


Abstract
Amyloid-β (Aβ) deposition is a pathologic hallmark of Alzheimer disease (AD). Although the typical spatial distribution pattern of Aβ deposition in early AD mainly involves regions distant from the hippocampus, the predominant clinical feature is impairment of hippocampus-dependent memory. We aimed at elucidating the relationship between neocortical Aβ load, regional neuronal function, and memory impairment. Methods: Thirty patients with early AD underwent combined 11C-Pittsburgh compound B (11C-PIB) and 18F-FDG PET and memory assessments. Composite measures of hemispheric Aβ load were calculated by volume-weighted mean values of neocortical 11C-PIB binding. Voxelwise 18F-FDG uptake was used as a measure of regional glucose metabolism reflecting neuronal activity. We investigated the relationship between left- and right-hemispheric Aβ load and regional glucose metabolism (voxelwise analyses). In addition, we assessed the correlations of hemispheric Aβ load (region-of-interest–based analyses) and regional glucose metabolism (voxelwise analysis) with memory performance. Analyses were corrected for age and sex. Results: Higher Aβ load in the left hemisphere was associated with reduced glucose metabolism of the left medial temporal lobe (MTL; r2 = 0.38) and correlated with worse wordlist recall (r = −0.37; partial correlation controlled for sex and age). Furthermore, wordlist recall correlated with regional glucose metabolism in the bilateral MTL and precuneus–posterior cingulate cortex and right lingual gyrus (r2 = 0.24). Conclusion: We demonstrated an association between the left-hemispheric Aβ load and impairment of the left MTL in AD at 2 different levels: regional hypometabolism and verbal memory. This correlation suggests that neocortical amyloid deposition is connected to or even drives neuronal dysfunction and neurodegeneration of the MTL, which is associated with impaired episodic memory processing as a clinical core symptom of AD.
The amyloid cascade hypothesis of Alzheimer disease (AD) posits that cortical amyloid-β (Aβ) deposition is related to downstream neuronal dysfunction and cognitive impairment (1). Aβ plaques presumably do not directly impair cognition but rather promote tau pathology in distant vulnerable neurons, which in turn leads to cognitive deficits (2). Intriguingly and at present not fully understood, the spatial distribution of Aβ deposition in early AD mainly involves regions that are distant from the hippocampus, although the predominant clinical feature is impairment of hippocampus-dependent memory (3). With the advent of Aβ PET imaging, most commonly using the radiotracer 11C-Pittsburgh compound B (11C-PIB (4)), quantitative in vivo assessment of the Aβ load in the human brain has become feasible. Furthermore, PET imaging of regional cerebral glucose metabolism with 18F-FDG represents an established marker of neuronal activity and, consequently, neuronal dysfunction and neurodegeneration (1). In concert with measures of episodic memory, these in vivo biomarkers may crucially contribute to our understanding of pathologic mechanisms underlying cognitive changes in early AD.
Previous studies assessing the relationship between the amount of amyloid deposition and memory functions in vivo yielded inconclusive results: when groups of nondemented individuals and AD patients were combined, negative correlations have been detected, primarily driven by the huge group differences in each of the 2 measures (5–10). This finding mainly recapitulates the established knowledge of increased Aβ and decreased memory capabilities in AD. Examining nondemented individuals (healthy individuals or patients with mild cognitive impairment [MCI]) led to conflicting results. Although significant correlations between memory performance and Aβ load have been detected in some cohorts of nondemented individuals (8,11–15), they could not be found in others (9,13,14,16–19). When such correlations within groups of AD patients were evaluated, most studies did not find significant results (7,9,15,16,20–22). This may in part be explained by the fact that the rate of cerebral Aβ accumulation is apparently highest before the onset of dementia and decreases in symptomatic AD stages (1), resulting in little variability of amyloid burden within the groups of AD patients despite variable degrees of cognitive impairment. Only a few studies thus far have investigated amyloid deposition, glucose metabolism, and memory in the same patient (4,5,16,20,21,23) and yielded inconclusive results. Although memory deficits were typically related to hypometabolism, Aβ load and glucose metabolism were found to be negatively correlated—predominantly in parietal, frontal, and posterior cingulate or precuneus cortex—in most (4,5,16,20,23), but not all, studies (21). Importantly, the relationship between Aβ load and glucose metabolism in these studies was assessed within predefined regions of interest, permitting detection of local but not remote effects.
