromethanesulfonyl-β-d-glucopyranoside as a precursor for the nucleophilic synthesis of 18F-FDM. The precursor was radiofluorinated with 18F-KF/Kryptofix222, followed by removal of the protecting groups with an acid. 18F-FDM was purified by preparative high-performance liquid chromatography and then subjected to in vitro evaluation regarding phosphorylation by hexokinase as well as uptake and metabolism in AH109A tumor cells. The in vivo properties of 18F-FDM were examined in Donryu rats bearing AH109A tumor cells by biodistribution studies and imaging with a small-animal PET system. Results: We radiosynthesized 18F-FDM in sufficient radiochemical yields (50%–68%) with excellent purities (97.6%–98.7%). 18F-FDM was phosphorylated rapidly by hexokinase, resulting in 98% conversion into 18F-FDG-6-phosphate within 30 min. Tumor cells showed significant uptake of 18F-FDM with time in vitro, and uptake was dose-dependently inhibited by d-glucose. 18F-FDM injected into tumor-bearing rats showed greater uptake in tumors (2.17 ± 0.32 percentage injected dose per gram [%ID/g]) than in the brain (1.42 ± 0.10 %ID/g) at 60 min after injection. PET studies also revealed the tumor uptake of 18F-FDM (quasi–standardized uptake value, 2.83 ± 0.22) to be the same as that of 18F-FDG (2.40 ± 0.30), but the brain uptake of 18F-FDM (1.89 ± 0.13) was ≈30% lower than that of 18F-FDG (2.63 ± 0.26). Conclusion: We prepared 18F-FDM with good radiochemical yield and purity by an SN2 reaction. We demonstrated that 18F-FDM had adequate tumor cell uptake by a metabolic trapping mechanism and can afford high-contrast tumor images with less uptake in the brain, indicating that 18F-FDM has almost the same potential as 18F-FDG for PET tumor imaging, with better advantages with regard to the imaging of brain tumors.
Fluorine-18–labeled 2-deoxy-2-fluoro-d-glucose (18F-FDG, Fig. 1A), a glucose analog, is taken up into tumor cells through membrane transport by glucose transporter 1 (GLUT1), phosphorylated by hexokinase, and then trapped in tissues (1–3). Rapid and high accumulation of 18F-FDG in animal tumors was first described by Som et al. (4) and later by Fukuda et al. (5). Several experimental and clinical studies from our research team (6) have been conducted to reveal the usefulness of 18F-FDG PET in cancer imaging. 18F-FDG has been used widely as a PET tracer for tumor imaging, and the usefulness of 18F-FDG PET in clinical oncology has been well documented (7–9).

Chemical structures: 18F-FDG (A), 18F-FDM (B), precursor-1 for 18F-FDM synthesis (C), precursor-2 for 18F-FDM synthesis (D), and precursor for 18F-FDG synthesis (E).
2-Deoxy-2-18F-fluoro-d-mannose (18F-FDM; Fig. 1B) is a derivative of d-mannose and an isomer of 18F-FDG. Previously, we showed that the uptake of 18F-FDM in tumors was as high as that of 18F-FDG and that the uptake in the normal brain was 30% lower than that of 18F-FDG, with faster blood clearance (5). However, the detailed mechanism of action of 18F-FDM uptake in tumors has not been undertaken. Whether 18F-FDM can be used as a PET radiopharmaceutical for cancer imaging, with results almost comparable or superior to 18F-FDG, has not been elucidated. With regard to use as a tumor-imaging agent, lower uptake in the normal brain is an attractive property of 18F-FDM and could be a potential application for brain tumor imaging by 18F-FDM PET. Additionally, it is also of interest whether 18F-FDM shows uptake in the same manner as 18F-FDG in inflamed tissue.
To conduct further studies on 18F-FDM, however, the establishment of a practical synthetic method is necessary. Classically, 18F-FDM has been obtained as a by-product of 18F-FDG synthesis by electrophilic fluorination (10). Today, nucleophilic displacement reactions are used in the large-scale synthesis of 18F-FDG from protected mannose analogs that have a triflate group as a leaving group (11). This approach also seems to be applicable for the large-scale synthesis of 18F-FDM. Actually, 18F-FDM had been synthesized from a glucose triflate protected with stable protecting groups by an SN2 (18F-nucleophilic substitution) reaction with 18F-fluoride in good yields (12,13), but the method requires hydrolysis by a strong acid at high temperature for deprotection after fluorination.
In the present study, we prepared a new precursor for 18F-FDM that can be radiolabeled by an SN2 reaction and optimized the conditions and procedure of radiosynthesis. We then used it for evaluation of in vitro uptake and metabolism in tumor cells, in vivo biodistribution, and PET imaging using tumor- and inflammation-bearing rats to revalidate the utility of 18F-FDM for cancer imaging.