Given the puzzling spatial separation of neocortical amyloid deposition from hippocampal dysfunction and degeneration as reflected by biomarkers (6) and cognitive assessment in AD, the present study was undertaken to gain further insights into local and distant effects of Aβ pathology on neuronal functioning and cognitive performance. We chose a novel approach to assess the relationship between regional glucose metabolism (on a voxel-by-voxel level) and hemispheric Aβ load. Because the earliest and most extensive Aβ deposition in AD is observed in the neocortex (3), the current study focused on neocortical Aβ load. We investigated the relationship between Aβ load, regional glucose metabolism, and memory performance in a group of AD patients including patients with early diagnoses, because we presume that Aβ load might show greater variation among these individuals. This variation would result in a greater chance to detect significant correlations of Aβ load with regional glucose metabolism and cognition. We hypothesized that in our sample of AD patients greater neocortical amyloid burden would be related to reduced medial temporal glucose metabolism and impaired memory performance.
MATERIALS AND METHODS
Patients
Data from 30 AD patients who underwent combined 11C-PIB and 18F-FDG PET examinations were analyzed after the patients gave written informed consent, as approved by the local ethics committee. Patients were clinically diagnosed as early AD based on National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association criteria for probable AD (24). They were recruited from a specialized memory clinic of the University of Freiburg Medical Center. Handedness data were available from 26 of 30 patients. All were right-handed except for 1 left-handed patient.
Neuropsychologic Test Data
Patients were examined with the neuropsychologic assessment battery of the Consortium to Establish a Registry for Alzheimer Disease by experienced clinical neuropsychologists within a median of 38 d before or after PET imaging. We report here episodic memory data, because episodic memory impairment is the cognitive hallmark of AD.
Acquisition and Analysis of 11C-PIB and 18F-FDG PET Data
PET data were acquired as previously described (25). In brief, all patients underwent 11C-PIB and 18F-FDG PET examinations on the same ECAT EXACT 922/47 PET system (Siemens-CTI). Both scans were acquired on the same day in 14 patients (18F-FDG PET about 2 h after 11C-PIB injection; 1-h break in between) and within 1 wk in 12 patients. In the remaining 4 patients, the time gap was 8, 9, and 16 d and 6 mo. For each acquisition, the patient’s head was gently restrained with an elastic tape. Its position was carefully monitored and, if necessary, manually corrected throughout data acquisition (using the scanner laser beams and reference skin marks). After a 5-min 68Ge/68Ga transmission scan for attenuation correction, dynamic 11C-PIB PET acquisition lasting 60 min (frame length ranging from 20 to 300 s) started with an intravenous bolus injection of 486 ± 91 MBq of 11C-PIB. Datasets were reconstructed by filtered backprojection (Shepp filter, 5 mm in full width at half maximum). 11C-PIB radiochemical synthesis was performed as previously described (26) (precursor provided by ABX GmbH). Radiochemical purity was greater than 95% and specific activity at the time of injection greater than 74 GBq/μmol in each patient. 18F-FDG PET scans were obtained 40 min after intravenous injection of 307 ± 14 MBq of 18F-FDG. All patients fasted for at least 6 h before tracer injection, with plasma glucose level being within the reference range. Four 5-min frames were collected in 2-dimensional mode and reconstructed by filtered backprojection with calculated attenuation correction. Summed 18F-FDG PET datasets (20 min total scan time) were used for further data analyses.
Pharmacokinetic analyses of 11C-PIB data were done using the PMOD software package (version 3.0; PMOD Technologies Ltd.) as follows (25): parametric images of regional 11C-PIB binding potential (BPND; corresponding to regional Aβ load) and relative ligand delivery (R1; relative measure of regional cerebral blood flow (25)) were generated using the 2-step simplified reference tissue model (27). The cerebellum was chosen as a reference region without relevant specific 11C-PIB binding (4). To apply this model, k2′ (i.e., efflux rate constant of 11C-PIB from the reference region) was individually estimated for each patient using the simplified reference tissue model (28) on regional time–activity curves as previously described (25). For further analyses, all datasets were spatially normalized to the MNI (Montreal Neurologic Institute) PET template. This was done by first normalizing the 18F-FDG dataset and copying individual normalization parameters to the patient’s parametric 11C-PIB BPND dataset after the latter had been automatically coregistered to the former by use of the corresponding R1 image. Left- and right-hemispheric Aβ loads were assessed by first performing a volume-of-interest (VOI) analysis using a predefined VOI set and then averaging regional BPND estimates of frontal, orbitofrontal, posterior cingulate gyrus or precuneus, parietal, and lateral temporal cortex VOIs of each hemisphere (volume-weighted by VOI volumes) (25).
Statistical Analyses
We chose to test the predictive values of left- and right-hemispheric Aβ load separately and not combined as global 11C-PIB as usually done. The rationale behind this was that we considered lateralized memory functions (verbal vs. nonverbal) to be more specifically impaired by amyloid deposition within the same hemisphere and less by more distant transcallosal effects of Aβ.
Relationship Between Hemispheric Aβ Load and Regional Glucose Metabolism
The software SPM8 (http://www.fil.ion.ucl.ac.uk/spm/software/spm8/) was used to perform whole-brain, voxelwise regression analyses on spatially normalized 18F-FDG PET data after all datasets had been smoothed by an isotropic 10-mm gaussian filter and normalized to individual cerebellar uptake values. Two separate analyses were run, with the first including left-hemispheric Aβ load and the second including right-hemispheric Aβ load as predictor variables. Sex and age were included as nuisance variables. Results were considered significant if the P values were less than 0.001 at the voxel level and less than 0.05 at the cluster level, corrected for familywise errors (FWEs).
Relationship Between Aβ Load and Memory Performance
Two separate analyses were performed testing partial correlations between left-hemispheric Aβ load and verbal memory and between right-hemispheric Aβ load and visuospatial memory using SPSS 19 (http://www.spss.com/). In each of the analyses, we controlled for sex and age. Results were considered significant if the P value was less than 0.05 (1-tailed).
Relationship Between Memory Performance and Regional Glucose Metabolism
An additional regression analyzed the relationship between verbal memory performance and 18F-FDG uptake throughout the whole brain in a voxelwise fashion using SPM8. Sex and age were controlled for. Results were considered significant if the P value was less 0.001 at the voxel level and less than 0.05 at the cluster level, corrected for FWEs.
RESULTS
Twenty-seven of 30 clinically diagnosed AD patients were classified as 11C-PIB–positive (global 11C-PIB BPND value above the institutional cutoff of 0.35), and the remaining 3 patients showed no appreciable 11C-PIB binding. The mean (±SD) 11C-PIB BPND values in 11C-PIB–positive and 11C-PIB–negative AD patients were 1.07 ± 0.28 and 0.16 ± 0.05, respectively. Demographics and cognitive test data are summarized in Table 1.
Demographics and Cognitive Test Data from Patients Included in Imaging Analysis, Group Mean ± SD
Relationship Between Aβ Load and Regional Glucose Metabolism
Whole-brain analyses revealed that left-hemispheric Aβ load significantly predicted regional glucose metabolism in the left posterior medial temporal lobe (MTL) (peak voxel MNI coordinates [−24 −43 −14 mm], z = 3.53, P < 0.001 at the voxel level, P < 0.05 FWE-corrected at the cluster level), with higher Aβ load being associated with lower glucose metabolism (Fig. 1; r2 = 0.38). There were no other clusters of significant correlation.