MATERIALS AND METHODS
Precursors for 18F-FDM Synthesis
18F-FDM radiosynthesis was undertaken using 2 types of precursor: 4,6-O-benzylidene-3-O-ethoxymethyl-2-O-trifluoromethanesulfonyl-1-O-methyl-β-d-glucopyranoside (precursor-1; Fig. 1C) and 1,3,4,6-tetra-O-acetyl-2-O-trifluoromethanesulfonyl-α-d-glucopyranoside (precursor-2; Fig. 1D). Precursor-2 was prepared according to the literature (14). Precursor-1 was synthesized by trifluoromethanesulfonylation of the 2-hydroxyl group of 4,6-O-benzylidene-3-O-ethoxymethyl-1-O-methyl-β-d-glucopyranoside (15). The structure was determined by electrospray ionization mass spectroscopy and nuclear MR spectroscopy. Mass spectra were obtained on a JMS-DX303 system (JEOL). 1H-NMR (nuclear magnetic resonance) and 13C-NMR spectra were recorded on an Avance III 600 system (Bruker). For precise assignment, the spectra of gradient-selected heteronuclear multiple-bond correlation and pulse gradient-selected heteronuclear multiple-quantum correlation were also recorded. MS m/z: 339.1[M+H]+. 1H-NMR (600 MHz, CDCl3) δ 1.21 (3H, m), 3.44–3.51 (3H, m), 3.60 (4H, m), 3.69 (1H, t, 9.0 Hz), 3.77–3.83 (3H, m), 3.95 (1H, d, 1.2 Hz), 4.35–4.39 (2H, m), 4.81 (1H, d, 7.2 Hz), 4.92 (1H, d, 6.6 Hz), 5.54 (1H, s), 7.35–7.49 (5H, m), 13C-NMR (150 MHz, CDCl3) δ 14.8, 57.5, 64.2, 66.4, 69.7, 74.0, 79.6, 82.0, 96.5, 101.6, 104.4, 126.1, 126.2, 128.2, 129.1, 137.1.
Radiosynthesis
No-carrier-added 18F-fluoride was produced by the 18O(p, n)18F reaction on enriched 18O-H2O (Taiyo Nippon Sanso) with an HM-12 cyclotron (Sumitomo Heavy Industries) installed in the Cyclotron and Radioisotope Center (CYRIC) of Tohoku University. The specific activity (SA) of 18F-fluoride was in the range of 74–740 GBq/μmol at the end of bombardment.
Radiofluorination using the 18F-fluoride and precursor-1 and precursor-2 was performed by a conventional SN2 reaction with activated 18F-KF/Kryptofix222 (16,17). Briefly, the aqueous 18F− contained in the K2CO3 solution (1.88–3.47 GBq) and Kryptofix222 (15 mg) were placed in a brown vial. Then, water was removed by azeotropic means with acetonitrile by heating at 110°C and helium gas flow. After drying, the activated 18F-KF/Kryptofix222 was reacted with the precursor in CH3CN (1.0 mL) at 110°C (sealed condition) for 10 min. CH3CN was then removed by helium gas flow. Deprotection of the hydroxyl groups was undertaken by the addition of aqueous 6 M HCl, followed by a 20-min reaction at 110°C. The crude product was neutralized with 4 M NaOH and 400 mM Na2HPO4 and then subjected to semipreparative high-performance liquid chromatography (HPLC; column: YMC Pack ODS-A, 250 × 20 mm, 5 μm [YMC Co. Ltd.]; mobile phase: 0.9% aqueous NaCl; flow rate: 6.0 mL/min). 18F-FDG prepared by a conventional method (11) for a clinical 18F-FDG PET study at the CYRIC was used for biologic experiments without further purification by HPLC.
Radiochemical purities were determined by radio–thin-layer chromatography (radio-TLC) on silica gel aluminum sheets impregnated with monosodium phosphate (60F254; Merck) with CH3CN/H2O, 95/5 (v/v) as eluent. The development process to separate 18F-FDG and 18F-FDM was performed according to the literature (18). After development, the plate was dried and exposed to a BAS imaging plate (Fuji Film). Autoradiograms of radio-TLC were obtained using a BAS-5000 phosphor imaging instrument (Fuji Film).
Uptake of 18F-FDM into Tumor Cells In Vitro
Nonadherent rat hepatoma cells AH109A (19) were cultured with RPMI-1640 medium containing l-glutamine and d-glucose (Wako Chemical Industries) and used for uptake studies (1.5–1.9 × 106 cells/mL of medium). The medium was replaced with d-glucose–free RPMI-1640 medium (Gibco) 3–4 h before experimentation.
18F-FDM in saline (100 μL; 1.29 MBq/mL at start of experimentation; radiochemical purity, >96%) was added to a 96-well plate. Suspended AH109A cells in 100 μL of d-glucose–free RPMI-1640 medium at a final density of 0.75 × 106 cells/mL were added to the wells and incubated at 37°C with continuous stirring. Cells were harvested at predetermined times through suction filtration with a vacuum manifold (Millipore) at 20, 60, 120, and 180 min. Trapped cells were washed three times with phosphate-buffered saline (200 μL/well). Filters holding the trapped cells were then separated, and radioactivity was measured with a γ-counter (AccuFLEXγ7000; Hitachi Aloka Medical). The radioactivity of 18F-FDM in tumor cells was expressed as percentage injected dose per 106 cells (%ID/106 cells).
Uptake into Tumor Cells Under d-glucose Load
18F-FDM or 18F-FDG in 50 μL of saline (1.38 MBq/mL at the start of experimentation; radiochemical purity, >96%) was mixed with d-glucose in 50 μL of medium at a final concentration of 0, 0.1, 1.0, 5.0, and 10 mM and put into a 96-well plate. Then, 100 μL of AH109A cells suspended in d-glucose–free RPMI-1640 medium at a density of 0.95 × 106 cells/mL were added to the wells and incubated at 37°C for 2 h with continuous stirring. Tumor cells were trapped and washed, and the radioactivity was measured as described above. Half-maximal inhibitory concentration (IC50) values for the uptake inhibition by d-glucose were calculated by fitting the data to a sigmoidal dose–response regression curve with GraphPad Prism software (version 5.0; GraphPad Software).