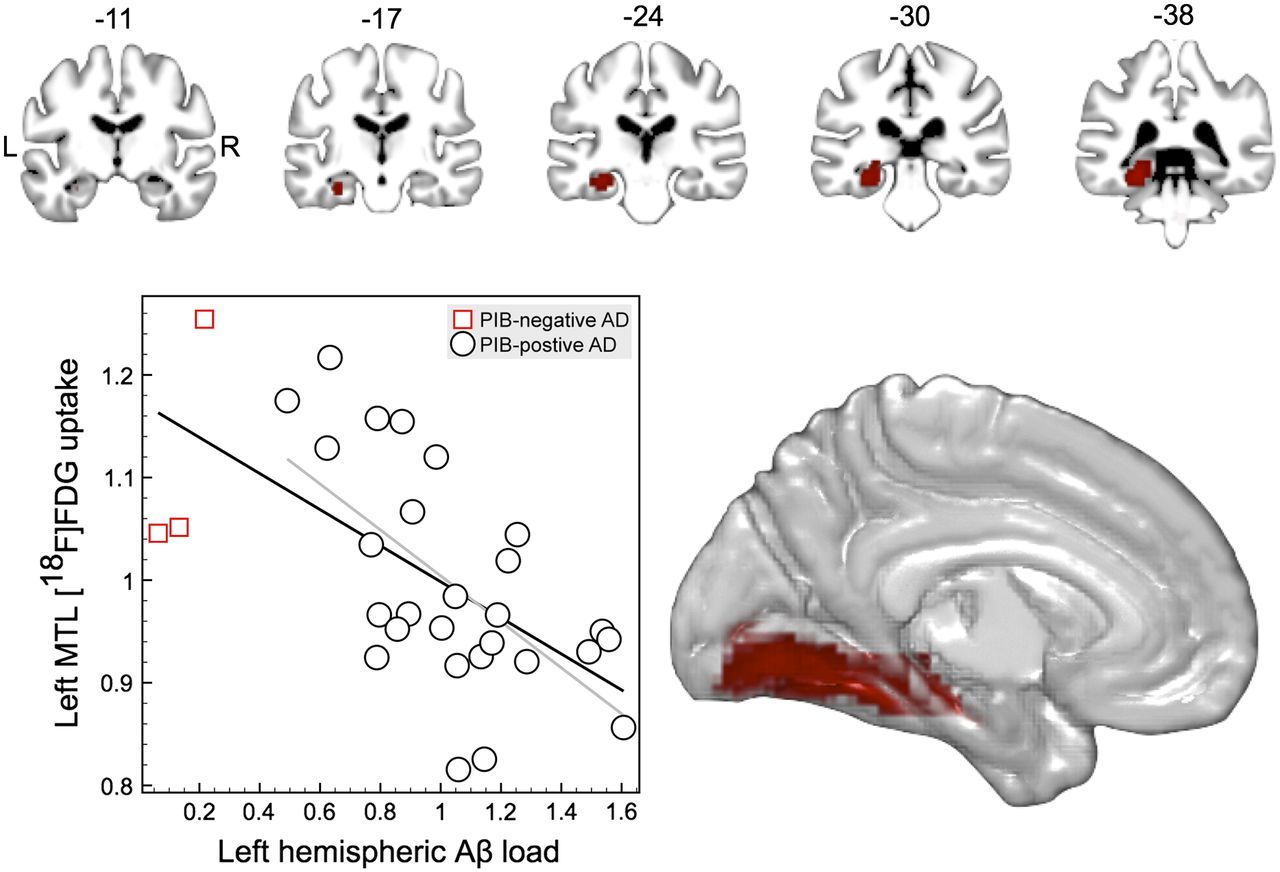
Left-hemispheric Aβ load predicted left posterior MTL glucose metabolism (P < 0.05, FWE-corrected at cluster level; P < 0.001 at voxel level; overlay on standard brain in MNI space, averaged over 82 healthy elderly subjects). Scatterplot depicting individual values of composite estimates of left-hemispheric neocortical Aβ load (BPND; x-axis; see “Methods” section for details) and normalized 18F-FDG uptake of left MTL (y-axis; peak MNI coordinates [−24 −43 −14 mm]), including linear fit for entire group of patients (black line; r2 = 0.378) and 11C-PIB–positive AD patients (gray line; r2 = 0.379).
Right-hemispheric Aβ load did not significantly predict regional glucose metabolism in any region. The aforementioned effect of Aβ load of the left hemisphere on left MTL metabolism was replicated in confirmatory analyses including Mini-Mental Status Examination scores as confounds, albeit weaker (z = 3.21; P < 0.001 at the voxel level; peak coordinates [−24 −43 −14]).
Relationship Between Aβ Load and Memory Performance
Partial correlation analyses revealed a significant negative correlation between Aβ load within the left hemisphere and verbal memory recall (r = −0.37, P < 0.05; Fig. 2), controlled for age and sex (Fig. 2).

Scatterplot depicting individual values of composite estimates of left-hemispheric neocortical Aβ load (BPND; x-axis; see “Methods” section for details) and verbal memory recall performance (y-axis; numbers of words recalled out of 10), including linear fit for entire group of patients (black line; partial correlation controlling for age and sex: r = −0.37, P < 0.05) and 11C-PIB–positive AD patients (gray line; r = −0.69, P < 0.01).
Right-hemispheric Aβ load was not significantly correlated with visuospatial memory recall, controlling for age and sex (r = 0.14, not significant).
Analyses in 11C-PIB–Positive Patients
Both imaging and behavioral data analyses were rerun excluding a subgroup of three 11C-PIB–negative patients from the former analyses who nevertheless fulfilled the diagnostic criteria for probable AD.
The relationship between left-hemispheric Aβ load and regional glucose metabolism of the left MTL was essentially replicated (z = 3.91; uncorrected P < 0.001 at the voxel level, FWE-corrected P < 0.05 at the cluster level; peak MTL voxel coordinates [−27 −43 −11 mm]; r2 = 0.49).
The correlation between verbal memory recall and left-hemispheric Aβ load in this subgroup markedly increased to an r of −0.69 (P < 0.01, controlled for age and sex; Fig. 2).
Relationship Between Memory Performance and Regional Glucose Metabolism
This analysis revealed no significant results at the a priori chosen threshold of P less than 0.001 (uncorrected) at the voxel level and P less than 0.05 (FWE-corrected) at the cluster level. Therefore, an exploratory, more lenient threshold of P less than 0.005 at the voxel level and cluster extent greater than 30 contiguous voxels was applied. 18F-FDG uptake was significantly related to verbal memory performance (Fig. 3) in 6 clusters: left anterior hippocampus (z = 2.92; peak voxel coordinates [−27 −10 −26]), bilateral posterior MTL (z = 3.07, [−24 −40 −5], and z = 3.55, [33 −43 −5]), bilateral precuneus and posterior cingulate cortex (PCC; z = 3.16, [3 −34 37]), right lingual gyrus (z = 3.52, [21 −76 4]), and right cerebellum (z = 3.28, [51 −67 −44]).

Regions that showed significant relationship between verbal memory performance and regional glucose metabolism (P < 0.005, k > 30). Overlay on standard brain in MNI space, averaged over 82 healthy elderly subjects. Scatterplot depicting individual values of normalized 18F-FDG uptake averaged over left MTL clusters (y-axis; peak MNI coordinates [−27 −10 −26 mm] and [−24 −40 −5]) and verbal memory recall performance (y-axis; numbers of words recalled out of 10), including linear fit for entire group of patients (black line; r2 = 0.24) and 11C-PIB–positive AD patients (gray line; r2 = 0.38).
The left posterior MTL region overlaps with the region that showed a significant relationship with Aβ load (see the “Results” section; Figs. 1 and 3).