Metabolism Studies
Enzymatic phosphorylation of 18F-FDM and 18F-FDG was examined with yeast hexokinase (Saccharomyces cerevisiae; Sigma-Aldrich). Seventy-four kilobecquerels of 18F-FDM or 18F-FDG (in 10 μL of saline) were added to a mixture of yeast hexokinase (8 μL, 100 U/mL), adenosine triphosphate (40 μL, 40 μM), MgCl2 (20 μL, 170 mM), and Tris-buffered phosphate-buffered saline (102 μL) and incubated at 37°C. The enzymatic reaction was stopped by the addition of aqueous HClO4 (100 μL, 1.0 M) at 2, 3, 5, 7.5, 30, and 60 min after the start of incubation. The solution was neutralized and used for analyses by radio-TLC on silica gel–coated aluminum sheets (60F254; Merck) with ethanol/AcNH4 (1 M), 1:1 (v/v) as eluent. After development, autoradiograms of radio-TLC were obtained by the method as described above.
To examine metabolism in tumor cells, 74 kBq of 18F-FDM (in 50 μL of saline) were added to AH109A cells (100 μL, 0.75 × 106 cells/mL) and incubated at 37°C. Cells were harvested by centrifugation (500g, 5 min, 4°C) 10, 60, and 120 min after incubation. Their radioactivities were measured by a γ-counter. Metabolites were extracted by 50% ethanol and analyzed by radio-TLC as described above.
Biodistribution Study of 18F-FDM
The Ethics Committee for Experimental Research in Animals of Tohoku University approved the study protocol. AH109A cells (which were maintained in ascites form in Donryu rats) were used for preparing tumor-bearing rats. Tumor cells in ascites (5 × 105 cells) were inoculated subcutaneously into the right flank of Donryu rats. After 9 d, by which time the tumor had grown to ≈1–2 cm in diameter, 18F-FDM in saline (1.11 MBq/300 μL) was injected into the lateral tail vein. Rats were killed by cervical dislocation after heart puncture to obtain blood samples 60 and 120 min after injection (5 rats at each time point). Tumors and organs were removed shortly thereafter and weighed. Radioactivity was measured using a γ-counter. Tissue uptake was expressed as percentage injected dose per gram of tissue (%ID/g).
PET of Tumor- and Inflammation-Bearing Rats
AH109A cells (5 × 106 cells) were inoculated in the right flank of male Donryu rats (225–268 g). Two or 3 d after cell inoculation, 200 μL of telepine oil were injected into the left flank. After 7–9 d, by which time the tumor had grown to a diameter of ≈2–3 cm, rats were anesthetized with 2% isoflurane (flow rate, 2 mL/min) at 4 min before PET. Rats were starved overnight before PET. Whole-body PET of rats was undertaken using a small-animal PET imaging system with a spatial resolution of 1.5 mm (Clarivivo-PET; Shimadzu) (20). Acquired data were decay-corrected and images reconstructed using the 3-dimensional dynamic row-action maximum likelihood algorithm. Attenuation correction was not performed. After PET, rats were placed onto an animal bed and moved to an animal CT scanner (Clarivivo-CT; Shimadzu). CT images were taken under the same geometry as that used for PET. The next day, PET with 18F-FDG was undertaken using the same rats. Three rats were used for the PET study.
We calculated tissue radioactivity per voxel divided by injected activity per body weight of the animals to be used as a parameter of tissue uptake. We expressed it as quasi–standardized uptake value (qSUV) because attenuation correction was not applied for the data.
Statistical Analysis
The statistical significance of differences in the mean values of qSUV between 18F-FDM and 18F-FDG was determined using the Student t test. A P value of less than 0.05 was considered significant.
RESULTS
18F-FDM Radiosynthesis
The reaction conditions and results of 18F-FDM synthesis using precursor-1 are summarized in Table 1. With respect to the reaction time, 18F-fluorination for 10–30 min and deprotection for 20–30 min afforded 18F-FDM in good-to-excellent radiochemical yields (50%–68%), with sufficient radiochemical purities (97.6%–98.7%). An epimerized product, 18F-FDG, was not found in the final solution of 18F-FDM. When precursor-2 was used for 18F-FDM synthesis, the yields were less than 1%. The SA of 18F-FDM (which was not measured in the present study) was estimated to be greater than 37 GBq/μmol (end of synthesis) because no-carrier-added 18F-fluoride (>74 GBq/μmol) was used and the total synthesis time was less than 110 min.
Reaction Conditions and Yields for 18F-FDM Synthesis from Precusor-1
Uptake of Tracers into Tumor Cells In Vitro
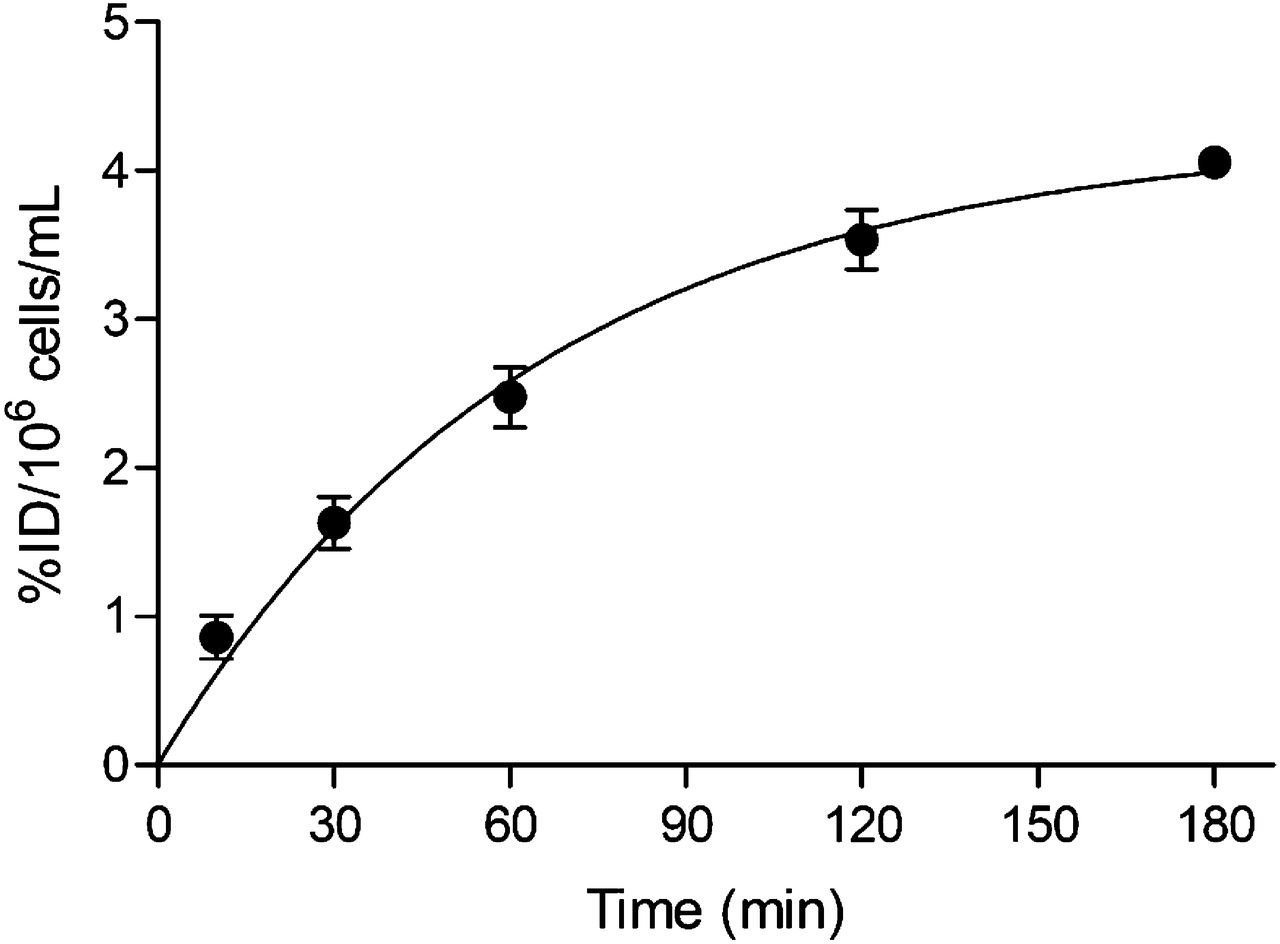
Uptake of 18F-FDM into tumor cells increased with increasing uptake time and reached 44 %ID/106 cells 180 min after the start of incubation (Fig. 2). Although we did not confirm the time for reaching a plateau for uptake, we adopted 120 min as the incubation time for the following uptake study according to the amount of d-glucose. Figure 3 illustrates the inhibition of 18F-FDM or 18F-FDM by d-glucose load. Uptake of either tracer was inhibited dose-dependently by the d-glucose load. The IC50 values by d-glucose were 1.95 × 10−1 mM for 18F-FDM and 4.47 × 10−2 mM for 18F-FDG.

In vitro uptake of 18F-FDM into AH109A cells.

Inhibition of uptake of tracers in tumor cells by d-glucose. IC50 values due to inhibition by d-glucose were 1.95 × 10−1 mM for 18F-FDM and 4.47 × 10−2 mM for 18F-FDG.
Metabolism in AH109A Tumor Cells
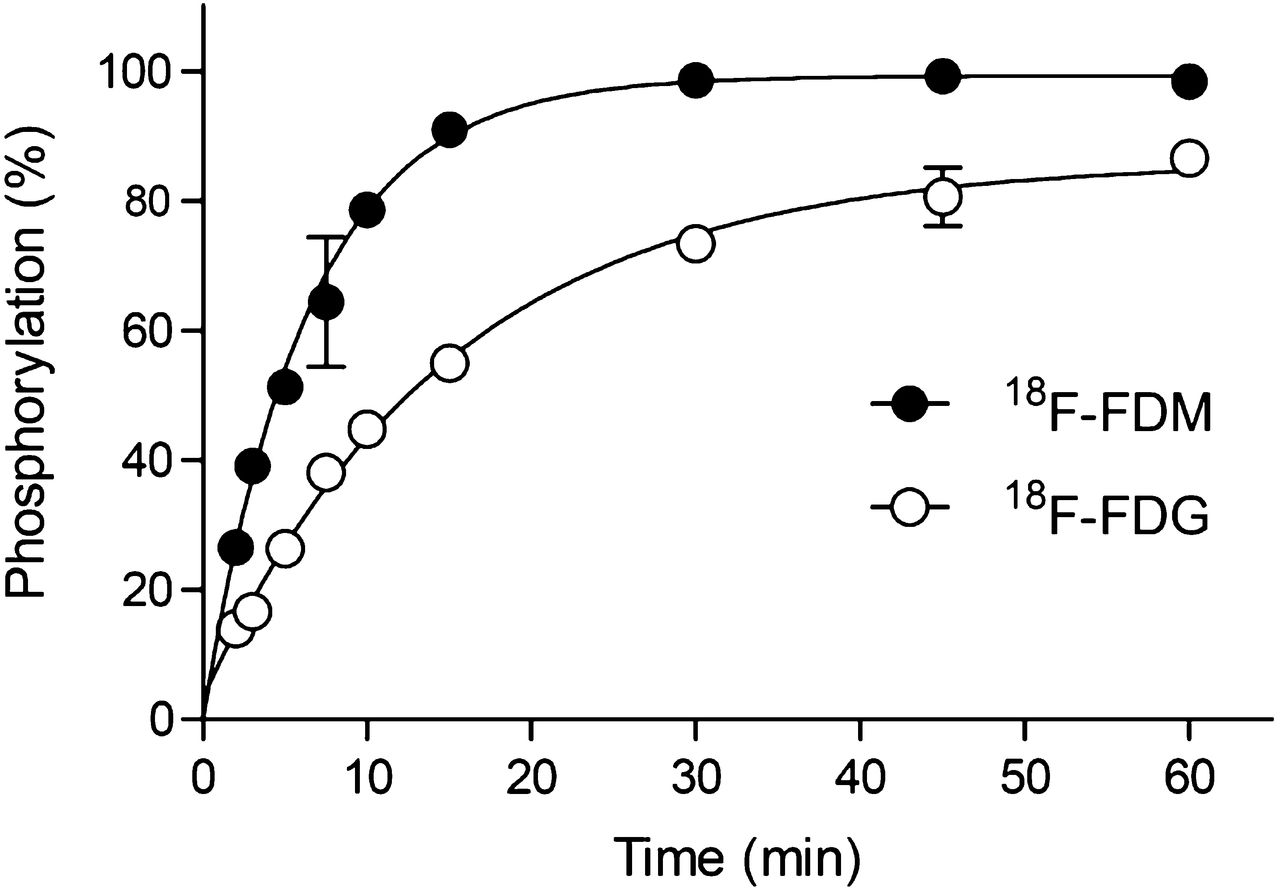
After incubation of 18F-FDG with yeast hexokinase, a metabolite showing a retention factor (Rf) value of 0.63, 18F-FDG-6-phosphate (18F-FDG-6-P), was observed. The fraction of 18F-FDG-6-P increased with time