DISCUSSION
We investigated the relationships between Aβ load and regional glucose metabolism and memory in a sample of AD patients with a wider range of disease stages, including early stages (Mini-Mental Status Examination score 27/30). In contrast to earlier studies, left- and right-hemispheric Aβ loads were calculated as separate variables and related to regional glucose metabolism and lateralized memory processes to increase the sensitivity of the analyses. Most importantly, evaluation of the predictive value of the Aβ load of each hemisphere on regional glucose metabolism permitted the identification of remote effects of hemispheric Aβ load on neuronal dysfunction. Finally, we used quantitative estimates of hemispheric Aβ load that were calculated from neocortical 11C-PIB BPND values gained from pharmacokinetic modeling of dynamic PET data. Unlike commonly used standardized uptake value ratios, estimates of BPND should be less biased by concomitant cerebral blood flow changes. Given the tight coupling between regional glucose metabolism and cerebral blood flow, the present approach should thus be the most suitable noninvasive Aβ load outcome measure available and ideal to investigate possible associations between Aβ load and neuronal functioning.
The magnitude of amyloid deposition in the left cerebral hemisphere was related to left MTL hypometabolism and verbal, but not visuospatial, memory deficits. Right-hemispheric Aβ load did not significantly predict regional hypometabolism. Aβ load in the right hemisphere was not associated with verbal memory (supplemental data; supplemental materials are available at http://jnm.snmjournals.org) or visuospatial memory deficits. Notably, the spatial resolution of 18F-FDG PET is limited, and spatial smoothing further degrades the spatial resolution. Therefore, MTL substructures (e.g., parahippocampal gyrus and hippocampus) have been subsumed under the term MTL in the present study.
Our findings strongly suggest that higher left-hemispheric neocortical Aβ load is associated with increasing left MTL dysfunction. This is supported by significant correlations between left-hemispheric Aβ load and left MTL metabolism (directly reflecting neuronal activity) and between left-hemispheric Aβ load and impairment of verbal episodic memory (a function typically subserved by left MTL structures). In line with the notion that Aβ deposits exert a detrimental, possibly remote, effect on MTL functioning, which in turn leads to memory deficits, we also detected clusters of significant correlation between regional glucose metabolism and verbal memory performance in the left MTL. These were located in direct proximity to or overlapped (in particular considering PET resolution) with the correlation cluster between left-hemispheric Aβ load and regional glucose metabolism.
Importantly, Aβ deposition in 11C-PIB–positive AD patients was greatest in characteristic neocortical regions not including the MTL, which is consistent with published histopathologic (3) and imaging data (6,29). The mechanism by which neocortical amyloid deposition leads to remote dysfunction of hippocampal archicortex and adjacent MTL neocortex is currently under debate. One hypothesis is that synaptic dysfunction due to oligomeric Aβ leads to tau pathology and dying-back of neurons connected to these areas of excess Aβ load. It has been demonstrated that neocortical Aβ plaques can exert a remote effect on MTL neurofibrillary tangle accumulation (30). This might explain the spatial separation of tau pathology, which is earliest and most profound in the MTL (31), from neocortical amyloid deposition (2,3). Dysregulation of axonal transport might be one mechanism underlying this spatial separation (2,30). Importantly, posterior midline cortices (precuneus and PCC) are among the regions with the highest Aβ load as observed in PET studies including the current study, and Aβ load of the PCC predicts conversion of MCI to AD, which is accompanied by increasing hippocampal atrophy (32). In an animal model, it has been shown that experimentally induced PCC dysfunction leads to functional disconnection with the hippocampus (33). The hippocampal/MTL region is strongly connected with posterior midline cortex via the descending cingulate bundle. Therefore, intrahemispheric fiber tracts, among them the cingulate bundle, may connect distant amyloid pathology with left hippocampal dysfunction.