and almost reached a plateau at 60 min (Fig. 4). In the case of 18F-FDM, a metabolite showing an Rf value of 0.63 was identified and considered to be 18F-FDM-6-P because FDM is known to be a good substrate for hexokinase (2). The fraction of 18F-FDM-6-P increased with a faster initial speed than that by 18F-FDG and reached a plateau at 30 min (Fig. 4).

Phosphorylation of 18F-FDM and 18F-FDG by yeast hexokinase. Percentages of 6-phosphorylation versus incubation time are shown. 18F-FDM showed more rapid 6-phosphorylation than 18F-FDG.
Figure 5 shows the profiles of metabolites analyzed by radio-TLC 10, 60, and 120 min after incubation with AH109A cells and 18F-FDM. In addition to 18F-FDM, 4 metabolites were observed. The Rf value of the major metabolite was 0.62 and coincided with the Rf value of the metabolite produced by hexokinase. Therefore, the metabolite was considered to be 18F-FDM-6-P. The fraction of this metabolite reached 43.4% at 10 min and remained constant until 120 min (41.0%) (Table 2). The other 3 metabolites were not identified.
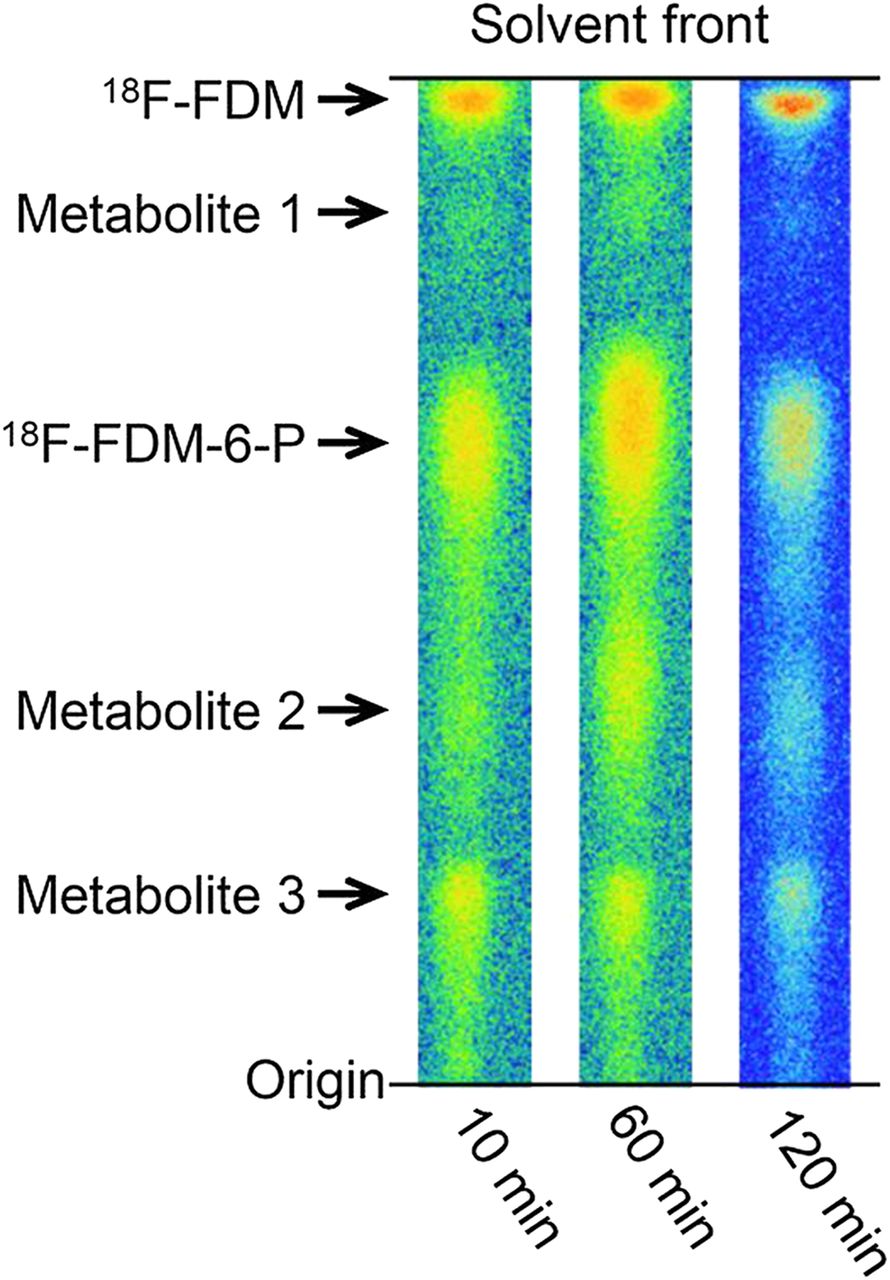
Radio-TLC autoradiograms of 18F-FDM metabolites obtained 10, 60, and 120 min after incubation with AH109A tumor cells in vitro.
Metabolism of 18F-FDM in AH109A Tumor Cells
Biodistribution Study
Table 3 illustrates the result of the biodistribution study. The tumor uptake of 18F-FDM was the highest among the tissues: 2.17 ± 0.32 and 2.09 ± 0.20 %ID/g at 60 and 120 min, respectively. Relatively high uptake was observed in the brain (1.42 ± 0.10 %ID/g) at 60 min. Uptake in the other normal organs was less than 0.6 %ID/g at 60 min after injection. Levels in the blood were low (0.11 ± 0.02 and 0.06 ± 0.01 %ID/g at 60 and 120 min, respectively). The tumor-to-blood and tumor-to-muscle ratios were 19.0 and 18.8, respectively, at 60 min after injection. The tumor-to-brain ratio was 1.5 and 1.6 at 60 and 120 min after injection, respectively.
Biodistribution of 18F-FDM in AH109A Tumor Cell–Bearing Donryu Rats
PET of Tumor- and Inflammation-Bearing Rats
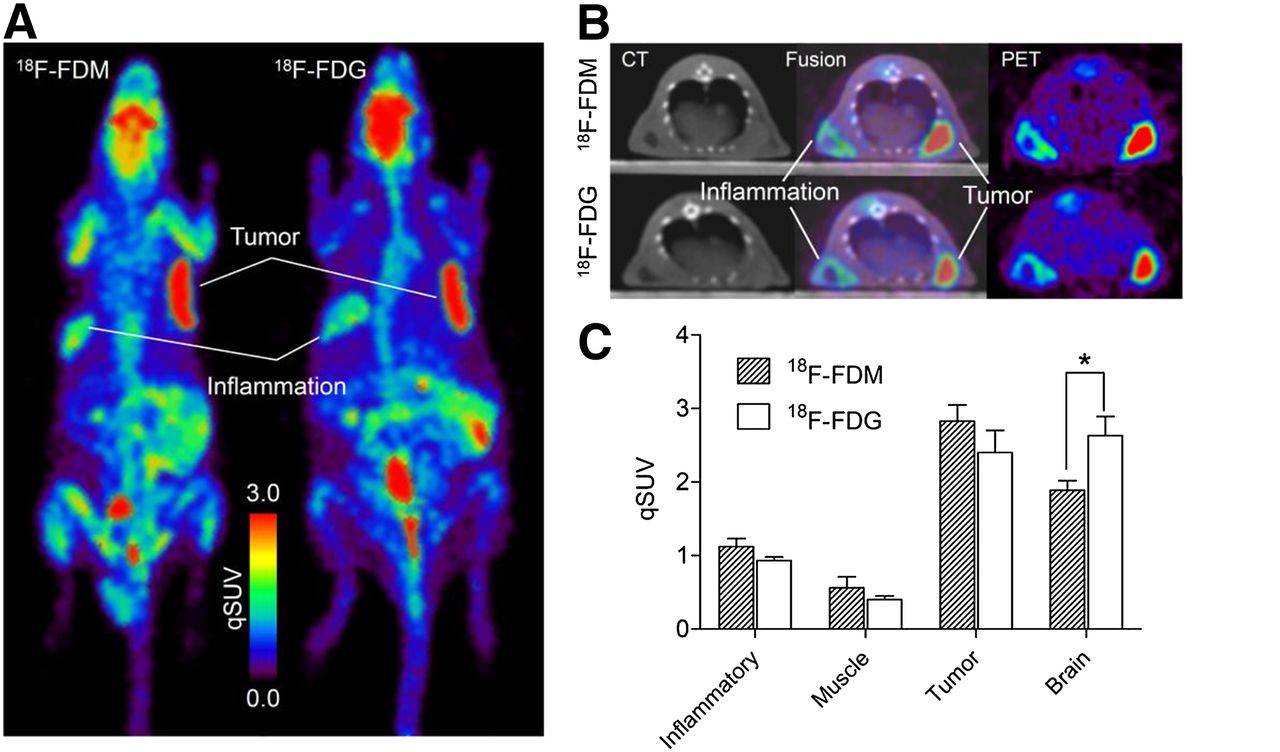
The results of PET of Donryu rats with tumors and inflammation at 55–65 min after the injection of 18F-FDM and 18F-FDG are summarized in Figure 6. Tumors were visualized clearly in maximum-intensity-projection images, with fewer counts in the inflammation model with 18F-FDM or 18F-FDG (Fig. 6A). Uptake of 18F-FDM in the brain was less than that of 18F-FDG. Transaxial images also demonstrated high uptake in tumors, whereas relatively low uptake in inflammatory tissue with a central low-count area was noted (Fig. 6B). qSUVs in tumors and tissues are shown in Figure 6C and Table 4. Tumor uptake of 18F-FDM and 18F-FDG by qSUV was 2.83 ± 0.22 and 2.40 ± 0.30, respectively, with no significant differences between the 2 values. Uptake of 18F-FDM in the brain (1.89 ± 0.13) was significantly (P < 0.05) lower than that of 18F-FDG (2.63 ± 0.26). Uptake in chemically induced inflammatory tissues was 1.12 ± 0.11 and 0.93 ± 0.03 in 18F-FDM and 18F-FDG, respectively. These values were less than half those of tumor uptake, and there was no significant difference between the 2 tracers. There was no significant difference between inflammation-to-muscle ratios for 18F-FDM (2.00) and 18F-FDG (2.33).