In this study, we observed an association between left-hemispheric Aβ load and verbal memory deficits. Interestingly, this finding was not mirrored by an association between right-hemispheric Aβ load and visuospatial memory deficits, although it is widely accepted that verbal and visuospatial memory processes are highly lateralized in the brain (34). It has been pointed out that in AD the spatial distribution of amyloid deposition across the brain shows a remarkable resemblance to the default network of the brain. Furthermore, a causal link has been proposed between the high metabolism of default network regions and their increased vulnerability for amyloid pathology (35), consistent with the observation that greater neuronal activity drives local Aβ deposition (36). Interestingly, the default network has been described to be asymmetrically connected to the MTL, with a stronger connection to the left than the right hippocampus (37). This preponderance of functional coupling to the left hemisphere presumably reflects underlying structural connectivity and might thus explain a greater impact of neocortical Aβ on the left than the right MTL function. The impact of lateralized rather than global amyloid deposits on memory function has also been shown in nondemented elderly patients, where left-hemispheric temporal (but not right-hemispheric or global) Aβ deposition negatively correlated with memory scores (11). In a study of AD patients by Cohen et al. (38), Aβ load of a region of interest comprising the bilateral precuneus was inversely related to frontal and parietal 18F-FDG uptake, the latter showing a clear left-lateralized distribution. 18F-FDG uptake of the MTL, however, was not negatively correlated with Aβ load of the precuneus or any other region. This discrepancy to our findings might be due to the fact that metabolism in this study has been tested for correlations with several single regions but not the cumulated Aβ load of an entire hemisphere.
Moreover, the lateralized effect observed in the present study might in part be explained by an asymmetry of 11C-PIB binding, with right-hemispheric 11C-PIB binding (BPND; mean, 1.10; range, 0.65–1.57) being higher and less variable than left-hemispheric 11C-PIB binding (mean, 1.04; range, 0.49–1.61) in our group of 11C-PIB–positive AD patients. This may favor the detection of left-hemispheric correlation patterns. Whether this is a consistent pattern that also applies to other cohorts needs to be addressed in further studies.
The present study has several limitations: first, we evaluated data from patients whose dementia was clinically diagnosed. Histologic data were unavailable, leaving uncertainty about the pathology underlying the clinical syndrome. About 10% of clinically diagnosed AD patients in the current study were 11C-PIB–negative, consistent with reports from other studies (20). However, the main effect reported here also held true when only 11C-PIB–positive patients were considered. Furthermore, the use of biopsy or postmortem tissue would also allow for a more direct verification of the present results using in vitro techniques like histology, immunohistochemistry, and autoradiography for assessment of neuronal and synaptic integrity and Aβ and also tau pathology. Second, although all patients in this study received MR imaging or CT for diagnostics, uniform structural MR imaging data were not available, and thus partial-volume correction was not possible. An effect of Aβ deposition on MTL atrophy in our patients seems likely, as has been proposed by the cascade hypothesis of AD (1) and evidenced recently in MCI patients with 11C-PIB PET and voxel-based morphometry (39). However, regional hypometabolism measured by 18F-FDG PET is an especially sensitive marker for AD-related changes, and regional hypometabolism precedes and exceeds atrophy in dementia (40). Third, the number of patients included in the present study is rather small. Thus, replication of the present results in a larger patient population is warranted (e.g., using the Alzheimer Disease Neuroimaging Initiative database).
CONCLUSION
We demonstrated an association between the left-hemispheric Aβ load and impairment of the left MTL in AD at 2 different levels: regional hypometabolism and verbal memory. Our findings suggest that neocortical amyloid deposition is connected to or even drives neuronal dysfunction and neurodegeneration of the MTL, which is associated with impaired episodic memory processing as a clinical core symptom of AD.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked \x{201c}advertisement\x{201d} in accordance with 18 USC section 1734. Michael Hüll has received speaker honoraria from Pfizer Inc., Merz Pharmaceuticals, and Novartis and served as PI in clinical trials for AD sponsored from Pfizer, Medivation, and NOSCIRA. Philipp T. Meyer received research grants from GE Healthcare and payments for lectures by Siemens AG and consultancy by Bayer-Schering AG. Wolfgang A. Weber received research support from Bayer HealthCare AG and Philips Medical Systems. Lars Frings and Timo S. Spehl report no disclosures. No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Oct. 7, 2013.
- © 2013 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication January 21, 2013.
- Accepted for publication May 17, 2013.





{kind=link}
{kind=link}
{kind=link}