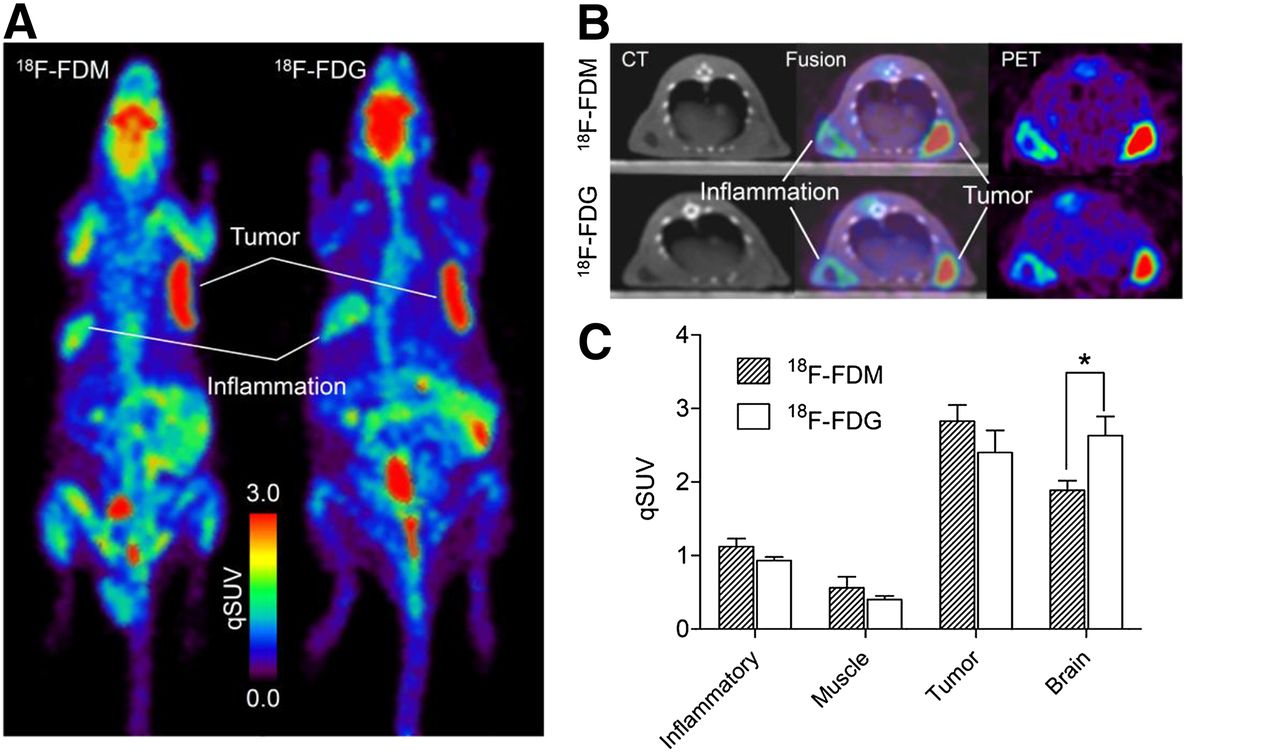
PET images of tumor- and inflammation-bearing rats and comparison with qSUVs. (A) Representative maximum-intensity-projection images of 18F-FDM and 18F-FDG. (B) Representative transaxial PET and CT images of rats. (C) Tissue uptake (qSUV) of 18F-FDM and 18F-FDG (n = 3, *P < 0.05).
Uptake of 18F-FDM and 18F-FDG into Tissues and Tumors as Measured by PET
DISCUSSION
When one considers that 18F-FDG can be prepared from an O-acetylated mannose triflate (Fig. 1E) in sufficient yield, a corresponding O-acetylated glucose triflate (precursor-2, Fig. 1D) could be a candidate precursor of 18F-FDM. However, the present study revealed that only a trace amount of 18F-FDM was obtained from precursor-2. On the basis of the SN2 reaction mechanism, the axial leaving group of the 18F-FDG precursor is suitable for the nucleophilic attack of fluoride, whereas the equatorial leaving group of precursor-2 is not. In addition, as suggested in a study using tetrabutylammonium fluoride and precursor-2 (14), an unwanted side reaction between 18F-fluoride and the acetoxy groups neighboring the triflate group might predominate in the 18F-FDM synthesis. To avoid this side reaction, we considered that the hydroxyl-protecting groups adjacent to the triflate group should be stable to 18F-fluoride. Therefore, we prepared 4,6-O-benzylidene-3-O-ethoxymethyl-1-O-methyl-2-O-trifluoromethanesulfonyl-β-d-glucopyranoside, precursor-1, as a new precursor for 18F-FDM synthesis.
Using this precursor, we synthesized 18F-FDM in satisfactory yields (50%–68%) that were nearly comparable with those seen with 18F-FDG synthesis. The estimated SA of 18F-FDM in the present study (>37 GB/μmol) was much higher than that reported in 1 study (6.7–67 MBq/μmol) (5). However, in that study, the FDM concentration of injection (0.55–5.5 μg/mL) was far lower than the blood-glucose level in rats (≈1 mg/mL (21)). Therefore, the difference in SAs between 18F-FDM might have had little effect on the in vivo experiments. The method used in the present study afforded improved isolated yields of 18F-FDM and a shorter synthesis time in comparison with the results reported by Luxen et al. (13). The results of the present study suggest that the synthesis method using a new precursor enables preparation of a sufficient amount of 18F-FDM for routine clinical use.
Uptake in AH109A cells was inhibited by d-glucose in a dose-dependent manner. These findings suggested that the mechanism of uptake of 18F-FDM was related to GLUT1, similar to the mechanism of d-glucose and 18F-FDG. Additionally, the in vitro enzyme assay clearly demonstrated that 18F-FDM is also a good substrate for hexokinase 18F-FDG, as reported previously (1,2). However, the IC50 value obtained by d-glucose for 18F-FDM was greater than that for 18F-FDG, and the fraction of 18F-FDG-6-P increased with a slightly slower initial speed than that by 18F-FDM. We speculate that these effects on 18F-FDG were caused by unlabeled ingredients such as pyranose derivatives in the 18F-FDG injection (22) because the 18F-FDG injection was used without HPLC purification. In the case of in vivo experiments, the ingredients would have little effect because of the effect of blood glucose; Figure 6C shows no difference in qSUV in muscle between 18F-FDM and 18F-FDG.
We then revealed that 18F-FDM was metabolized to the major metabolite of 18F-FDM-6-P and other, unknown metabolites in tumor cells. These results suggest that 18F-FDM accumulates in tumor cells through a metabolic trapping mechanism such as the one seen with 18F-FDG. With respect to 18F-FDG metabolism, 18F-fluorodeoxy-6-phospho-d-gluconolactone and 18F-fluorodeoxy-6-phospho-d-gluconate were observed in squamous carcinoma and mammary carcinoma in mice in addition to the major metabolite of 18F-FDG-6-P at 1–3 h after injection (23). Furthermore, conversion of FDG-6-P into FDM-6-P or vice versa within sarcoma cells (24) or colon26 cells (25) has been reported. Therefore, further metabolism studies for 18F-FDM in tumors and in normal tissues are necessary.
The results of our biodistribution study were almost consistent with our previous biodistribution studies using the same rat strain and AH109A tumor cells (Table 5) (5), but tumor uptake in the present study was slightly lower than that in the previous study. One reason might be due to the differences of rat substrains used in the present study. We previously used the Suzuki-A Donryu rat, a substrain of the Donryu rat in which AH109A cells grew well. The Suzuki-A Donryu rat strain is no longer available, so we used conventional Donryu rats in the present study. The biodistribution studies demonstrated the highest 18F-FDM uptake in AH109A tumor cells, relatively high uptake in the brain, and lower uptake in the other normal organs, indicating that 18F-FDM could be an effective tumor-imaging agent. However, uptake of 18F-FDM in the liver was slightly higher than that of 18F-FDG (Table 5), suggesting a potential disadvantage of 18F-FDM for the imaging of liver tumors using PET.
Data Comparison of Uptake of 18F-FDM and 18F-FDG into Tissues and Tumors
Braun et al. (26) measured cerebral metabolic rates for glucose in baboons with PET using 18F-FDM. There was a 20% reduction in glucose use, compared with 18F-FDG, if 18F-FDM was used as a glucose analog. Wienhard et al. (27) also reported that the lumped constant for 18F-FDM was 20% lower than that for 18F-FDG in the human brain. The PET study described here also confirmed that the uptake of 18F-FDM in the brain was significantly lower (28%) than that of 18F-FDG, whereas 18F-FDM showed no significant difference in tumor uptake, compared with that seen with 18F-FDG. Therefore, a relatively lower uptake in normal brain tissue without change in tumor uptake could be an advantage of 18F-FDM for the visualization of brain tumors as compared with imaging using 18F-FDG. In particular, low-grade glioma often exhibits low uptake of 18F-FDG in tumor tissue similar to or less than that in the normal brain, resulting in a decrease in the sensitivity of lesion detection (28). However, given that 18F-FDM shows 30% lower uptake in normal brain and the same uptake in tumor in comparison to 18F-FDG, the tumor–to–normal brain ratio of 18F-FDM will become higher by ≈40% than that of 18F-FDG. It would be worth verifying whether 18F-FDM PET improves the sensitivity of detection of brain tumors.
With regard to the imaging of inflammation, the qSUVs of 18F-FDM and 18F-FDG were less than half of the uptake seen in tumors. However, this finding does not indicate that 18F-FDM has an advantage for the differential diagnosis between cancer and inflammation. Macroautoradiography of inflammatory tissue revealed that 18F-FDM accumulated as much as 18F-FDG in inflammatory cells and granulation tissues (data not shown). Relatively low uptake in PET might be due to a partial-volume effect because these inflammatory and granulation tissues were thin.
CONCLUSION
We successfully synthesized 18F-FDM by a nucleophilic substitution reaction in high radiochemical yield and purity. 18F-FDM showed high accumulation in tumors, lower uptake in the brain than that of 18F-FDG, and rapid excretion from the blood, indicating promising characteristics for cancer imaging. Additionally, we elucidated (at least in part) the mechanism of cellular uptake and metabolism of 18F-FDM. 18F-FDM has almost the same excellent potential as 18F-FDG for PET in oncology, with an advantage of better imaging of brain tumors. We are continuing further research of 18F-FDM for clinical use, including development of an automated radiosynthesis system and estimation of acute toxicity and radiation doses.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This study was supported by a Grant-in-Aid for Scientific Research (grant nos. 19390312 and 23650610) from the Japanese Ministry of Education, Culture, Sports, Science and Technology and supported in part by a Grant-in-Aid for Cancer Research from the Japanese Ministry of Health, Labour and Welfare and by the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation. No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Jul. 10, 2013.
- © 2013 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication September 23, 2012.
- Accepted for publication February 19, 2013.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.